Restrictive Cardiomyopathy: Practice Essentials, Background, Pathophysiology (original) (raw)
Overview
Practice Essentials
Restrictive cardiomyopathy (RCM) is a rare disease of the myocardium and is the least common of the three clinically recognized and described cardiomyopathies. [1, 2] It is characterized by diastolic dysfunction with restrictive ventricular physiology, whereas systolic function often remains normal. Atrial enlargement occurs due to impaired ventricular filling during diastole, but the volume and wall thickness of the ventricles are usually normal. RCM accounts for approximately 5% of all cases of diagnosed cardiomyopathies. [3]
Signs and symptoms
Symptoms may include the following:
- Gradually worsening shortness of breath
- Progressive exercise intolerance
- Orthopnea
- Fatigue
- Weight loss, cardiac cachexia
- Paroxysmal nocturnal dyspnea
- Abdominal discomfort or liver tenderness
- Chest pain, primarily in patients with amyloidosis or due to angina
- Palpitations
Physical examination
Search for extracardiac manifestations of a systemic disorder that may cause secondary restrictive cardiomyopathy (eg, hemochromatosis, amyloidosis, sarcoidosis, or scleroderma).
General examination findings:
- Increased jugular venous pressure
- Decreased pulse volume
- Ascites and pitting edema of the lower extremities
- Hepatomegaly due to fluid or amyloid infiltration
- Easy bruising, periorbital purpura, macroglossia, and other systemic findings (eg, carpal tunnel syndrome) may be evidence of amyloidosis
Cardiovascular system examination findings:
- Normal S1 and S2 heart sounds, with a normal S2 split
- Loud early diastolic filling sound (S3)
- A fourth heart sound (S4) is almost never present
- Murmurs due to mitral and tricuspid valve regurgitation (usually not hemodynamically significant)
Respiratory system examination findings:
- Breath sounds are often decreased due to pleural effusions, frequently bilateral and large in amyloidosis
- Crepitations or rales are rarely heard
See Clinical Presentation for more detail.
Diagnosis
Establishing the diagnosis of RCM and excluding constrictive pericarditis are imperative. The workup in a patient with suspected restrictive cardiomyopathy may include the following:
- Complete blood cell (CBC) count
- Blood gas analysis
- Serum electrolyte, blood urea nitrogen (BUN), and creatinine levels
- Liver function profile
- Serum iron concentrations and other possible indicators of hemochromatosis
- Serum brain natriuretic peptide (BNP) levels
- Radiography
- Angiography
- Echocardiography
- Cardiac catheterization
- Electrocardiography
- Radionuclide imaging
- Cardiovascular magnetic resonance imaging (CMRI)
- Computed tomography (CT) scanning to exclude diagnosis of restriction
- Ventricular biopsy
- Liver biopsy for diagnosis of hemochromatosis
See Workup for more detail.
Management
RCM has no specific treatment. However, therapies directed at individual causes of RCM have been proven to be effective.
Pharmacologic therapy may include:
- Beta blockers
- Amiodarone
- Cardioselective calcium channel blockers
- Diuretics
- Anticoagulants for patients with atrial fibrillation
- Digoxin (use with caution)
- Melphalan (for antiplasma cell therapy in systemic amyloidosis)
- Chemotherapy (in amyloidosis)
- Corticosteroids, cytotoxic agents (eg, hydroxyurea), and interferon (in Loeffler endocarditis)
- Chelation therapy or therapeutic phlebotomy (in hemochromatosis)
Other treatments:
- Pacemaker implantation
- Endomyocardectomy
- Cardiac transplantation
- Novel therapies (eg, RNA interference or gene silencing molecules that target abnormal protein production in familial amyloidosis)
See Treatment for more detail.
