Rfam: Family: tRNA (RF00005) (original) (raw)
2215 structures 14413 species 5336929 sequences
Description: tRNA
Loading page components…
Summary
Clan
This family is a member of clan (CL00001), which contains the following 7 members:
alpha_tmRNA beta_tmRNA cyano_tmRNA mt-tmRNA tmRNA tRNA tRNA-Sec
Wikipedia annotation Edit Wikipedia article
The Rfam group coordinates the annotation of Rfam families in Wikipedia. This family is described by a Wikipedia entryTransfer RNA. More...
The Wikipedia text that you see displayed on our web site was retrieved from Wikipedia. This means that the information we display is a copy of the information from the Wikipedia database. The button above ("Edit wikipedia entry") takes you to the edit page for the article directly within Wikipedia.
Before you edit for the first time
Wikipedia is a free, online encyclopedia. Although anyone can edit or contribute to an article, Wikipedia has some strong editing guidelines and policies, which promote the Wikipedia standard of style and etiquette. Your edits and contributions are more likely to be accepted (and remain) if they are in accordance with this policy.
You should take a few minutes to view the following pages:
Things you should know
How your contribution will be recorded
Anyone can edit a Wikipedia entry. You can do this either as a new user or you can register with Wikipedia and log on. When you click on the "Edit Wikipedia entry" button, your browser will direct you to the edit page for this entry in Wikipedia. If you are a registered user and currently logged in, your changes will be recorded under your Wikipedia user name. However, if you are not a registered user or are not logged on, your changes will be logged under your computer’s IP address. This has two main implications. Firstly, as a registered Wikipedia user your edits are more likely seen as valuable contribution (although all edits are open to community scrutiny regardless). Secondly, if you edit under an IP address you may be sharing this IP address with other users. If your IP address has previously been blocked (due to being flagged as a source of 'vandalism') your edits will also be blocked. You can find more information on this and creating a user account at Wikipedia.
If you have problems editing a particular page, contact us atrfam-help@ebi.ac.uk and we will try to help.
Information we would like to see added
We would value contributions that are referenced directly to the primary literature. Information on structure and function will be especially valuable.
Adding references is explained by this Wikipedia how-to article.
For a good example of what is possible in wikipedia, look at the Hammerhead Ribozyme entry.
Does Rfam agree with the content of the Wikipedia entry ?
Rfam has chosen to create Wikipedia entries for all of our RNA families and to open them up to community annotation. While the original Wikipedia article that we import was (in most cases) generated from Rfam annotations, the Wikipedia article you see now may bear little resemblance to that original text. The Wikipedia community does monitor edits to try to ensure that (a) the quality of article annotation increases, and (b) vandalism is very quickly dealt with. However, we would like to emphasise that Rfam does not curate the Wikipedia entries and we cannot guarantee the accuracy of the information on the Wikipedia page.
Contact us
If you have problems editing or experience problems with these pages please contact us.
If you are interested in contributing to a wide range of articles relating to RNA, see the Wikiproject RNA page.
This page is based on a Wikipedia article. The text is available under the Creative Commons Attribution/Share-Alike License.
Sequences
Alignment
There are various ways to view or download the seed alignments that we store. You can use a sequence viewer to look at them, or you can look at a plain text version of the sequence in a variety of different formats. More...
Viewing
You can choose from two different sequence viewers:
a Java applet developed at the University of Dundee. You will need Java installed before running jalview
HTML
an HTML page showing the seed alignment in blocks. We do not store separate alignments with species or "name/start-end" labels, but you can switch between these different labels within the block viewer
Downloading
You can download (or view in your browser) a text representation of an Rfam seed alignment in various formats:
- Stockholm
- Pfam
- Gapped FASTA
- Ungapped FASTA
View options
You can view Rfam seed alignments in your browser in various ways. Choose the viewer that you want to use and click the "View" button to show the alignment in a pop-up window.
Formatting options
You can view or download Rfam seed alignments in several formats. Check either the "download" button, to save the formatted alignment, or "view", to see it in your browser window, and click "Generate".
Download
Download agzip-compressed, Stockholm-format file containing the seed alignment for this family. You may find RALEE useful when viewing sequence alignments.
Submit a new alignment
We're happy receive updated seed alignments for new or existing families. Submit your new alignment and we'll take a look.
Secondary structure
This section shows a variety of different secondary structure representations for this family. More...
In this page you can view static images showing the secondary structure of this family using a variety of colouring schemes:
Conservation (cons): this plot colours each character by how well conserved it is. A site with 100% sequence conservation is coloured red, 0% is violet.
