Tumour class prediction and discovery by microarray-based DNA methylation analysis (original) (raw)
Abstract
Aberrant DNA methylation of CpG sites is among the earliest and most frequent alterations in cancer. Several studies suggest that aberrant methylation occurs in a tumour type-specific manner. However, large-scale analysis of candidate genes has so far been hampered by the lack of high throughput assays for methylation detection. We have developed the first microarray-based technique which allows genome-wide assessment of selected CpG dinucleotides as well as quantification of methylation at each site. Several hundred CpG sites were screened in 76 samples from four different human tumour types and corresponding healthy controls. Discriminative CpG dinucleotides were identified for different tissue type distinctions and used to predict the tumour class of as yet unknown samples with high accuracy using machine learning techniques. Some CpG dinucleotides correlate with progression to malignancy, whereas others are methylated in a tissue-specific manner independent of malignancy. Our results demonstrate that genome-wide analysis of methylation patterns combined with supervised and unsupervised machine learning techniques constitute a powerful novel tool to classify human cancers.
INTRODUCTION
Aberrant DNA methylation within CpG islands is among the earliest and most common alteration in human malignancies leading to abrogation or overexpression of a broad spectrum of genes (1). In addition, abnormal methylation has been shown to occur in CpG-rich regulatory elements in intronic and coding parts of genes for certain tumours (2). In contrast to the specific hypermethylation of tumor suppressor genes, an overall hypomethylation of DNA can be observed in tumour cells. This decrease in global methylation can be detected early, well before the development of frank tumour formation (3,4). A correlation between hypomethylation and increased gene expression has been reported for many oncogenes (5,6).
Using genome-wide, gene-non-specific approaches, several groups have demonstrated that methylation patterns vary among different tumour types. Using restriction landmark genomic scanning, Costello and co-workers were able to show that methylation patterns are tumour type specific (7). Highly characteristic DNA methylation patterns could also be shown for breast cancer cell lines (8). Gene-specific approaches with a limited number of genes have also resulted in the demonstration of unique hypermethylation profiles in several cancer types (9,10).
Recently, several groups have shown that precise determination of tumour class can be achieved by microarray-based expression analysis. Golub and co-workers screened the expression levels of almost 7000 genes, between 10 and 100 of which were then shown to be sufficient to distinguish between acute lymphoblastic leukaemia (ALL) and acute myeloid leukaemia (AML) (11). In a similar approach, Alizadeh and co-workers discovered as yet unknown subclasses of diffuse large B cell lymphoma with significant differences regarding response to therapy and disease outcome (12). As for mRNA expression profiles, genome-wide methylation patterns represent a molecular fingerprint of cancer tissues and therefore tumour class prediction and discovery should also be feasible using methylation profiles.
For this purpose, a high throughput approach to assess the methylation status of many genes in parallel is needed. Currently used approaches which allow genome-wide assessment of CpG dinucleotides all employ the digestion of genomic DNA with methylation-sensitive enzymes, thereby limiting analysis to sites for which methylation-sensitive enzymes are available (7,13–15). In addition, most of these techniques are highly labour intensive and cannot be automated. Gene-specific approaches are primarily based on sodium bisulphite treatment of the DNA, which converts all unmethylated cytosines to uracil, whereas methylated cytosines are protected. Several methods can then be employed to distinguish between the resulting sequence variants (10,16,17), some of which have been applied to larger patient populations (9,10). However, none of these techniques can be used to analyse many genes in parallel.
Here we present a novel microarray-based assay which is suited to methylation analysis of large numbers of genes and CpG dinucleotides in parallel. Methylation profiles of different tumour types were generated and analysed using supervised and unsupervised learning algorithms. Tumours included 17 samples obtained from patients with ALL or cell lines thereof, eight samples from patients with AML or respective cell lines, 10 primary prostate carcinomas and nine primary clear cell kidney carcinomas. MACS-sorted B and T cells from 13 healthy donors as well as 10 samples representing benign prostate hyperplasia (BPH) and nine samples of tumour-free kidney tissue served as normal controls. Control tissues from prostates and kidney were taken from areas of tumour-carrying prostates confirmed to be free of tumour contamination by histological analysis of adjacent sections. Up to 232 CpG dinucleotides located in CpG-rich regions of the promoters, intronic and coding sequences of 56 different genes were evaluated for methylation status. All genes were randomly selected from a panel of genes representing different pathways associated with tumourigenesis. Two of the selected genes were located on the X chromosome.