![]()
Background
Restrictive cardiomyopathy (RCM) is a rare disease of the myocardium and is the least common of the three clinically recognized and described cardiomyopathies. [1, 2] It is characterized by diastolic dysfunction with restrictive ventricular physiology, whereas systolic function often remains normal. Atrial enlargement occurs due to impaired ventricular filling during diastole, but the volume and wall thickness of the ventricles are usually normal. RCM accounts for approximately 5% of all cases of diagnosed cardiomyopathies. [3]
RCM may be idiopathic or secondary to other diseases (ie, amyloidosis and endomyocardial disease with or without hypereosinophilia). The course of RCM varies, depending on the pathology and treatment. RCM has been found to be a significant cause of heart failure with preserved ejection fraction, although there is a high clinical overlap between RCM and other forms of heart failure. [4] RCM therefore presents a diagnostic challenge, and multiple modalities are usually required to make a final diagnosis.
See the images below.
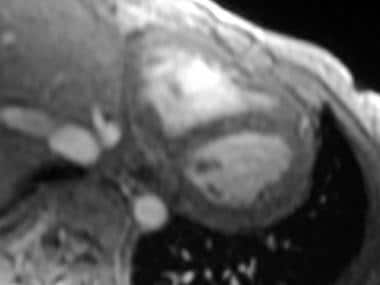
Restrictive cardiomyopathy. Axial double inversion-recovery magnetic resonance image of the heart in a 30-year-old woman with sarcoidosis demonstrates a normal pericardium.
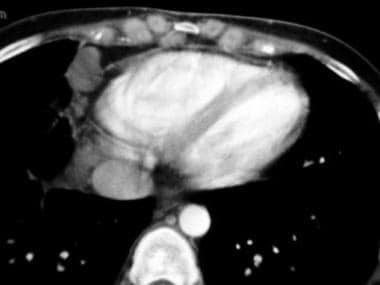
Restrictive cardiomyopathy. Axial contrast-enhanced computed tomography scan through the heart (same patient as in the previous image) shows a thin pericardium without calcification. Note the cardiophrenic and internal mammary lymph nodes. The patient had extensive mediastinal and hilar adenopathy, as well as interstitial lung changes.
![]()
Pathophysiology
Restrictive cardiomyopathy (RCM) can be idiopathic or secondary to a heart muscle disease that manifests as restrictive physiology. [1, 5] Both inherited and acquired forms of the disease exist and affect men and women equally. Increased stiffness of the myocardium causes ventricular pressures to rise precipitously with small increases in volume. Thus, accentuated filling occurs in early diastole and terminates abruptly at the end of the rapid filling phase. When pressure tracings are taken at this point, they show a characteristic diastolic “dip-and-plateau” or “square-root” pattern, both similar to constrictive pericarditis. [6]
Patients typically have reduced compliance (increased diastolic stiffness), and the left ventricle cannot fill adequately at normal filling pressures. Reduced left ventricular filling volume leads to a reduced cardiac output. Early in the disease process, systolic function usually remains normal. Wall thickness may be increased in cases of infiltrative processes such as amyloidosis, but the increase is usually not as pronounced as that observed in hypertrophic cardiomyopathy.
As the disease progresses, a variable reduction in systolic function may develop with symptoms of reduced cardiac output, such as fatigue and lethargy, becoming evident. Increased filling pressures can manifest as pulmonary and systemic congestion. RCM affects both ventricles and therefore may cause signs and symptoms of both left-sided and right-sided heart failure. Some patients may have complete heart block as a consequence of fibrosis encasing the sinoatrial or the atrioventricular nodes.
RCM is one of the cardiomyopathies that is known to have a genetic cause, although only a few RCM-causing mutations have been described. [7, 8, 9] The Heart Failure Society of America (HFSA) issued updated guidelines on the genetic evaluation of cardiomyopathy in 2010. [10]
Based on pathology findings, RCM can further be classified as obliterative (ie, thrombus-filled ventricles) or nonobliterative. Idiopathic (primary) RCM is nonobliterative, as progressive fibrosis of the myocardium occurs but no thrombus forms. This entity also is said to lack specific histopathologic changes.
Obliterative RCM is very rare. It may result from the end stage of the eosinophilic syndromes, in which an intracavitary thrombus fills the left ventricular apex and hampers the filling of the ventricles. The fibrosis of the endocardium may extend to involve the atrioventricular valves and cause regurgitation. Two forms of endomyocardial fibrosis (EMF) exist—an active inflammatory eosinophilia and chronic EMF.
![]()
Etiology
Restrictive cardiomyopathy (RCM) may be caused by various local and systemic disorders; many of them are rare and unlikely to be observed in the United States.
