Analysis of microsatellite-stable gastrointestinal cancer with high tumour mutational burden: a retrospective cohort study (original) (raw)
. Author manuscript; available in PMC: 2023 Oct 25.
Abstract
Background:
Genomic signatures contributing to high tumor mutation burden (TMB-H) independent from mismatch-repair deficiency (dMMR)/microsatellite instability-high (MSI-H) status are not well-studied. We aimed to characterize molecular features of TMB-H microsatellite stable (MSS) gastrointestinal (GI) tumors.
Methods:
Molecular alterations of 48606 GI tumor from CARIS with next generation sequencing were compared among MSS&TMB-H, dMMR/MSI-H, MSS&TMB-low(L), using Chi-square or Fisher’s exact tests. Anti-tumor immune response within tumor environment was predicted by analysing the infiltration of immune cells and immune signatures using TCGA database. Kaplan–Meier method and the log-rank test were used to evaluate the impact of gene alterations on the efficacy of immune checkpoint inhibitors (ICIs) in MSS GI cancers from CARIS, MSKCC and PUCH.
Findings:
MSS&TMB-H was observed in 3·29% of patients, while dMMR/MSI-H, MSS&TMB-L were observed in 4·67% and 92·03% respectively. Gene mutations in SMAD2, MTOR, NFE2L2, RB1, KEAP1, TERT, and RASA1 may impair anti-tumor immune response despite TMB-H, while mutations in 16 other genes (CDC73, CTNNA1, ERBB4, EZH2, JAK2, MAP2K1, MAP2K4, PIK3R1, POLE, PPP2R1A, PPP2R2A, PTPN11, RAF1, RUNX1, STAG2, XPO1) were related to TMB-H with enhanced anti-tumor immune response independent of dMMR/MSI-H, constructing a predictive model (modified TMB, mTMB) for ICIs. Patients with any mutation in mTMB showed a superior survival benefit from ICIs (nivolumab or pembrolizumab) in MSS GI cancers in the CARIS cohort (n=95, median overall survival: 18.8 vs 7.03 months, _p_=0·044). In addition, Chr11q13 amplification (e.g., CCND1, FGFs) was more prevalent in the MSS&TMB-H tumors.
Interpretation:
Not all the mutations related to TMB-H can enhance anti-tumor immune response. More composite biomarkers should be investigated (e.g., mTMB signature) to tailor ICIs. Our data also provide novel insights for the combination of targeting CCND1/FGFs and ICIs.
Funding
National Cancer Institute, Gloria Borges WunderGlo, Dhont Family, Gene Gregg Pancreas Research, San Pedro Peninsula Cancer Guild, Daniel Butler Research, Victoria and Philip Wilson Research, Fong Research, Ming Hsieh Research, Shanghai Sailing Program, China National Postdoctoral Program for Innovative Talents, China Postdoctoral Science Foundation, National Natural Science Foundation of China.
Introduction
The introduction of immune checkpoint inhibitors (ICIs) targeting programmed cell death protein 1/ligand 1 (PD-1/PD-L1) has revolutionized cancer therapy, providing robust and durable responses in a subset of patients with cancer. The variability of response to ICIs highlights the unmet need for identifying and validating predictive biomarkers. As a proxy for the expression of tumor-specific neoantigens, high tumor mutational burden (TMB-H) was reported to be associated with durable responses based on KEYNOTE-158 trial, accelerating the pan-cancer approval by the United States Food and Drug Administration (FDA) for pembrolizumab to treat patients with TMB-H [≥10 mutations (mut) per megabase (Mb)] advanced tumors1. However, KEYNOTE-158 may not be representative of a common cancer type as the clinical benefit varied widely by tumor histology, and assessment was limited to overall response rate (ORR) rather than survival advantage. These concerns limit the generalizability of TMB as a robust tissue-agnostic marker.
Microsatellite instability-high (MSI-H) is well recognized as a positive predictor for ICI efficacy; however, over 90% gastrointestinal (GI) tumors are microsatellite stable (MSS). Studies in patients with MSS GI cancers have shown mixed results for TMB-H as a predictive biomarker for ICI efficacy, including use of the FDA-approved threshold of 10 mut/Mb. Xu et al revealed that a higher ORR was observed in TMB-H gastric cancer (GC) than in TMB-Low (L) GC, without consideration of mismatch-repair deficiency (dMMR)/MSI-H status2. However, a retrospective analysis of 251 GI tumors (derived from a database of 1678 MSS solid tumors) from Memorial Sloan Kettering Cancer Center (MSKCC) revealed no significant association between TMB-H and clinical benefit [including ORR, progression-free survival (PFS), overall survival (OS)] 3. Besides, Kim’s study4, EPOC1603 study5, and Marabelle’s study1 did not observe a significant association between TMB and ORR in MSS GC, colorectal cancer (CRC), and anal cancer respectively. These data suggest caution in using TMB as a predictive biomarker for ICI efficacy in MSS GI tumors despite the FDA tumor-agnostic approval.