MATERIALS AND METHODS
High molecular weight chromosomal DNA from six human B cell precursor leukaemia cell lines (380, ACC 39; BV-173, ACC 20; MHH-Call-2, ACC 341; MHH-Call-4, ACC 337; NALM-6, ACC 128; REH, ACC 22) and six T cell leukaemia cell lines (CCRF-CEM, ACC 240; JURKAT, MOLT-17, ACC 36; P12-ICHIKAWA, ACC 34; RPMI-8402, ACC 290; KARPAS-299, ACC 31) was obtained from the Deutsche Sammlung von Mikroorganismen und Zellkulturen (Braunschweig, Germany). DNA prepared from five human AML cell lines, CTV-1, HL-60, Kasumi-1, K-562 (human chronic myeloid leukaemia in blast crisis) and NB4 (human acute promyelocytic leukaemia), were provided by the Department of Hematology, Oncology and Tumor Immunology (Charité, Berlin, Germany). T cells and B cells from peripheral blood of eight healthy individuals were isolated using a magnetically activated cell separation system (MACS; Miltenyi, Bergisch-Gladbach, Germany) following the manufacturer’s recommendations. As determined by FACS analysis, CD4+ T cells were >73% pure and CD19+ B cells >90% pure. Chromosomal DNA from the purified cells was isolated using a QIAamp DNA minikit (Qiagen, Hilden, Germany) according to the recommendations of the manufacturer. DNA samples of cloned bisulphite-treated DNA fragments of unmethylated and up-methylated exon 14 of the Factor VIII gene were kindly provided by Jörn Walter (MPI, Berlin). DNA was isolated at time of diagnosis from the peripheral blood or bone marrow samples of five ALL patients and three AML patients. The prostate and kidney carcinoma specimens and the method of DNA isolation have been described previously (18,19). Prostate carcinomas included four pT2cG2 tumours and five >pT3N1G3 tumours. All were histopathologically verified and represented the major tumour component. All clear cell renal carcinomas were pT2G2 tumours. Ten BPH samples were taken from areas of tumour-carrying prostates confirmed to be free of tumour contamination by histological analysis of adjacent sections.
Bisulphite treatment and PCR amplification
Bisulphite treatment of genomic DNA was done as described earlier (20), with minor modifications. Genomic DNA was digested with _Mss_I (MBI Fermentas, St Leon-Rot, Germany) prior to modification by bisulphite. For PCR amplification of the bisulphite-treated sense strand of the genes used for class prediction and discovery the primers were designed according to the guidelines of Clark and Frommer (21), ensuring preferred amplification of DNA fragments with complete bisulphite conversion. For leukaemia classification CpG sites from the following genes were analysed: ELK1, CSNK2B, l-myc, CD63, CDC25A, TUBB2, CD1A, CDK4, n-myc, AR and c-MOS. For classification of the solid tissues a total of 232 CpG dinucleotides located in regulatory regions of the following genes were analysed: hMLH1/MLH1, MGMT, N33, CDH3, c-MOS, TSP, VEGF, EGFR, CDC25A, ERBB2, c-fos, c-myc, n-myc, l-myc, c-abl, ELK1, NCL, CSNK2B, SOD1, UBB, OAT, CRIP1, MRP5, APOA1, APOC2, CSF1, CD1R3, ADCYAP1, EGR4, ATP5G1, TNF-b, GP1BB, DAPK1, DBCCR1, PR, WT1, CTLA4, DAD1, HSPA2, MC2R, AFP, TGFB1, NF1, TGFA, POMC, ME491/CD63, IL13, UNG, ATP5A1, HLA-F, ATP6, G6E, NEU1, RD, C4B and HLA-DNA. Up to 16 fragments were amplified using multiplex PCR (mPCR). The following protocol was used for single PCR reactions (modifications for mPCR are indicated in parentheses): 10 ng DNA used as template DNA for all PCR reactions; 12.5 or 40 pmol (CY5-labelled) each primer (mPCR, 1 pmol), 0.5–2 U Taq polymerase (HotStarTaq; Qiagen, Hilden, Germany) and 1 mM dNTPs (mPCR, 3.2 mM dNTPs) were incubated with the reaction buffer supplied with the enzyme in a total volume of 20 µl (mPCR, 25 µl). After activation of the enzyme (15 min, 96°C) the incubation times and temperatures were 95°C for 1 min followed by 34 (mPCR, 38) cycles (95°C for 1 min, annealing temperature for 45 s, 72°C for 75 s) and 72°C (mPCR, 65°C) for 10 min.
Microarray procedure
Oligonucleotides with a C6-amino modification at the 5′-end were spotted with 4-fold redundancy on activated glass slides (11). For each analysed CpG position two oligonucleotides, N2–16CGN2–16 and N2–16TGN2–16, reflecting the methylated and non-methylated status of the CpG dinucleotides, were spotted and immobilised on the glass array. Oligonucleotides were designed such that they matched only the bisulphite-modified DNA fragments; this is important to exclude signals arising from incomplete bisulphite conversion. The oligonucleotide microarrays representing up to 232 CpG sites were hybridised with a combination of up to 56 Cy5-labelled PCR fragments as described earlier (22). Subsequently, the fluorescent images of the hybridised slides were obtained using a GenePix 4000 microarray scanner (Axon Instruments). Hybridisation experiments were repeated at least three times for each sample.
Statistical methods
For class prediction we used a support vector machine (SVM) on a set of selected CpG sites. First we ranked the CpG sites for a given separation task by the significance of the difference between the two class means. The significance of each CpG was estimated by a two sample _t_-test (23). Then a SVM was trained on the most significant CpG positions, where the optimal number of CpG sites depends on the complexity of the separation task. Our implementation of the SVM used the Sequential Minimal Optimization algorithm to find the 1-norm soft margin separating hyperplane (24). The box constraint was set to C = 10 and a linear kernel was used. Generalisation performance was estimated by averaging over 50 cross-validation runs on randomly permutated samples partitioned into eight groups, i.e. selection of the most significant CpG sites and training of the SVM were performed on training sets of seven groups and the eigth group was used as an independent test set. The significance value for the class prediction in Table 1 represents the probability that the SVM classifies the same data points at least as well as observed if the tissue classes were assigned randomly. The significance value was estimated by sampling the distribution of cross-validation errors over 50 random shuffles of the labels, keeping the initial class priors. A Gaussian distribution was fitted to these 50 error estimates and used to calculate the probability of random generation of separations at least as good as the observed one.