According to World Health Organization (WHO) guidelines, the term “cardiomyopathy” refers to diseases of the myocardium that are idiopathic (ie, primary cardiomyopathies). However, secondary infiltrative myocardial diseases, which are actually cardiac manifestations of systemic diseases, often are grouped together with cardiomyopathies. [11]
The etiologies of RCM may be grouped into broad categories as follows:
The specific pathophysiologies of the more common causes of RCM will be described in detail below.
Primary (idiopathic) RCM
Both genetic and sporadic cases of primary (idiopathic) RCM have been described. This is a rare condition that can present in children and adults, [13] and males and females are affected equally. However, the prognosis appears to be worse in children than in adults. Genetic cases show autosomal dominant inheritance with incomplete penetrance. The mutation appears to occur in the genes encoding sarcomeric proteins, including troponin I, troponin T, alpha cardiac actin, and beta-myosin heavy chain. [13] A history of familial RCM is reported in approximately 30% of RCM cases. [3]
A subset of patients has heart muscle disease of unknown cause that is manifested by heart failure and restrictive hemodynamics, but without significant ventricular hypertrophy, endocardial thickening or fibrosis, associated eosinophilia, or other diagnostically distinct histopathologic changes.
Children require relatively high filling pressures for maintenance of systolic output, and the therapeutic margin between volume depletion (leading to low output) and volume overload (leading to congestive heart failure) is narrow. An observational study suggests that poor left ventricular function may be a hallmark for pediatric restrictive cardiomyopathy even in the presence of normal diastolic parameters. [14]
In addition to the presenting symptoms of right- and left-side heart failure, as many as one third of patients with idiopathic RCM may present with thromboembolic complications. Pathologically, these patients have strikingly dilated atria, which may account for the increased cardiothoracic ratio on chest radiography. Echocardiography shows bilateral atrial enlargement with normal ventricular size but significant diffuse left ventricular hypertrophy, especially with amyloidosis. Histologic features include interstitial fibrosis, which is minimal in some patients and extensive in others.
Eosinophilic cardiomyopathy (Loeffler endocarditis) and EMF
EMF is the most common global cause of RCM, affecting an estimated 12 million people worldwide. This condition is observed in equatorial Africa and, less frequently, in tropical and subtropical Asia and South America.
Severe prolonged eosinophilia from any cause (eg, allergic, autoimmune, parasitic, leukemic, or idiopathic) can lead to eosinophilic infiltration of the myocardium. Eosinophilic cardiomyopathy, also known as Loeffler endocarditis, begins with an acute inflammatory phase characterized by fever and pancarditis. Left ventricular and right ventricular thrombus formation occurs in the intermediate phase and, after months to years, the final stage includes development of endocardial fibrosis. The intracytoplasmic granular content of activated eosinophils is believed to be responsible for the toxic damage to the heart. [13]
EMF was originally believed to be the end stage of eosinophilic endomyocarditis. However, chronic EMF is currently considered a separate entity because it does not exhibit eosinophilia. EMF demonstrates pathology that is similar to that of Loeffler endocarditis.
Both EMF and Loeffler endocarditis are categorized as types of obliterative RCM. Intraventricular thrombus formation leads to obliteration of the ventricular cavity in the late stages. Echocardiography may show endomyocardial thickening, ventricular apical obliteration, and tethering of mitral and tricuspid leaflets. [13]
The prognosis is poor for patients with diffuse involvement of the heart, but localized lesions involving the valves are amenable to surgical repair or removal and replacement.