Covariation (cov): this plot colours each base-pair according to how much the corresponding nucleotides are co-varying. A base-pair position at which every pair of nucleotides is co-variant with respect to every other pair in the alignment gets a score of 2 and is coloured red. Conversely, a base-pair position at every pair is anti-co-variant with respect to every other pair (e.g. lots of mutations to non-canonical pairs) gets a score of -2 and is coloured violet. Further information on this metric can be found in thisdocument.
Sequence entropy (ent): this plot colours each character by how under- or over-represented the residues at the site are. Sites where one or more nucleotides are over-represented while the other nucleotides are either non-existent or near the background frequencies, receive positive scores; sites where all the nucleotides are under-represented receive negative scores. Further information on this metric can be found in this document.
Fraction of canonical basepairs (fcbp): this plot colours each base-pair by the percentage of canonical basepairs (A:U, C:G, G:U) which are found in the corresponding position in the alignment. A pair of sites with 100% canonical pairs is coloured red, a site with 0% is violet.
Maximum parse of the covariance model (maxcm): this plot takes the covariance model for the family and generates the sequence with the maximum possible score for that model. Each character is coloured by how many bits it contributes to the total score.
Sequence: for most of the above cases, the representative sequence used for the backbone is the most informative sequence (MIS). Any residue that has a higher frequency than than the background frequency is projected into the IUPAC redundancy codes.
Normal: this plot simply colours each stem loop
R-chie (rchie): arc diagrams showing secondary structure, calculated using theR-chie package. The consensus secondary structure is visualized as arc diagrams on top of each diagram, where a basepair in an arc, connect two columns of the block of sequences below. The block of sequences below represent the multiple sequence alignment of the Rfam seed, where each sequence is a horizontal strip. Sequences in the alignments are ordered so sequences that best fit the structure are on top, and those that do not fit as well are towards the bottom. For seed alignments for over 500 sequences, 500 random sequences were chosen. Rfam entries without sturcture have a blank plot. Colour information can be found on the R-chieFAQ.
You can also view the secondary structure in the VARNA applet. The applet is shown in a separate pop-up window.
Acknowledgements
The bulk of the code for generating these graphics was kindly supplied by Andreas Gruber and Ivo Hofacker. The statistics were implemented by Rfam.
The VARNA applet is developed byYann Ponty et al:
The R-chie arc diagrams were calculated usingR-chie:
You can view the secondary structure of the family using theVARNA applet. You can see more information about VARNA iselfhere.
Current Rfam structure
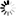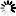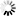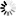 Loading...
Loading...
R-scape optimised structure
Loading...
- Colours
- Statistically significant basepair with covariation
- 97% conserved nucleotide
- 90% conserved nucleotide
- 75% conserved nucleotide
- 50% conserved nucleotide
- Nucleotides
- R: A or G
- Y: C or U
Tip: The diagrams are interactive:
you can pan and zoom to see more details
or hover over nucleotides and basepairs.
R-scape is a method for testing whether covariation analysis supports the presence of a conserved RNA secondary structure. This page shows R-scape analysis of the secondary structure from the Rfam seed alignment and a new structure with covariation support that is compatible with the same alignment.
To find out more about the method, see the R-scape paper by_Rivas et al., 2016_. The structures are visualised using R2R.
Move your mouse over the image to show details and click to show full image.

- Arc colours
- 100% canonical basepair
- 50%
- 0%
- Nucleotide colours
- Valid basepairing
- Two-sided covariation
- One-sided covariation
- Invalid
- Unpaired
- Gap
- Ambiguous
Species distribution
We are unable to display the sunburst tree for this family.
- There are too many sequences in this alignment
The tree shows the occurrence of this RNA across different species. More...
Species trees
For the majority of our families we provide an interactive tree representation, which allows you to select specific nodes in the tree and view the selected sequences as an alignment.
Unfortunately we have found that there are problems viewing the interactive tree when the it becomes larger than a certain limit. Furthermore, we have found that Internet Explorer can become unresponsive when viewing some trees, regardless of their size. We therefore show a text representation of the species tree when the size is above a certain limit or if you are using Internet Explorer to view the site.
If you are using IE you can still load the interactive tree by clicking the "Generate interactive tree" button, but please be aware of the potential problems that the interactive species tree can cause.
Interactive tree
For all of the sequence regions (RNA annotations) in a full alignment, we count the total number that are found in the alignment. This total is shown in the purple box.
We also count the number of unique sequences on which each RNA is found, which is shown in green.Note that an RNA annotation may appear multiple times on the same sequence, leading to the difference between these two numbers (think of repeats like tRNA where the same RNA is found in tandem along a single sequence).
Finally, we group sequences from the same organism according to the NCBI code that is assigned byUniProt, allowing us to count the number of distinct sequences on which the RNA is found. This value is shown in the pink boxes.