Increasing evidence reveals that the quality of neoantigens may predict ICI efficacy more accurately than the quantity. Genetic mutations may have an impact on peptide presentation, intratumoural heterogeneity and the tumor immune microenvironment (TIME), all of which further influence the immunogenicity of neoantigens6. However, this complex biological process is not included by the current TMB scoring system. Thus, improved understanding of molecular features of MSS GI tumors with TMB-H may provide novel insights for patient selection, the biology of the tumor-immune interaction, and the development for more rational immunotherapy combinations in the future. In this study, we aimed to explore the impact of molecular features associated with TMB-H on TIME and the survival benefit of ICI treatment in MSS GI tumors.
Materials and Methods
Tumor Samples
A total of 48606 samples with pathologically confirmed gastrointestinal cancer were collected in the US by a commercial Clinical Laboratory Improvement Amendments (CLIA)-certified laboratory (Caris Life Sciences, Phoenix, USA) (referred as CARIS). Formalin-fixed paraffin embedded tissue (FFPE) were prepared at each patient’s submitting pathology department according to each facility’s own standard protocols. The tissue blocks were cut into 4 μm sections using microtomes. All the samples with TMB and MSI status were included in our study. International cases and tumors of non-GI cancer types were excluded. In addition, cases tested with a platform but yielded with no informative results (indeterminate due to low coverage for the particular gene) were excluded from the denominators when calculating the prevalence. According to local or national treatment guidelines, 95 patients receiving pembrolizumab or nivolumab were explored for survival analysis retrospectively in the CARIS database (2016.6-2021.4). All tests, results and the validation data associated with this study have met the CLIA/ The College of American Pathologists and International Organization for Standardization requirements. (https://www.ncbi.nlm.nih.gov/gtr/tests/560898/performance-characteristics/; https://www.ncbi.nlm.nih.gov/gtr/labs/505822/). Public databases [Peking University Cancer Hospital (PUCH) and MSKCC] were used for ICI-related survival analysis, while TCGA cohort for non-ICI survival analysis. Study design is shown in Figure S1 and Supplementary method.
Ethics approval
Our study was conducted according to Institutional Review Board (IRB) guidelines. Since this is a retrospective biomarker study with all data deidentified, our study is considered exempt from IRB approval (Sponsor Protocol No.: CCC-001-0320). The most common exemption for informed consent requirement cited was that the research falls under 45 CFR 46.104(d)(4), shown as below:
“The research involves the use of identifiable private information/biospecimens; and information, which may include information about biospecimens, is recorded by the investigator in such a manner that the identity of the human subjects cannot readily by ascertained directly or through identifiers linked to the subjects, the investigator does not contact the subjects, and the investigator will not reidentify subjects.”
The collections for clinical data and tissues were approved by TCGA-specific institutional review boards (IRB)7, MSKCC8, and PUCH9.
Next-generation sequencing (NGS)
NGS was performed on genomic DNA isolated from FFPE tumor samples using the NextSeq or NovaSeq 6000 platforms (Illumina, Inc., San Diego, CA). For NextSeq sequenced tumors, a custom-designed SureSelect XT assay was used to enrich 592 whole-gene targets (Agilent Technologies, Santa Clara, CA). For NovaSeq sequenced tumors, more than 700 clinically relevant genes at high coverage and high read-depth (>700x) were used, along with another panel designed to enrich for an additional >20,000 genes at lower depth (>200x). All variants were detected with > 99% confidence based on allele frequency and amplicon coverage, with an average sequencing depth of coverage of > 500 and an analytic sensitivity of 5%. Prior to molecular testing, tumor enrichment was achieved by harvesting targeted tissue using manual microdissection techniques. Genetic variants identified were interpreted by board-certified molecular geneticists (Oberley M is the leader of pathologists and geneticists from CARIS) and categorized as ‘pathogenic,’ ‘likely pathogenic,’ ‘variant of unknown significance,’ ‘likely benign,’ or ‘benign,’ according to the American College of Medical Genetics and Genomics standards. When assessing mutation frequencies of individual genes, ‘pathogenic,’ and ‘likely pathogenic’ were counted as mutations. The copy number alteration of each exon is determined by calculating the average depth of the sample along with the sequencing depth of each exon and comparing this calculated result to a pre-calibrated value. TMB-H was defined as ≥10 mut/Mb, according to the Friends of Cancer Research TMB Harmonization Project10. Genomic features were also compared when 20 and 50 mut/Mb were used as the cutoff for TMB-H (Figure S5). More details for fusion detection, immunohistochemistry staining (IHC) and Chromogenic In Situ Hybridization (CISH) were descripted in the supplementary method.
dMMR/MSI-H
A combination of multiple test platforms was used to determine the MSI or MMR status, including fragment analysis (FA, Promega, Madison, WI), immunohistochemistry staining (IHC, MLH1, M1 antibody; MSH2, G2191129 antibody; MSH6, 44 anti-body; and PMS2, EPR3947 antibody; Ventana Medical Systems, Inc., Tucson, AZ) and NGS (7,000 target microsatellite loci were examined and compared to the reference genome hg19 from the University of California). The three platforms generated highly concordant results and in the rare cases of discordant results, the MSI or MMR status of the tumor was determined in the order of IHC, FA and NGS.