Table 1. Cross-validation results for several classifications.
| Class 1 | Class 2 | Optimal CpG number | Accuracy of prediction (%) | Standard deviation (%) | Significance of prediction | ||
|---|---|---|---|---|---|---|---|
| Female | 14 | Male | 7 | 2 | 91 | 2 | 2.6E–4 |
| Healthy T and B cells | 13 | T-ALL/B-ALL | 17 | 60 | 94 | 3 | 5.3E–9 |
| T-ALL/B-ALL | 17 | AML | 8 | 5 | 94 | 4 | 3.9E–5 |
| BPH | 10 | Prostate carcinoma | 10 | 9 | 72 | 6 | 2.1E–2 |
| Healthy kidney | 9 | Kidney carcinoma | 9 | 2 | 92 | 5 | 3.8E–4 |
| Prostate | 20 | Kidney | 18 | 3 | 92 | 2 | 2.2E–5 |
For class discovery we applied hierarchical clustering with Ward’s minimum variance method, using the Bhattacharyya distance between methylation patterns as the distance measure (25).
RESULTS
Microarray-based methylation analysis
In order to allow sequence-specific distinction of the methylated from the unmethylated state of CpG dinucleotides by hybridisation analysis, total DNA from all samples was bisulphite treated, converting all unmethylated cytosines to uracil, whereas methylated cytosines were conserved (17). Regions of interest were then amplified by PCR using fluorescently labelled primers converting originally unmethylated CpG dinucleotides to TG and conserving originally methylated CpG sites. Primers were designed complementary to DNA segments containing no CpG dinucleotides. This allowed unbiased amplification of both methylated and unmethylated alleles in one reaction. For analyses including more than 11 genes, mPCR reactions were used amplifying at least 12 fragments in one reaction. All PCR products performed on an individual sample were mixed and hybridised to glass slides carrying a pair of immobilised oligonucleotides for each CpG position. Each of these detection oligonucleotides was designed to hybridise to the bisulphite-modified sequence around one CpG site, which was either originally unmethylated (TG) or methylated (CG). Hybridisation conditions were selected to allow detection of the single nucleotide differences between the TG and CG variants. Log ratios for the two signals were calculated based on comparison of intensity of the fluorescent signals. Sensitivity for detection of methylation changes was determined using artificially up- and down-methylated DNA fragments mixed at different ratios. For each of these mixtures, a series of experiments was conducted to define the range of CG:TG ratios that corresponds to varying degrees of methylation at each of the CpG sites tested. In Figure 1 the results for two CpG positions located in exon 14 of the human Factor VIII gene are shown as examples. For the mixtures of 3:0, 2:1, 1:2 and 0:3 the degree of methylation of the individual CpG sites could safely be distinguished. The results clearly demonstrate that our assay is capable of quantifying methylation at individual CpG sites with high resolution and the log ratio of the CG and TG intensities faithfully reflects the methylation status of the different CpG sites. In the following we use this ratio to describe methylation patterns at the investigated CpG sites, which reliably detects differential methylation between the investigated tumour types (Figs 2 and 3). The degree of methylation of a single CpG position can then be exactly quantified by calibration of the microarray, as shown in Figure 1.
Figure 1.

Methylation analysis and quantification of two CpG dinucleotides in exon 14 of the human Factor VIII gene. For calibration purposes a series of hybridisations was performed with mixtures of artificially up- and down-methylated DNA fragments of the Factor VIII exon 14 gene. Down- and up-methylated DNA fragments were mixed in the ratios 0:3, 1:2, 2:1 and 3:0, representing methylation statuses of 100, 66, 33 and 0%, respectively. (A) Methylation detection by oligonucleotide microarray hybridisation. The fluorescence signals of the CG and TG versions of the Factor VIII exon 14 oligonucleotides F8-5 (TTATTAACGGGAAATAAT and TTATTAATGGGAAATAAT) and F8-3 (AATAAGTTCGAAATAGAA and AATAAGTTTGAAATAGAA) are shown, which were generated by samples reflecting methylation statuses of 0, 33, 66 and 100%. The hybridisation signals are shown as a false colour image with the colours blue, green and yellow indicating fluorescence signal ranges at 635 nm of 200–800, 800–2000 and 2000–8000, respectively. (B) Quantification of methylation measurements. For each CpG position two kinds of detection oligomers were used. Oligomers that hybridise if the CpG was methylated are referred to as CG oligomers and oligomers that hybridise if the CpG was unmethylated are referred to as TG oligos. For the four kinds of compounds 59, 36, 40 and 63 identical slides were made. The log ratio of the CG and TG oligomer hybridisation intensities was calculated and then averaged for experimental sub-groups each containing three identical experiments. The density function of the CG:TG ratios shows that measured values for the different mixtures are well separated and therefore allow high resolution detection of the methylation level of a single CpG. This is an essential prerequisite for methylation-dependent class prediction or class discovery. Taking into account only the 100 and 0% methylated DNA and averaging for the 22 CpG sites investigated in the calibration experiments, the average error for methylation detection is 4%. The log ratios are not grouped symmetrically around zero but are shifted towards negative values. We assume that the energetically different effects of G-T and A-C mismatches allow hybridisation of the methylated allele to the oligonucleotide representing the unmethylated more easily than vice versa.