Secondary RCM
Infiltrative cardiomyopathy
Infiltrative cardiomyopathies are characterized by deposition of abnormal substances (ie, amyloid proteins, noncaseating granulomas, iron) within the heart tissue. Infiltration causes the ventricular walls to stiffen, leading to diastolic dysfunction. Disease occurs in a wide variety of age groups and, given the systemic nature of the underlying disease, extracardiac manifestations are common. Restrictive physiology predominates in the early stages, causing conduction abnormalities and diastolic heart failure. Adverse remodeling may lead to systolic dysfunction and ventricular arrhythmias in advanced cases. [15]
Confirmatory evidence of infiltrative cardiomyopathy is often obtained by endomyocardial biopsy, echocardiography, or cardiac magnetic resonance imaging (CMRI). Depending on the etiology and extent of involvement, medications, device therapy, and transplantation can be effective, although treatment is largely supportive in many cases. [15]
Amyloidosis
Amyloidosis is the most common cause of RCM in the United States. Due to advancements in noninvasive diagnostic modalities, relatively recent studies have shown that there may be a higher prevalence of amyloidosis among elder patients with heart failure with preserved ejection fraction than previously recognized. [4]
Amyloidosis is characterized by the multisystem deposition of proteins known as amyloid fibrils, and it typically presents as a systemic disorder, with infiltration of the liver, kidneys, bowel, nerves, skin, and tongue. [13] Cardiac involvement is common and the major source of associated morbidity and mortality. The myocardial wall thickens and becomes firm, rubbery, and noncompliant as amyloid accumulates in tissues. These changes lead to abnormalities of contractility, conduction, and coronary blood flow. Interestingly, amyloid deposition in the bundle branches is rare. Biventricular diastolic dysfunction causes intracardiac pressures to rise, and it may progress to systolic dysfunction in advanced disease. [15] The heart typically does not collapse when removed from the chest during autopsy.
Amyloidosis is classified into the following four major clinical types based on the composition of amyloid protein:
- Primary or amyloid light-chain (AL): This is the most common form, often associated with multiple myeloma; prognosis is poor, with a median 1-year survival from diagnosis.
- Secondary amyloidosis or amyloid A (AA): This type is secondary to chronic diseases, especially inflammatory conditions.
- Senile amyloidosis or wild-type transthyretin (wt-TTR) amyloidosis: This form is seen in 25-36% of patients older than 80 years; it is caused by deposition of wt-TTR. [16] The median survival is 6 years.
- Familial amyloidosis or hereditary mutant TTR (m-TTR)/hereditary transthyretin-derived (ATTR) amyloidosis: This type is a systemic autosomal dominant disorder due to tissue deposition of various proteins. Cardiac involvement is rare.
The cardiac involvement in primary amyloidosis is most commonly associated with restrictive physiology.
In the early stages of the disease, typical restrictive hemodynamics may not be evident; however, in more advanced cases, typical restrictive hemodynamics are more likely. Restrictive diastolic dynamics strongly predict cardiac death in patients with amyloidosis. Studies have shown that patients with elevated cardiac biomarkers such as troponin (Tn) and B-type natriuretic peptide (BNP) have a worse prognosis. [15]
On histologic examination, amyloid may deposit within any part of the heart, including the myocardium, vessels, endocardium, valves, epicardium, and parietal pericardium. The ventricular walls are typically thickened, sometimes with disproportionate septal thickening, and may mimic the appearance of hypertrophic cardiomyopathy. Atrial dilatation develops as a consequence of increased ventricular filling pressures and restrictive physiology. [17] Involvement of the valves may create regurgitant lesions, but a hemodynamically and clinically significant degree of regurgitation is unusual.