We use the NCBI species tree to group organisms according to their taxonomy and this forms the structure of the displayed tree.Note that in some cases the trees are too large (have too many nodes) to allow us to build an interactive tree. In these few cases if you do really need to see a representation of the tree for this entry, please contact us and we will be happy to discuss ways to generate it for you.
You can use the tree controls to manipulate how the interactive tree is displayed:
- show/hide the summary boxes
- highlight species that are represented in the seed alignment
- expand/collapse the tree or expand it to a given depth
- select a sub-tree or a set of species within the tree and view them graphically or as an alignment
- save a plain text representation of the tree
Loading...
Please note: for large trees this can take some time. While the tree is loading, you can safely switch away from this tab but if you browse away from the family page entirely, the tree will not be loaded.
Trees
This page displays the predicted phylogenetic tree for the alignment. More...
These trees were generated using either a maximum likelihood approach or neighbour-joining. If the number of sequences in the alignment was less than or equal to 64 then the maximum likelihood approach of Rivas and Eddy was used [1]. For families with more than 64 sequences in the alignment the neighbour-joining approach with F84 distances as implemented in phylip was used [2].
[2] PHYLIP (Phylogeny Inference Package) version 3.6: J. Felsenstein Distributed by the author Department of Genome Sciences, University of Washington, Seattle.
Note: You can also download the data file for theseed tree.
We do not have tree information for this family. This is most likely due to the size of the family and the number of species covered. For very large families it is too computationally expensive to calculate trees and the resulting tree images would be too large to display in a browser.
Structures
For those sequences which have a structure in theProtein DataBank, we generate a mapping between EMBL, PDB and Rfam coordinate systems. The table below shows the structures on which the tRNA family has been found.
Loading structure mapping...
Motif matches
There are 1 motifs which match this family.
This section shows the Rfam motifs that match sequences within the seed alignment of this family. Users should be aware that the motifs are structural constructs and do not necessarily conform to taxonomic boundaries in the way that Rfam families do. More...
Motifs in this context are defined as recurring RNA sequences and/or secondary structures found within larger structures that can be modeled by either a covariance model (CM) or a profile HMM. The motifs models come from release 0.3 of the RMfam database available atgithub.
To annotate the family with a motif model, the seed sequence was first filtered using a 0.9 fraction identity cut-off. The filtered seed was then scanned using Infernal cmscan (v1.1) with a concatenated CM file containing each of the motifs. Significance of hits between a seed sequence and the CM was based on a gathering threshold that was individually set for each motif. Only motifs where more than two and at least 10% of seed sequences scored higher than the gathering threshold were included for the next stage of processing. These subsets of motifs were then rescanned against the entire (non-filtered) seed to generate matches.
Number of Hits: the number of sequences in the family seed that score above the gathering threshold from motif.
Fraction of Hits: the fraction of sequences in the family seed that score above the gathering threshold from motif.
Sum of Bits: the sum of the bit scores of matches between the motif and the family seed sequence.
Image: plot illustrating where on the consensus secondary structure matches occur between seed sequences and the motif model.
| Original order | Motif Accession | Motif Description | Number of Hits | Fraction of Hits | Sum of Bits | Image |
|---|---|---|---|---|---|---|
| 7 | RM00024 | T-loop | 567 | 0.594 | 8155.1 | |
References
This section shows the database cross-references that we have for this Rfam family.
Literature references
- Hou YM Trends Biochem Sci 1993;18:362-364. The tertiary structure of tRNA and the development of the genetic code. PUBMED:8256282
- Lowe TM, Eddy SR Nucleic Acids Res 1997;25:955-964. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. PUBMED:9023104
External database links
| Gene Ontology: | GO:0030533 (triplet codon-amino acid adaptor activity); |
|---|---|
| Sequence Ontology: | SO:0000253 (tRNA); |
Curation and family details
This section shows the detailed information about the Rfam family. We're happy to receive updated or improved alignments for new or existing families. Submit your new alignment and we'll take a look.
Curation
| Seed source | Eddy SR |
|---|---|
| Structure source | Published; PMID:8256282 |
| Type | Gene; tRNA; |
| Author | Eddy SR , Griffiths-Jones SR, Mifsud W , Griffiths-Jones SR, Mifsud W |
| Alignment details | Alignment Number ofsequences full 5,335,975 seed 954 |
Model information
| Build commands | cmbuild -F CM SEED cmcalibrate --mpi CM |
|---|---|
| Search command | cmsearch --cpu 4 --verbose --nohmmonly -T 22.00 -Z 2958934 CM SEQDB |
| Gathering cutoff | 29.0 |
| Trusted cutoff | 29.0 |
| Noise cutoff | 28.9 |
| Covariance model | Download |