Figure 2.

(A) Methylation patterns of leukaemia samples and controls as described by the log ratio of the CG and TG signal intensities. The colour represents the distance from the mean between the two investigated groups (calculated as the mean of the group means). Hypermethylation corresponds to red, mean methylation level to black and hypomethylation to green. The labels on the left of the plot are gene and CpG identifiers. The labels on the right give the significance of the difference between the means of the two groups. Each row corresponds to a single CpG and each column to the methylation levels of one sample. The 15 CpG sites with the most significant differences between the two classes are shown. Classifications shown are male/female, healthy/ALL and AML/ALL. For male/female separation only non cell lines were used. As expected, the majority of significant CpG dinucleotides come from the two X chromosome genes (ELK1 and AR). (B) Class prediction of leukaemia samples and healthy controls. The plots show a SVM trained on the two most significant CpG sites for the respective discrimination using all available samples as training data. Circled points are the support vectors defining the borderline (white) between the area of the first (green) and the area of prediction of the second class (blue). The colour intensity corresponds to the prediction strength. Classifications shown are male/female, healthy/ALL and AML/ALL.
Figure 3.

(A) Methylation patterns of solid tissues as described by the log ratio of the CG and TG signal intensities. The colour represents the distance from the mean between the two investigated groups (calculated as the mean of the group means). Hypermethylation corresponds to red, mean methylation level to black and hypomethylation to green. The labels on the left of the plot are gene and CpG identifiers. The labels on the right give the significance of the difference between the means of the two groups. Each row corresponds to a single CpG and each column to the methylation levels of one sample. The 15 CpG sites with the most significant differences between the two classes are shown. Classifications shown are BPH/prostate carcinoma, healthy kidney/kidney carcinoma, BPH and prostate carcinoma/healthy kidney and kidney carcinoma. (B) Class prediction of solid tissues. The plots show a SVM trained on the two most significant CpG sites for the respective discrimination using all available samples as training data. Circled points are the support vectors defining the borderline (white) between the area of the first (green) and the area of prediction of the second class (blue). The colour intensity corresponds to the prediction strength. Classifications shown are BPH/prostate carcinoma, healthy kidney/kidney carcinoma, BPH and prostate carcinoma/healthy kidney and kidney carcinoma.
Supervised learning methods
We addressed the problem of class prediction using a supervised machine learning method called SVM (24,26), which has already been successfully applied to the analysis of microarray gene expression data (27,28). The SVM constructs an optimal discriminant between two classes of given training samples. A supervised learning technique, such as SVM, has the advantage that it exploits the prior knowledge represented by data labels.
In order to quantify the contribution of single CpG sites to the prediction of classes, we initially ranked all CpG sites according to their discriminatory power for the investigated sample classes using a two sample _t_-test. Then the SVM was trained using an increasing number of CpG sites in the order of their ranking. The number of CpG sites where the prediction error is minimised depends on which particular classes were to be separated from each other. This is explained by the fact that, on the one hand, the exclusion of certain CpG positions from the analysis potentially leads to a loss of information. On the other hand, as suggested by statistical learning theory (26), for a limited number of samples the reliability of the class predictor generally increases with decreasing number of free parameters, in our case number of CpG sites. The inherent, task-specific complexity of the classification problem determines where these two counteracting effects are balanced and class prediction achieves its optimal performance. This suggests that a proper feature selection in genome-wide analyses not only improves class prediction (29), but also provides information about the complexity of the problem.
Validation of the learning algorithm
A cross-validation method (30,31) was used to evaluate the prediction performance of the SVM and the feature selection algorithm. For each classification task, the samples were partitioned into eight groups of approximately equal size. Initially all CpG positions were ranked according to their discriminatory power using seven groups as the training samples. Then the SVM was trained on the same seven sample groups. The predictor obtained by this method was then tested on the remaining group of independent test samples. The number of correct classifications was counted over eight runs for the feature selection and SVM algorithm for all possible choices of the independent test group without using any knowledge obtained from the previous runs. To obtain a reliable estimate for the accuracy, i.e. the probability of correct classification of a previously unknown sample, the number of correct classifications was averaged over 50 different partitionings of the samples into eight groups. Note that the above described cross-validation procedure evaluates the accuracy using practically all possible combinations of training and independent test sets. It therefore gives a better estimate of the prediction accuracy than simply using one training sample set and then one independent test set (31).
Methylation-based class prediction
As a first test case we chose the distinction of males from females, evaluating all available peripheral blood samples at 81 different CpG positions. In females one X chromosome becomes methylated upon inactivation (32). Therefore, females are expected to exhibit higher degrees of methylation at X chromosome genes and distinction between female and male should be feasible by determining the methylation status of X chromosome genes. In fact, male and female primary samples could be classified with an accuracy of 91% based on two CpG dinucleotides in the ELK1 gene (Fig. 2 and Table 1). Furthermore, 14 of 16 CpG dinucleotides from X chromosome genes of our panel were among the 17 most significant markers for gender, showing high specificity of hybridization. Notably, cell lines could not be classified into one of the two groups, possibly because of considerable chromosomal aberrations.