Cardiac biopsy is needed to confirm the diagnosis if doubt remains after noninvasive tests. Classically, the deposition of insoluble fibrillary protein displays as apple-green birefringence under polarized light microscopy with Congo Red staining. [13]
Classic two-dimensional echocardiography findings include an increased left ventricular and right ventricular wall thickness, normal or small left ventricular cavity size with preserved ejection fraction, and biatrial enlargement. Up to one third of patients can present with normal left ventricular wall size. Pericardial effusion and thickening of both valves and papillary muscles is common. A granular and speckled appearance of the ventricular myocardium is suggestive of amyloidosis, but it is no longer considered specific. [15]
Other less common forms of infiltrative RCM include:
- Sarcoidosis: Presence of noncaseating granulomas (cardiac involvement: 25-30%)
- Hemochromatosis and iron overload cardiomyopathy: Excess deposition of iron
- Fabry disease: Lysosomal storage disorder with accumulation of glycosphingolipid
- Danon disease: Deficiency in lysosome-associated membrane protein 2 (LAMP2)
- Friedreich ataxia: Mutation of frataxin gene (FXN)
Treatment-induced RCM
Postirradiation fibrosis
Radiation-induced myocardial and endocardial fibrosis is a cause of noninfiltrative RCM. Fibrosis causes endothelial cell damage and subsequent microvascular dysfunction. An increase in total collagen concentration leads to decreased distensibility of the ventricular tissue. Radiation affects the coronary vessels, valves, and pericardium. [13] This complication of radiotherapy, as with pericardial constriction, is evident several years after treatment. Differentiating between constriction and restriction may be particularly difficult in these patients because the two conditions may coexist. Echocardiography may show normal left ventricular wall thickness with abnormal left ventricular filling, valvular calcification, and pericardial constriction. [13]
Drug induced
Drug-induced RCM is a rare disorder that has been described with long-term use of the antimalarial medications chloroquine and hydroxychloroquine. Common findings include conduction abnormalities and valvular thickening. [13]
![]()
Epidemiology
Idiopathic restrictive cardiomyopathy (RCM) is observed primarily in the United States. Loeffler endocarditis is common in the temperate zone, whereas chronic endomyocardial fibrosis (EMF) is observed exclusively in tropical and subtropical Africa, Asia, and South America. EMF occurs most commonly in children and young adults in Uganda and Nigeria [18] ; this condition may account for up to one fourth of deaths due to cardiac disease in those areas.
![]()
Prognosis
Restrictive cardiomyopathy (RCM) has the poorest prognosis among all types of heart muscle diseases, with 2- and 5- year mortality rates of 50% and 70%, respectively, and the highest rate of sudden cardiac death. Due to restrictive physiology, patients with RCM ultimately develop heart failure and pulmonary hypertension. [3]
The course of RCM varies depending on the pathology, and treatment is often unsatisfactory. The prognosis is generally poor in the adult population, as RCM shows progressive deterioration. The natural history of RCM is especially poor in children with heart failure. Adults experience a prolonged course of heart failure and may have complications of cardiac cirrhosis and thromboembolism. Patients whose condition is refractory to supportive therapy usually die of low-output cardiac failure unless cardiac transplantation is an option.
Complications
Complications of RCM may include the following:
- Thromboembolism
- Dysrhythmias
- Cardiac cirrhosis
- Progressive deterioration of cardiac function
![]()
- Kushwaha SS, Fallon JT, Fuster V. Restrictive cardiomyopathy. N Engl J Med. 1997 Jan 23. 336 (4):267-76. [QxMD MEDLINE Link].
- Merlo M, Abate E, Pinamonti B, et al. Restrictive cardiomyopathy: clinical assessment and imaging in diagnosis and patient management. In: Pinamonti B, Sinagra Gianfranco, eds. Clinical Echocardiography and Other Imaging Techniques in Cardiomyopathies. Cham, Switzerland: Springer; 2014. 185-206. [Full Text].
- Huby AC, Mendsaikhan U, Takagi K, et al. Disturbance in Z-disk mechanosensitive proteins induced by a persistent mutant myopalladin causes familial restrictive cardiomyopathy. J Am Coll Cardiol. 2014 Dec 30. 64 (25):2765-76. [QxMD MEDLINE Link].
- Gonzalez-Lopez E, Gallego-Delgado M, Guzzo-Merello G, et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J. 2015 Oct 7. 36 (38):2585-94. [QxMD MEDLINE Link].
- Schlant RC, Alexander RW, eds. The Heart. New York, NY: McGraw-Hill; 1994. 1637-45.
- Higano ST, Azrak E, Tahirkheli NK, Kern MJ. Hemodynamic rounds series II: hemodynamics of constrictive physiology: influence of respiratory dynamics on ventricular pressures. Catheter Cardiovasc Interv. 1999 Apr. 46 (4):473-86. [QxMD MEDLINE Link].
- Kostareva A, Kiselev A, Gudkova A, et al. Genetic spectrum of idiopathic restrictive cardiomyopathy uncovered by next-generation sequencing. PLoS One. 2016. 11 (9):e0163362. [QxMD MEDLINE Link].
- Towbin JA. Inherited cardiomyopathies. Circ J. 2014. 78 (10):2347-56. [QxMD MEDLINE Link].