Establishment of mTMB signature
Genes from panel A (genes with exclusively highest mutation rates in the MSS&TMB-H subgroup) and panel B (genes with similar mutation rates between dMMR/MSI-H and MSS&TMB-H subgroups, but significantly lower in the MSS&TMB-L subgroup) were explored for the potential association with antitumor immune response using the immune signature scores (calculated by averaging the expression value of included genes in the corresponding signature gene sets), and the infiltration of immune cells (estimated using CIBERSORT and xCell algorithms) (Supplementary method). Only genes with positive association with anti-tumor immune response in MSS GI tumors were further used to construct the predictive model for ICI treatment.
Statistical analysis
Statistical analysis was performed with R 3.5.0 (R Foundation for Statistical Computing, Vienna, Austria) and SPSS 26.0 (Chicago, IL). Comparisons in continuous data were evaluated using the Mann-Whitney U test. Categorical data were evaluated using the Fisher-Exact or Chi-square test with false discovery rate (q-value) controlled to 0·05 using Benjamini-Hochberg for multiple and pairwise comparison. Immune-related OS was defined as the time from initial immunotherapy treatment to the day of death or the end of follow-up. We used the Kaplan–Meier method to estimate survival functions and the log-rank test to compare survival distributions. In the sensitivity analysis, we included ICI-treated patients ranked top 95% from the longest to the shortest of follow-up time in the CARIS cohort. All p<0·05 was considered significant.
Role of the funding source
The funder of the study had no role in study design, data collection, data analysis, data interpretation, or writing of the report.
Results
The most common tumor type in the CARIS cohort was colorectal cancer (Figure S2). The overall prevalence of MSS&TMB-H in this GI-tumor cohort was 3·29% (1600/48606), while dMMR/MSI-H and MSS&TMB-L were observed in 4·66% (2272/48606) and 92·05% (44734/48606) respectively (Figure 1A). When broken into cancer types by anatomic site of origin, the prevalence of MSS&TMB-H ranged from 0·1% to 13·17%, with the highest in anal carcinoma (13·17%, 88/668), followed by esophageal squamous cell cancer (10·01%, 81/809), whereas the lowest was seen in gastrointestinal stromal tumors (0·1%, 1/1002) (Figure 1B).
Figure 1. The relationship between dMMR/MSI-H and TMB-H status in GI cancers.
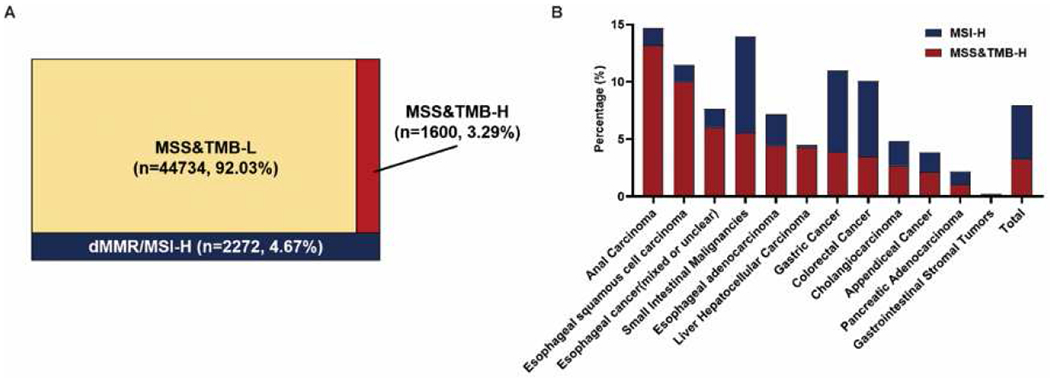
A. An overview of the distribution of MSI and TMB status in GI cancers. Of 48606 total patients, 1600 patients are MSS&TMB-H, 2272 patients are dMMR/MSI-H, 44734 patients are MSS&TMB-L. TMB-H cutoff: 10 mut/Mb. B. Distribution of MSI and TMB status amongst various tumor histologies (n=12) in GI cancers. Abbreviations: MSS, microsatellite stability; MSI-H, microsatellite instability-High; dMMR, mismatch-repair deficiency; TMB-H/L, tumor mutation burden-high/low; GI cancer, gastrointestinal cancer.