We then trained the learning algorithm (SVM) to recognise the difference between T and B cell leukaemias (from both patient samples and cell lines) and CD19+ B cells and CD4+ T cells obtained from healthy donors. Information was obtained for 81 CpG dinucleotides originating from 11 genes. Samples could be classified with 85% accuracy using only the two most informative CpG positions. Remarkably, the accuracy could be increased to 94% by including a total of 60 CpG positions in the analysis. Individual CpG sites were ranked according to their discriminatory power, showing that the decision between healthy T and B cells and ALL was primarily based on CpG sites located in intron 1 of the CDK4 gene and the coding sequence of the c-MOS oncogene, but CpG sites located in regulatory regions of other genes also contributed significantly (Table 1 and Fig. 2). Compared to the healthy group, the tumour cells consistently showed relative hypermethylation of these particular CpG sites.
In a next step, we tried to distinguish the two classes of leukaemia on the same set of genes. Again, the feature selection algorithm and the SVM were initially presented with a training set of AML and ALL samples (from both patient samples and cell lines) with the class information attached. Previously not included samples could then be classified into one of the classes with an accuracy of 81% when using two CpG positions. The optimal number of CpG sites in this case was calculated to be five, increasing the accuracy to 94% (Table 1 and Fig. 2). The most informative CpG sites were located in intron 1 of the CDK4 gene and the promoter region of CSNK2B. The overall degree of methylation at these sites was reproducibly higher in ALL than in AML cells. However, the CpG sites in the CDK4 gene contributing information to this particular decision were different from those in the same gene distinguishing between ALL and healthy lymphocytes. This clearly shows that in some cases, different CpG sites within one cluster can contribute independent information, with different CpG sites potentially answering questions on different aspects of a phenotype.
There is evidence that CpG islands of several tissue-specific genes become methylated in cell lines (33). Hence, it is important to rule out that the above classifications were based on cell line-specific artefacts rather than on disease-specific changes. We therefore grouped leukaemia cell lines into one and primary leukaemia samples into another class. Classification was possible with an accuracy of 56% based on two CpG positions and 76% using 51 CpG sites. Importantly, the CpG sites that contributed most to the distinction of cell lines from primary patient samples were neither informative in the classifications of healthy T and B cells versus T and B cell leukemia nor in the distinction between the two types of leukemia, excluding the possibility that these classifications are based merely on cell line-specific markers.
In a next step, we addressed the more complex problem of distinguishing different solid tissues. Tissue samples of prostate carcinomas, BPH, clear cell renal carcinomas and healthy kidney were obtained at time of surgery, i.e. in a routine clinical situation (18,19). A panel of genomic fragments from 56 different genes was assessed for their methylation status. Methylation patterns of malignant neoplasia and benign hyperplasia of the prostate and the results of the SVM are shown in Figure 3A and B. Although methylation patterns seem to differ substantially between the two conditions (Fig. 3A), only the inclusion of at least nine CpG dinucleotides allows a statistically significant separation. The most informative CpG positions for this discrimination are located within the TGF-α and POMC genes.
Clear cell renal carcinomas could be distinguished quite easily from their healthy counterparts based on only two CpG sites with an accuracy of 92%. These highly informative CpG sites were located in the regulatory sequences of HLA-F and APOC2.
In a number of clinical situations, e.g. for typing of disseminated tumour cells, the tissue of origin is not obvious and needs to be determined. We therefore tried to identify tissue-specific markers which would allow the distinction between prostate and kidney cells. For this purpose, methylation patterns of all prostate tissues were compared to all kidney tissues. Differentially methylated sites, which allow a clear distinction between the two tissue types independent of disease, were identified in the regulatory regions of apolipoprotein C2 and the platelet glycoprotein Ib. The correct tissue type could be determined with an accuracy of 92% if healthy or diseased test samples were presented to the algorithm. This indicates that the methylation pattern of these CpG sites is highly tissue specific and is conserved throughout progression to malignancy.
In general, the marker CpGs we found showed higher methylation levels in tumour as opposed to control samples, confirming the general observation of hypermethylation of specific CpG islands during tumour formation (1). Interestingly, in the case of renal carcinoma we found lower methylation levels in tumour samples compared to controls for the informative CpG sites, located in the regulatory sequences of HLA-F and APOC2. This observation might reflect the overall hypomethylation of genomic DNA seen in cancers (3,4).
Our results indicate that tumour- as well as tissue-specific methylation patterns exist independently. Combined with learning algorithms, these specific methylation fingerprints can be used for class prediction of different human tumours and healthy tissues.
Class discovery
Today’s diagnostic entities still constitute highly heterogeneous groups of diseases. We therefore explored whether tumour classes can be discovered by analysis of methylation patterns. All leukaemia samples and controls used in the experiments described above were subjected to an unsupervised learning method called hierarchical clustering. Methylation patterns derived from these samples were presented to the algorithm without any class information attached. The algorithm was designed to recognise major and minor differences between individual samples and to display them in a tree according to their similarity (Fig. 4). Samples derived from T and B cells of healthy donors were correctly grouped together. The second cluster contained all AML samples, whereas the majority of ALL samples were clustered in a third branch. The few misclassified samples did not differ in any of the available patient parameters. The results clearly demonstrate that tumour classes can be discovered without any prior knowledge solely based on analysis of methylation patterns.