- Peled Y, Gramlich M, Yoskovitz G, et al. Titin mutation in familial restrictive cardiomyopathy. Int J Cardiol. 2014 Jan 15. 171 (1):24-30. [QxMD MEDLINE Link].
- [Guideline] Lindenfeld J, Albert NM, Boehmer JP, et al, for the Heart Failure Society of America. HFSA 2010 Comprehensive Heart Failure Practice Guideline. J Card Fail. 2010 Jun. 16(6):e1-194. [QxMD MEDLINE Link].
- Davies MJ, Mann JM. Systemic pathology. The Cardiovascular System. Vol 10. New York, NY: Churchill Livingstone; 1995. 1409-16.
- Wald DS, Gray HH. Restrictive cardiomyopathy in systemic amyloidosis. QJM. 2003 May. 96 (5):380-2. [QxMD MEDLINE Link].
- Garcia MJ. Constrictive pericarditis versus restrictive cardiomyopathy?. J Am Coll Cardiol. 2016 May 3. 67 (17):2061-76. [QxMD MEDLINE Link].
- Sasaki N, Garcia M, Ko HH, Sharma S, Parness IA, Srivastava S. Applicability of published guidelines for assessment of left ventricular diastolic function in adults to children with restrictive cardiomyopathy: an observational study. Pediatr Cardiol. 2015 Feb. 36 (2):386-92. [QxMD MEDLINE Link].
- Bejar D, Colombo PC, Latif F, Yuzefpolskaya M. Infiltrative cardiomyopathies. Clin Med Insights Cardiol. 2015. 9 (suppl 2):29-38. [QxMD MEDLINE Link].
- Mankad AK, Shah KB. Transthyretin cardiac amyloidosis. Curr Cardiol Rep. 2017 Aug 24. 19 (10):97. [QxMD MEDLINE Link].
- Maleszewski JJ. Cardiac amyloidosis: pathology, nomenclature, and typing. Cardiovasc Pathol. 2015 Nov-Dec. 24 (6):343-50. [QxMD MEDLINE Link].
- Braunwald E, Abelmann WH, eds. Atlas of Heart Diseases. Vol 2. Philadelphia, PA: Current Medicine; 1994. 53-61.
- Niemann JI. Cardiomyopathies and pericardial disease. In: Tintinalli JE, Stapczynski JS, Ma OJ, et al, eds. Tintinalli's Emergency Medicine: A Comprehensive Study Guide. 8th ed. New York, NY: McGraw-Hill; 2016. 384.
- Castano A, Bokhari S, Maurer MS. Unveiling wild-type transthyretin cardiac amyloidosis as a significant and potentially modifiable cause of heart failure with preserved ejection fraction. Eur Heart J. 2015 Oct 7. 36 (38):2595-7. [QxMD MEDLINE Link].
- Goldstein JA. Differentiation of constrictive pericarditis and restrictive cardiomyopathy. ACC Ed Highlights. 1998 Fall. 14-22.
- Amaki M, Savino J, Ain DL, et al. Diagnostic concordance of echocardiography and cardiac magnetic resonance-based tissue tracking for differentiating constrictive pericarditis from restrictive cardiomyopathy. Circ Cardiovasc Imaging. 2014 Sep. 7 (5):819-27. [QxMD MEDLINE Link].
- Malik SB, Kwan D, Shah AB, Hsu JY. The right atrium: gateway to the heart--anatomic and pathologic imaging findings. Radiographics. 2015 Jan-Feb. 35 (1):14-31. [QxMD MEDLINE Link].
- Falk RH, Quarta CC. Echocardiography in cardiac amyloidosis. Heart Fail Rev. 2015 Mar. 20 (2):125-31. [QxMD MEDLINE Link].
- Leya FS, Arab D, Joyal D, et al. The efficacy of brain natriuretic peptide levels in differentiating constrictive pericarditis from restrictive cardiomyopathy. J Am Coll Cardiol. 2005 Jun 7. 45 (11):1900-2. [QxMD MEDLINE Link].