Gene mutations were more common in the dMMR/MSI-H subgroup, followed by MSS&TMB-H and MSS&TMB-L subgroups (Figure S3). Frameshift mutations were enriched in the dMMR/MSI-H subgroup, while nonsense mutations were enriched in the MSS&TMB-H subgroup (Figure S4). The MSS&TMB-H subgroup carried the exclusively highest mutation rates in POLE (MSS&TMB-H vs dMMR/MSI-H vs MSS&TMB-L: 6·97%, 111/1593; vs 0·66%, 15/2270; vs 0·01%, 4/44625) , XPO1 (2·09%, 32/1530 vs 0·09%, 2/2179; vs 0·04%, 15/42620) , SMAD2 (4·77%, 75/1571; vs 2·83%, 64/2264; vs 1·38%, 606/43790), ERBB4 (1·14%, 18/1582; vs 0·40%, 9/2265; vs 0·16%, 69/44305), MAP2K1 (2·63%, 42/1599; vs 1·45%, 33/2272; vs 0·74%, 331/44709), MTOR (2·01%, 32/1592; vs 0·75%, 17/2267; vs 0·54%, 241/44518) , NFE2L2 (1·90%, 27/1422; vs 0·24%, 5/2111; vs 0·54%, 220/40647), and TERT (5·15%, 42/815; vs 0·94%, 9/953; vs 2·99%, 574/19177) (defined as Gene panel A), compared to dMMR/MSI-H and MSS&TMB-L subgroups (q<0·0001, Foldchange>1.5, Figure 2A). With the increase of the TMB cutoff, POLE mutations dominate the ultra-mutated cancers [mutation rate 77.94% (106/136) and 43.82% (110/251) for TMB≥50 and ≥20 mut/Mb respectively]in the MSS&TMB-H subgroup (Figure S5). The frequencies of gene mutation in PIK3R1, CDC73, JAK2, RB1, MAP2K4, CTNNA1, EZH2, KEAP1, RUNX1, PPP2R2A, PPP2R1A, STAG2, PTPN11, RAF1 and RASA1 (defined as Gene panel B) were similar between dMMR/MSI-H and MSS&TMB-H subgroups, but significantly lower in the MSS&TMB-L subgroup [_q_<0·0001, Foldchange>3, Figure 2B]. MSS&TMB-H subgroup exhibited significantly lower mutation rates in RNF43, ARID1A, MSH3, KMT2D, and ASXL1 (top 5) than dMMR/MSI-H subgroup, but significantly higher mutation rates than MSS&TMB-L subgroup (q<0·0001, Figure 2C).
Figure 2. Distinct genomic mutations among dMMR/MSI-H, MSS&TMB-H, MSS&TMB-L subgroups in GI cancers.
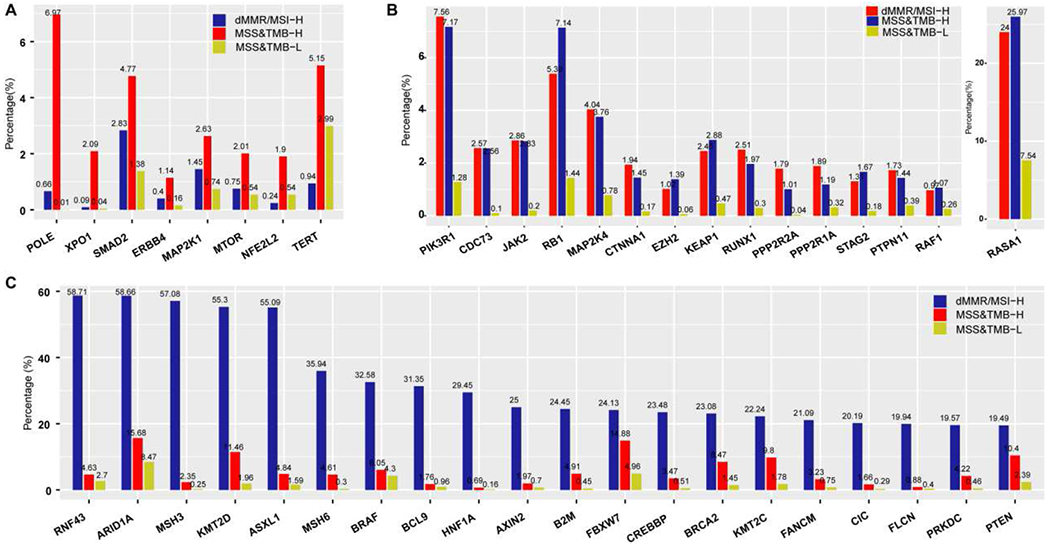
A. The landscape of gene mutations related to TMB-H, independent of dMMR/MSI-H in GI cancers (All q<0.0001, Foldchange>1.5, ranked by q value). B. The landscape of genes with similar mutation rates between dMMR/MSI-H and MSS&TMB-H subgroups, but significantly lower in the MSS&TMB-L subgroup (All q<0.0001, Foldchange>3, ranked by q value). C. The landscape of genes with significantly lower mutation rates in the MSS&TMB-H subgroup than in the dMMR/MSI-H subgroup, but significantly higher mutation rates than in MSS&TMB-L subgroup (All q<0.0001, ranked by Q value). Abbreviations: MSS, microsatellite stability; MSI-H, microsatellite instability-High; dMMR, mismatch-repair deficiency; TMB-H/L, tumor mutation burden-high/low; GI cancer, gastrointestinal cancer.