Figure 4.
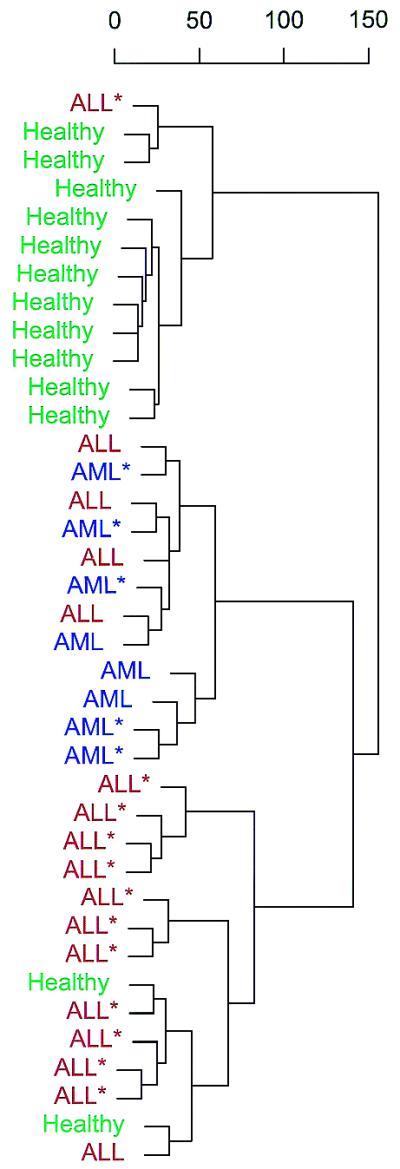
Class discovery. The figure shows a hierarchical clustering of all available samples. Healthy individuals are coloured green, patients with ALL red and patients with AML blue. Asterisks indicate cell line samples. The feature space consisted of all CpG sites except those from the two X chromosome genes. The diagnosis was unknown to the algorithm.
DISCUSSION
Taken together, we demonstrate that microarray-based methylation analysis combined with supervised and unsupervised learning techniques is highly effective in predicting known and discovering novel tumour classes. Our approach turns epigenetic studies into a genomics technology and makes targeted genome-wide methylation screening possible. In addition to mRNA expression arrays and proteomics this makes a new level of cellular information experimentally accesible in a high throughput manner.
For the purpose of class prediction, knowledge of the functional consequences of methylation of individual CpG dinucleotides is not required. Hypermethylation in the promoter regions of genes is usually associated with abrogation of expression, whereas the role of methylation in gene bodies is less clear but, as we and others have shown, also contains information (2). In any case, a knowledge of differing methylation status could be used to generate hypotheses on the functional context of a gene product. Hence, large-scale methylation analysis provides a valuable tool for the forthcoming post-genomic dissection of the complexities of genetic networks and the biological phenotypes emerging from them. We confirmed the general assumption that massively parallel analysis is in most cases superior to the use of low-dimensional data sets. Nevertheless, in some cases computational selection of informative features out of an initially high-dimensional space allows subsequent classification through a low-dimensional approach. The results of comparable studies will enable the development of a new generation of methylation-based biomarkers.
Microarray-based methylation analysis has several advantages compared to conventional techniques. Approaches which allow genome-wide assessment of CpG dinucleotides all employ the digestion of genomic DNA with methylation-sensitive enzymes. Major problems, therefore, are the limitation to sites for which methylation-sensitive enzymes are available, the inability to analyse a set of specific candidate genes, the occurence of false positives due to incomplete digestion and the large amount of high molecular weight DNA required. In addition, most of these techniques are highly labour intensive and cannot be automated.
Gene-specific approaches are primarily based on sodium bisulphite treatment of the DNA, which converts all unmethylated cytosines to uracil, whereas methylated cytosines are protected. Several methods can then be employed to distinguish between the resulting sequence variants. Methylation-specific PCR uses an individual primer designed for each of the CpG sites which is specific only to the originally methylated or unmethylated fragment (16). Methylation-specific PCR is highly sensitive but not quantitative, primer design is very labour intensive and false positives occur frequently. Other approaches are the unbiased amplification of both variants (10), which has the advantage of allowing quantification of the percentage of methylated alleles, and sequencing of bisulphite-modified DNA. Compared to these techniques, chip-based analysis allows a high degree of automation and profiling of many candidate gene-specific CpG sites in parallel. In addition, all currently available gene-specific approaches require several CpG positions in an area to be co-methylated, therefore an a priori assumption on the methylation status of selected CpG positions has to be made (10,16). We were able to demonstrate that our approach allows the analysis of many individual CpGs on one microarray without assuming co-methylation among them. This is important, as we have shown that in some cases the methylation status of different CpG sites within one cluster can contribute independent information, with different CpG sites potentially answering questions on different aspects of a phenotype.