- Selvaganesh M, Arul AS, Balasubramanian S, Ganesan N, Naina Mohammed S, Sivakumar GS, et al. An unusual ECG pattern in restrictive cardimyopathy. Indian Heart J. 2015 Jul-Aug. 67 (4):362-7. [QxMD MEDLINE Link].
- White JA, Fine NM. Recent advances in cardiovascular imaging relevant to the management of patients with suspected cardiac amyloidosis. Curr Cardiol Rep. 2016 Aug. 18 (8):77. [QxMD MEDLINE Link].
- Saeed M, Liu H, Liang CH, Wilson MW. Magnetic resonance imaging for characterizing myocardial diseases. Int J Cardiovasc Imaging. 2017 Sep. 33 (9):1395-414. [QxMD MEDLINE Link].
- Leviner DB, Hochhauser E, Arad M. Inherited cardiomyopathies--novel therapies. Pharmacol Ther. 2015 Nov. 155:36-48. [QxMD MEDLINE Link].
- Gursu HA, Varan B, Erdogan I. Use of oral budesonide in the management of protein-losing enteropathy due to restrictive cardiomyopathy. Cardiol Young. 2014 Aug. 24 (4):764-6. [QxMD MEDLINE Link].
- Miller LW, Guglin M. Evaluation of ventricular assist devices and cardiac transplantation. In: Baliga R, Haas G, eds. Management of Heart Failure. London, UK: Springer-Verlag; 2015.
- Sundararajan S, Thiruchelvam T, Hsia TY, Karimova A. New 15-mL ventricular assist device in children with restrictive physiology of the left ventricle. J Thorac Cardiovasc Surg. 2014 Jun. 147 (6):e79-80. [QxMD MEDLINE Link].
- Grupper A, Park SJ, Pereira NL, et al. Role of ventricular assist therapy for patients with heart failure and restrictive physiology: Improving outcomes for a lethal disease. J Heart Lung Transplant. 2015 Aug. 34 (8):1042-9. [QxMD MEDLINE Link].
- Robinson MR, Al-Kindi SG, Oliveira GH. Heart and heart-liver transplantation in patients with hemochromatosis. Int J Cardiol. 2017 Oct 1. 244:226-8. [QxMD MEDLINE Link].
- Uriel N, Vainrib A, Jorde UP, et al. Mediastinal radiation and adverse outcomes after heart transplantation. J Heart Lung Transplant. 2010 Mar. 29 (3):378-81. [QxMD MEDLINE Link].
- Hanna M. Novel drugs targeting transthyretin amyloidosis. Curr Heart Fail Rep. 2014 Mar. 11 (1):50-7. [QxMD MEDLINE Link].
- Madeira M, Teixeira R, Costa M, Goncalves L, Klein AL. Two-dimensional speckle tracking cardiac mechanics and constrictive pericarditis: systematic review. Echocardiography. 2016 Oct. 33 (10):1589-99. [QxMD MEDLINE Link].
- Zhang Y, He L, Cai J, et al. Measurements in pediatric patients with cardiomyopathies: comparison of cardiac magnetic resonance imaging and echocardiography. Cardiology. 2015. 131 (4):245-50. [QxMD MEDLINE Link].
- Topilsky Y, Pereira NL, Shah DK, et al. Left ventricular assist device therapy in patients with restrictive and hypertrophic cardiomyopathy. Circ Heart Fail. 2011 May. 4 (3):266-75. [QxMD MEDLINE Link].
- [Guideline] Hershberger RE, Lindenfeld J, Mestroni L, Seidman CE, Taylor MR, Towbin JA, et al. Genetic evaluation of cardiomyopathy--a Heart Failure Society of America practice guideline. J Card Fail. 2009 Mar. 15(2):83-97. [QxMD MEDLINE Link]. [Full Text].
- Fritschi S, Prothmann M, Schulz-Menger J. [Hypertrophic and restrictive cardiomyopathy. Differentiation by imaging] [German]. Herz. 2015 Jun. 40 (4):591-9. [QxMD MEDLINE Link].
- DePasquale EC, Nasir K, Jacoby DL. Outcomes of adults with restrictive cardiomyopathy after heart transplantation. J Heart Lung Transplant. 2012 Dec. 31 (12):1269-75. [QxMD MEDLINE Link].