Copy number amplifications were more prevalent in the MSS subgroup (Figure S6); tumors with MSS&TMB-H carried the significantly highest amplified genes in CCND1 (MSS&TMB-H vs dMMR/MSI-H vs MSS&TMB-L: 3·74%, 58/1552 vs 0·32%, 7/2192 vs 2·25%, 972/43280, q<0·0001), CCNE1 (2·59%, 40/1542 vs 0·19%, 4/2155 vs· 1·58%, 675/42842, q<0·0001), FGF19 (3·78%, 58/1534 vs 0·37%, 8/2167 vs 2·24%, 955/42688, q <0·0001), FGF3 (4·08%, 63/1518 vs 0·37%, 8/2135 vs 2·38%, 995/41739, q <0·0001), FGF4 (3·75%, 58/1548 vs 0·37%, 8/2184 vs 2·03%, 873/42942, q <0·0001), and PIK3CA (0·52%, 8/1539 vs 0, 0/2175 vs 0·15%, 64/42981, _q_=0·014), compared to dMMR/MSI-H and MSS&TMB-L subgroups (Figure 3). Most of them (67%, 4/6) are located on chromosome 11q13.
Figure 3. Distinct features of copy number amplifications among dMMR/MSI-H, MSS&TMB-H and MSS&TMB-L in GI cancers (Ranked by q value, q<0.05).
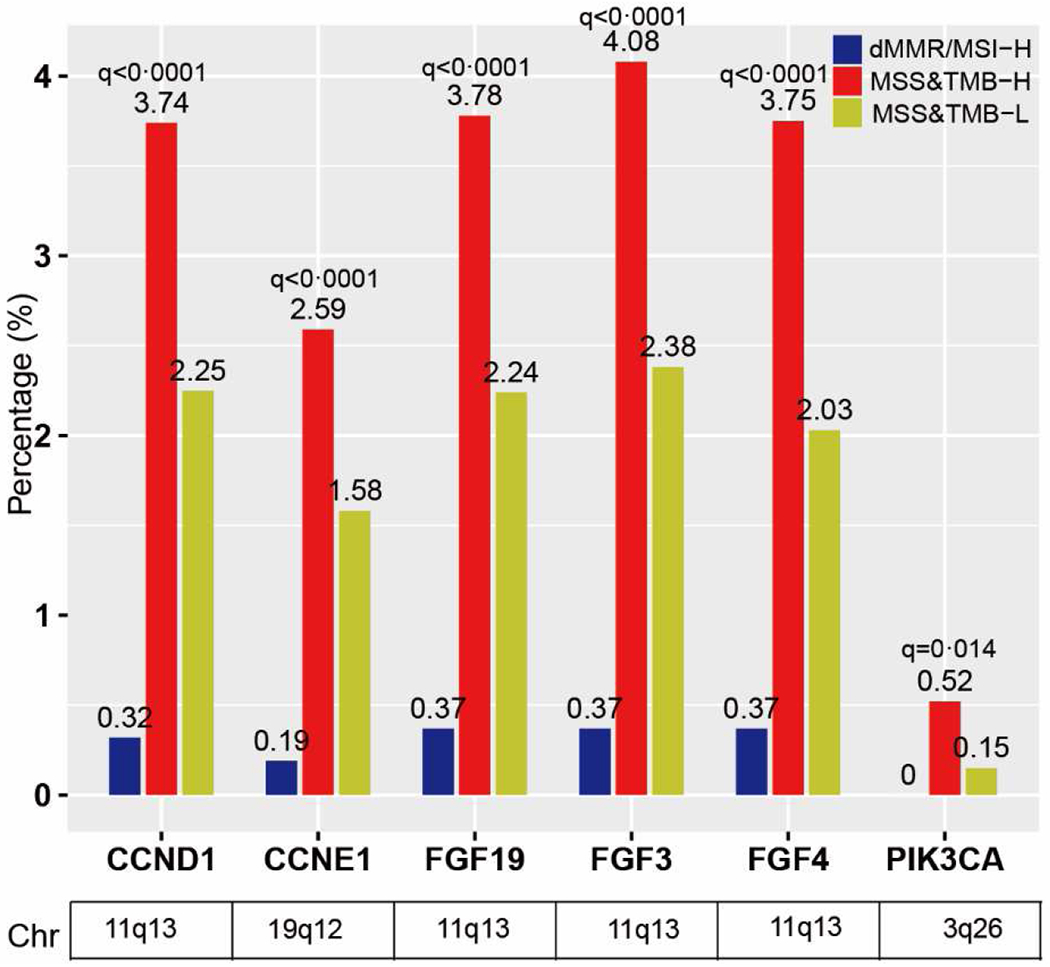
Abbreviations: MSS, Microsatellite stability; MSI-H, Microsatellite instability-high; dMMR, mismatch-repair deficiency; TMB-H/L, tumor mutation burden-high/low; GI cancer, gastrointestinal cancer.