Classification results were comparable to mRNA-based assays regarding accuracy, safety and reproducibility. However, in expression profiling signal intensities strongly depend on both the absolute and relative amounts of the different mRNA species and thus comparison between independent experiments is difficult. Hence, although signals are generally calibrated against those derived from housekeeping genes, there remains an inability of mRNA profiling to detect subtle changes in expression levels (34). In our approach, the CG:TG ratio is used as an internal calibration and thus the amount of probe hybridised to the slide does not influence the results. This greatly improves the comparability of the results and therefore enables the screening of larger populations, as is needed for example in multi-centre trials and prospective studies.
As we show, class prediction can be applied to solid tissues. Albeit we can demonstrate that our technique works on conventionally dissected specimens, sophisticated dissection techniques will have to be employed in many cases. In addition, most well-documented tissue samples can be obtained only as archived specimens. This strongly limits the amount and number of tissues available for expression analysis (35). Our approach has the potential to overcome these fundamental limitations: through the mere fact that stable DNA is the object of study, both extraction of material from archived samples and amplification from very few cells are possible (36). Hence, examination of methylation patterns in large numbers of archived specimens with well-documented, comprehensive clinical records becomes routinely possible, as well as application to microdissected specimens.
Supplementary Material
[Supplementary Data]
Acknowledgments
ACKNOWLEDGEMENTS
We thank Jörn Walter (Max Planck Insitute for Molecular Genetics, Berlin) for cloned fragments of artificially up- and down-methylated DNA of exon 14 of human Factor VIII. We would also like to thank Florian Winau (Max Planck Institute for Infection Biology) for discussions of medical questions, Thomas Hildmann (Epigenomics) for suggesting genes for the prostate and kidney gene panel and Tamás Ruján (Epigenomics) for his bioinformatics support.
REFERENCES
- 1.Jones P.A. (1996) DNA methylation errors and cancer. Cancer Res., 65, 2463–2467. [PubMed] [Google Scholar]
- 2.Chan M.F., Liang,G. and Jones,P.A. (2000) Relationship between transcription and DNA methylation. Curr. Top. Microbiol. Immunol., 249, 75–86. [DOI] [PubMed] [Google Scholar]
- 3.Christman J.K., Sheikhnejad,G., Dizik,M., Abileah,S. and Wainfan,E. (1993) Reversibility of changes in nucleic acid methylation and gene expression induced in rat liver by severe dietary methyl deficiency. Carcinogenesis, 14, 551–557. [DOI] [PubMed] [Google Scholar]
- 4.Pogribny I.P., Miller,B.J. and James,S.J. (1997) Alterations in hepatic p53 gene methylation patterns during tumor progression with folate/methyl deficiency in the rat. Cancer Lett., 115, 31–38. [DOI] [PubMed] [Google Scholar]
- 5.Hanada M., Delia,D., Aiello,A., Stadtmauer,E. and Reed,J.C. (1993) bcl-2 gene hypomethylation and high-level expression in B-cell chronic lymphocytic leukemia. Blood, 82, 1820–1828. [PubMed] [Google Scholar]
- 6.Feinberg A.P. and Vogelstein,B. (1983) Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature, 301, 89–92. [DOI] [PubMed] [Google Scholar]
- 7.Costello J.F., Fruhwald,M.C., Smiraglia,D.J., Rush,L.J., Robertson,G.P., Gao,X., Wright,F.A., Feramisco,J.D., Peltomaki,P., Lang,J.C., Schuller,D.E., Yu,L., Bloomfield,C.D., Caligiuri,M.A., Yates,A., Nishikawa,R., Su Huang,H., Petrelli,N.J., Zhang,X., O"Dorisio,M.S., Held,W.A., Cavenee,W.K. and Plass,C. (2000) Aberrant CpG-island methylation has non-random and tumour-type-specific patterns. Nature Genet., 24, 132–138. [DOI] [PubMed] [Google Scholar]
- 8.Huang T.H.-M., Perry,M.R. and Laux,D.E. (1999) Methylation profiling of CpG islands in human breast cancer cells. Hum. Mol. Genet., 8, 459–470. [DOI] [PubMed] [Google Scholar]
- 9.Esteller M., Corn,P.G., Baylin,S.B. and Herman,J.G. (2001) A gene hypermethylation profile of human cancer. Cancer Res., 61, 3225–3229. [PubMed] [Google Scholar]
- 10.Eads C.A., Danenberg,K.D., Kawakami,K., Saltz,L.B., Blake,C., Shibata,D., Danenberg,P.V. and Laird,P.W. (2000) MethyLight: a high-throughput assay to measure DNA methylation. Nucleic Acids Res., 28, e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Golub T.R., Slonim,D.K., Tamayo,P., Huard,C., Gaasenbeek,M., Mesirov,J.P., Coller,H., Loh,M.L., Downing,J.R., Caligiuri,M.A., Bloomfield,C.D. and Lander,E.S. (1999) Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science, 286, 531–537. [DOI] [PubMed] [Google Scholar]