- [Guideline] Yancy CW, Jessup M, Bozkurt B, et al, for the Writing Committee Members, American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation. 2013 Oct 15. 128(16):e240-327. [QxMD MEDLINE Link]. [Full Text].
- [Guideline] Ackerman MJ, Priori SG, Willems S, et al, for the Heart Rhythm Society (HRS), European Heart Rhythm Association (EHRA). HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Europace. 2011 Aug. 13(8):1077-109. [QxMD MEDLINE Link]. [Full Text].
- [Guideline] Ponikowski P, Voors AA, Anker SD, et al, for the European Society of Cardiology Authors/Task Force Members. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2016 Jul 14. 37(27):2129-200. [QxMD MEDLINE Link]. [Full Text].
- Ryan TD, Madueme PC, Jefferies JL, et al. Utility of echocardiography in the assessment of left ventricular diastolic function and restrictive physiology in children and young adults with restrictive cardiomyopathy: a comparative echocardiography-catheterization study. Pediatr Cardiol. 2017 Feb. 38 (2):381-9. [QxMD MEDLINE Link].
- Restrictive cardiomyopathy. Axial double inversion-recovery magnetic resonance image of the heart in a 30-year-old woman with sarcoidosis demonstrates a normal pericardium.
- Restrictive cardiomyopathy. Axial contrast-enhanced computed tomography scan through the heart (same patient as in the previous image) shows a thin pericardium without calcification. Note the cardiophrenic and internal mammary lymph nodes. The patient had extensive mediastinal and hilar adenopathy, as well as interstitial lung changes.
Author
Coauthor(s)
Asa William (Peter) Viccellio, MD Professor, Vice-Chair, Department of Emergency Medicine, State University of New York at Stony Brook
Asa William (Peter) Viccellio, MD is a member of the following medical societies: American College of Emergency Physicians
Disclosure: Nothing to disclose.
Specialty Editor Board
Francisco Talavera, PharmD, PhD Adjunct Assistant Professor, University of Nebraska Medical Center College of Pharmacy; Editor-in-Chief, Medscape Drug Reference
Disclosure: Received salary from Medscape for employment. for: Medscape.
Amin Antoine Kazzi, MD Professor of Clinical Emergency Medicine, Department of Emergency Medicine, American University of Beirut, Lebanon
Amin Antoine Kazzi, MD is a member of the following medical societies: American Academy of Emergency Medicine
Disclosure: Nothing to disclose.
Chief Editor
Henry H Ooi, MD, MRCPI Director, Advanced Heart Failure and Cardiac Transplant Program, Nashville Veterans Affairs Medical Center; Assistant Professor of Medicine, Vanderbilt University School of Medicine
Disclosure: Nothing to disclose.
Additional Contributors
Gary Edward Sander, MD, PhD, FACC, FAHA, FACP, FASH Professor of Medicine, Director of CME Programs, Team Leader, Root Cause Analysis, Tulane University Heart and Vascular Institute; Director of In-Patient Cardiology, Tulane Service, University Hospital; Visiting Physician, Medical Center of Louisiana at New Orleans; Faculty, Pennington Biomedical Research Institute, Louisiana State University; Professor, Tulane University School of Medicine
Gary Edward Sander, MD, PhD, FACC, FAHA, FACP, FASH is a member of the following medical societies: Alpha Omega Alpha, American Chemical Society, American College of Cardiology, American College of Chest Physicians, American College of Physicians, American Federation for Clinical Research, American Federation for Medical Research, American Heart Association, American Society for Pharmacology and Experimental Therapeutics, American Society of Hypertension, American Thoracic Society, Heart Failure Society of America, National Lipid Association, Southern Society for Clinical Investigation
Disclosure: Nothing to disclose.
Alan Vainrib, MD Fellow, Department of Cardiology, Stony Brook University Medical Center
Disclosure: Nothing to disclose.
Acknowledgements
The authors and editors of Medscape Reference gratefully acknowledge the contributions of previous authors Sarath Reddy, MD, Alan Forker, MD, Gunateet Goswami, MD, Nafisa Kuwajerwala, MD, Paul J Kaloudis, MD, and Andrew Wackett, MD, to the development and writing of the source articles.