Gene fusions in NTRK1, ALK, RET, and NTRK3 were rare in both MSS&TMB-H and MSS&TMB-L subgroups, but were exclusively highest in the dMMR/MSI-H subgroup. However, FGFR2 fusions were more prevalent in the MSS&TMB-L subgroup, than those in dMMR/MSI-H and MSS&TMB-H subgroups (Figure 4A). HER2 positivity evaluated by IHC and CISH was highest in the MSS&TMB-H subgroup, followed by the MSS&TMB-L and dMMR/MSI-H subgroups, which is also confirmed by copy number analysis (Figure 4B). PD-L1 expression was the highest in the dMMR/MSI-H subgroup, following by MSS&TMB-H and MSS&TMB-L subgroups (Figure 4C).
Figure 4. Differences of gene fusions, HER2 status and PD-L1 expression among dMMR/MSI-H, MSS&TMB-H and MSS&TMB-L subgroups in GI cancers.
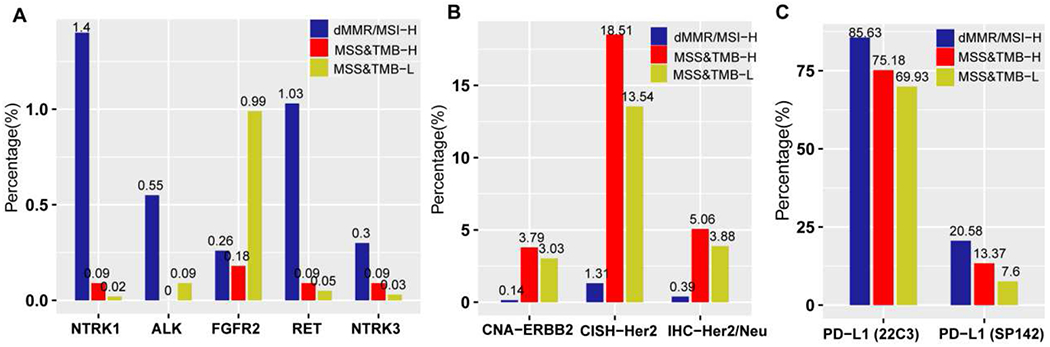
A. Comparison of actionable gene fusions among MSS&TMB-H, dMMR/MSI-H and MSS&TMB-L subgroups (All P<0.0001). B. Comparison of HER2 positivity evaluated by CISH, IHC and copy number analysis among MSS&TMB-H, dMMR/MSI-H and MSS&TMB-L subgroups (All P<0.0001). C. Comparison of PD-L1 expression among MSS&TMB-H, dMMR/MSI-H and MSS&TMB-L subgroups (All P<0.0001). For PD-L1 expression, Dako Link 48 platform for Dako 22C3 pharmDx kits and Ventana Benchmark Ultra platform for SP142 assay kit were used for gastroesophageal cancer and other GI cancers respectively. Abbreviations: MSS, microsatellite stability; MSI-H, microsatellite instability-High; dMMR, deficient mismatch repair; TMB-H/L, tumor mutation burden-high/low; IHC, immunohistochemical staining, IHC; CISH, Chromogenic In Situ Hybridization; Copy number amplification, CNA; GI cancer, gastrointestinal cancer.
Whether genes mutations in Gene Panel A and Panel B could enhance anti-tumor immune response remains unknown. Our data showed that NFE2L2 and TERT mutations significantly increased fraction of genome alterations (p<0.0001) and intratumour heterogeneity (_p_=0·002), respectively (Figure S7A), which may contribute to the resistance to the ICIs11. The immune cell infiltration analyses suggested that MTOR, KEAP1, NFE2L2, RASA1, RB1, and SMAD2 were predicted to be negatively associated with anti-tumor immune response [e.g. the decrease of natural killer (NK) T cells (NFE2L2, p<0·0001;); NK Cells Activated (_MTOR, ρ=0·006; SMAD2, p_=0·007); CD8+T (_RASA1, p_=0·019; _RB1, p_=0·002; _SMAD2, p_=0·003) and Type 1 T helper (Th1) cells (_NFE2L2, p_=0·025; _RB1, p_=0·018); the elevation of M2-like macrophages (_KEAP1, p_=0·001) and neutrophils (_MTOR, p_=0·022)] (Figure S7B-C). In addition, RB1 and NFE2L2 mutations are associated with the downregulation of immunoreactive signature scores (e.g., RB1: cytolytic activity, _p_=0·006, effective T cell score, _p_=0·016, T-cell-inflamed gene expression profile, _p_=0·007, lymphocyte infiltration signature score, _p_=0·002; NFE2L2: lymphocyte infiltration signature score, _p_=0·018) (Figure S7D).These results indicated that MTOR, TERT, KEAP1, NFE2L2, RB1, RASA1 and SMAD2 mutations may impair anti-tumor immune response in MSS GI tumors, despite the association with TMB-H. Therefore, we evaluated 16 other genes associated with active TIME (CDC73, CTNNA1, ERBB4, EZH2, JAK2, MAP2K1, MAP2K4, PIK3R1, POLE, PPP2R1A, PP2R2A, PTPN11, RAF1, RUNX1, STAG2, XPO1) to construct a modified TMB gene signature (mTMB) to further explore its impact on TIME and the efficacy of ICIs in the MSS GI tumor cohort. Interestingly, TMB in tumors with any mutation in MTOR, TERT, KEAP1, NFE2L2, RB1, RASA1 and SMAD2 were significantly lower than in tumors with any mutation in mTMB signature (Figure S8).