- 12.Alizadeh A.A., Eisen,M.B., Davis,R.E., Ma,C., Lossos,I.S., Rosenwald,A., Boldrick,J.C., Sabet,H., Tran,T., Yu,X. et al. (2000) Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature, 403, 503–511. [DOI] [PubMed] [Google Scholar]
- 13.Liang G., Salem,C.E., Yu,M.C., Nguyen,H.D., Gonzales,F.A., Nguyen,T.T., Nichols,P.W. and Jones,P.A. (1998) DNA methylation differences associated with tumor tissues identified by genome scanning analysis. Genomics, 53, 260–268. [DOI] [PubMed] [Google Scholar]
- 14.Toyota M., Ho,C., Ahuja,N., Jair,K.W., Li,Q., Ohe-Toyota,M., Baylin,S.B. and Issa,J.P. (1999) Identification of differentially methylated sequences in colorectal cancer by methylated CpG island amplification. Cancer Res., 59, 2307–2312. [PubMed] [Google Scholar]
- 15.Yan P.S., Perry,M.R., Laux,D.E., Asare,A.L., Caldwell,C.W. and Huang,T.H. (2000) CpG island arrays: an application toward deciphering epigenetic signatures of breast cancer. Clin. Cancer Res., 6, 1432–1438. [PubMed] [Google Scholar]
- 16.Herman J.G., Graff,J.R., Myohanen,S., Nelkin,B.D. and Baylin,S.B. (1996) Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc. Natl Acad. Sci. USA, 93, 9821–9826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frommer M., McDonald,L.E., Millar,D.S., Collis,C.M., Watt,F., Grigg,G.W., Molloy,P.L. and Paul,C.L. (1992) A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl Acad. Sci. USA, 89, 1827–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Santourlidis S., Florl,A.R., Ackermann,R., Wirtz,H.C. and Schulz,W.A. (1999) High frequency of alterations in DNA methylation in adenocarcinoma of the prostate. Prostate, 39, 166–174. [DOI] [PubMed] [Google Scholar]
- 19.Florl A.R., Loewer,R., Schmitz-Draeger,B.J. and Schulz,W.A. (1999) DNA methylation and expression of L1 LINE and HERV-K provirus sequences in urothelial and renal cell carcinoma. Br. J. Cancer, 80, 1312–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Olek A., Oswald,J. and Walter,J. (1996) A modified and improved method for bisulphite based cytosine methylation analysis. Nucleic Acids Res., 24, 5064–5066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clark S.J. and Frommer,M. (1997) Bisulphite genomic sequencing of methylated cytosines. In Taylor,G.R. (ed.), Laboratory Methods for the Detection of Mutations and Polymorphisms in DNA. CRC Press, Boca Raton, FL, pp. 151–161.
- 22.Chen D., Yan,Z., Cole,D.L. and Srivatsa,G.S. (1999) Analysis of (n – 1)mer deletion dequences in synthetic oligodesoxyribonucleotides by hybridisation to an immobilized probe array. Nucleic Acids Res., 27, 389–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mendenhall W. and Sincich,T. (1995) Statistics for Engineering and the Sciences. Prentice-Hall, NJ.
- 24.Christianini N. and Shawe-Taylor,J. (2000) An Introduction to Support Vector Machines and Other Kernel-Based Learning Methods. Cambridge University Press, Cambridge, UK.
- 25.Mardia K.V., Kent,J.T. and Bibby,J.M. (1979) Multivariate Analysis. Academic Press, London, UK.
- 26.Vapnik V.N. (1998) Statistical Learning Theory. John Wiley & Sons, New York, NY.
- 27.Brown M.P., Grundy,W.N., Lin,D., Cristianini,N., Sugnet,C.W., Furey,T.S., Ares,M.,Jr and Haussler,D. (2000) Knowledge-based analysis of microarray gene expression data by using support vector machines. Proc. Natl Acad. Sci. USA, 97, 262–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gaasterland T. and Bekiranov,S. (2000) Making the most of microarry data. Nature Genet., 24, 204–206. [DOI] [PubMed] [Google Scholar]
- 29.Weston J., Mukherjee,S., Chapelle,O., Pontil,M., Poggio,T. and Vapnik,V. (2001) Feature selection for SVMs. In Leen,T.K., Dietterich,T.G. and Tresp,V. (eds), Advances in Neural Information Processing Systems 13. MIT Press, Cambridge, MA.
- 30.Bishop C.M. (1995) Neural Networks for Pattern Recognition. Oxford University Press, New York, NY.
- 31.Ripley B.D. (1996) Pattern Recognition and Neural Networks. Cambridge University Press, Cambridge, UK.
- 32.Riggs A.D. (1975) X inactivation, differentiation and DNA methylation. Cytogenet. Cell Genet., 11, 9–25. [DOI] [PubMed] [Google Scholar]
- 33.Antequera F., Boyes,J. and Bird,A. (1990) High levels of de novo methylation and altered chromatin structure at CpG islands in cell lines. Cell, 62, 503–511. [DOI] [PubMed] [Google Scholar]
- 34.Lipshutz R.J., Fodor,S.P.A., Gingeras,T.R. and Lockhart,D.J. (1999) High density synthetic oligonucleotide arrays. Nature Genet., 21, 20–24. [DOI] [PubMed] [Google Scholar]
- 35.Bowtell D.D.L. (1999) Options available – from start to finish – for obtaining expression data by microarray. Nature Genet., 21 (suppl.), 25–32. [DOI] [PubMed] [Google Scholar]
- 36.Olek A. and Walter,J. (1997) The pre-implantation ontogeny of the H19 methylation imprint. Nature Genet., 17, 275–276. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
[Supplementary Data]