MSS GI tumors with any mutation in mTMB signature displayed significantly higher mutation counts (p<0·0001), indel (p<0·0001) and SNV (p<0·0001) neoantigens, B-cell receptor (BCR) richness (_p_=0·024) (Figure 5A-C), compared to mTMB wildtype tumors. Gene mutations in mTMB signature were also associated with high IFNγ signature score (_p_=0·007), high infiltration of activated NK cells (_p_=0·013), M1-like macrophages (_p_=0·00019) and Th1 cells (_p_=0·013), but low infiltration of M2-like macrophages (_p_=0·014) (Figure 5D-E, Figure S9).
Figure 5. The impact of gene mutations in mTMB signature on tumor immune environment and ICI efficacy in GI cancers.
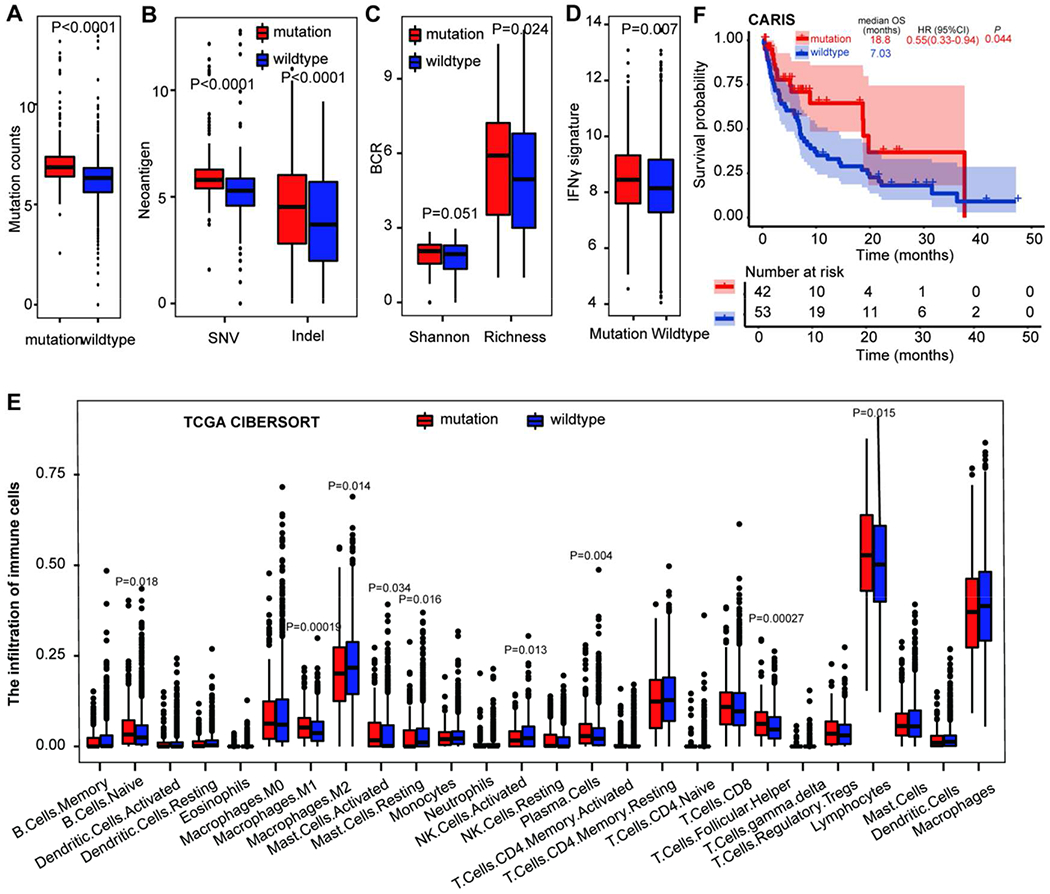
A. The difference of mutation counts between GI tumors with and without gene mutations in mTMB signature. B. The difference of neoantigens between GI tumors with and without gene mutations in mTMB signature. C. The difference of BCR repertoires between GI tumors with and without gene mutations in mTMB signature. D. The association of mTMB signature with immune signature in GI cancers. E. The impact of gene mutations in mTMB signature on the infiltration of immune cells in GI cancers using CIBERSORT. F. The impact of gene mutations in mTMB signature on the overall survival in patients with MSS GI cancers who received ICIs in the CARIS database. Abbreviations: MSS, microsatellite stability; MSI-H, microsatellite instability-High; TMB-H/L, tumor mutation burden-high/low; TCGA, The Cancer Genome Atlas; GI cancer, gastrointestinal cancer; mTMB, modified TMB; ICI, immune checkpoint inhibitor.
Basic characteristics for ICI cohorts were shown in Table S1, but ethnicity was not collected for our study. After a median follow-up of 6·5 (IQR 2·1–14·3) months, patients with any mutation in mTMB (n=42) exhibited significantly longer OS (18·8 [95%CI: 17·30-20·24] vs 7·03 [5·73-8·34] months, HR: 0·55 [0·33-0·94], _p_=0·044, Figure 5F), compared to mTMB wildtype tumors (n=53) in ICI-treated patients with MSS GI tumors in the CARIS cohort. This association remained significant in the sensitivity analysis (median follow-up: 6·73 [IQR 2·39-15·28] months; OS: 18·77 [95%CI 17.32·20.22] vs 7·03 [5·58-8·48] months, HR: 0·49 [0·27-0·92], _p_=0·024, Figure S10). A similar trend (HR: 0·47 [0·30-0·74], _p_=0·0018) was also observed in the MSKCC cohort (Figure S11A), but MSI-H bias for this cohort limited the analysis. No significant association between any mutation in mTMB signature and OS in the PUCH cohort (HR: 0·54 [0·19-1·54], _p_=0·32), probably due to the small sample size (Figure S11B). Of interest, at median follow-up of 17·27 (IQR 9·94-30·59) months, there was no significant association between gene mutations in mTMB signature and survival benefit in GI patients who never received ICI treatment (54·07 [95%CI: 41·24-55·96] vs· 48·6 [34·51-73·62] months, HR:0·94 [0·74-1·20], _p_=0·61, Figure S12, Table S2). The potential mechanism of the mTMB signature leading to the increased efficacy of ICI treatment is shown in Figure S13.
Discussion
To the best of our knowledge, this is the largest study to explore the molecular features of MSS GI tumors with TMB-H. In our study, the prevalence of TMB-H in MSS GI cancers was 3·29%, lower than Goodman’s study (43 different histologies)12. This discrepancy is probably due to the heterogeneity of population, the composition of cancer types, technological issues, and the different TMB algorithm. However, up to now, no sufficient evidence shows that MSS GI tumors with TMB⩾10 mutations/Mb can benefit from ICI treatment. Efforts to determine TMB accurately and adopt an optimal TMB threshold in a given cancer type are ongoing.
The correlation between TMB and the formation of immunogenic neo-antigens may partly depend on the mutational signatures, causing ICI response or resistance. Our study suggests that it may not be the quantity of mutations but the quality of mutation generating immunogenic neo-antigens. Not all the genes associated with TMB-H improve anti-tumor immune response. We found gene mutations in SMAD2, MTOR, KEAP1, NFE2L2, RB1, TERT and RASA1 were associated with negative ICI predictors, lower infiltration of immunoreactive cells or the inactivation of immune-related pathways, despite high TMB. SMAD2 is related to the restrained tumor-killing effect and the limited infiltration of immune effector cells13. The loss of RB1 can reduce immune cell mobilization and antigen presentation14. Mutations in KEAP1-NFE2L2 pathway may protect tumor cells from oxidative stress, promoting tumor growth and aggressiveness.
Although KEAP1-NFE2L2 mutations are associated with TMB-H, the deficient infiltration of CD4+ T cells, NK T cells and Treg cells were observed in KEAP1-NFE2L2 mutant non-small-cell lung cancers (NSCLCs)15. TERT mutations were reported to be a negative predictor in metastatic renal cell carcinoma16. The association of mutations in RASA1 and MTOR with TMB and ICI efficacy are not well studied so far. Based on our study, RASA1 mutations were related to lower infiltration of CD8+ T cells. MTOR mutations may reduce the infiltration of activated NK cells. Meanwhile, genes in mTMB signature were predicted to be associated with the enhanced anti-tumor immune response, indicated by the increase of the infiltration of active immune cells (e.g. M1-like macrophage, Th1 cells, NKT cells, and so on), which were further used to construct a mTMB signature. We observed that patients with any mutation in mTMB signature have a significant OS benefit when treated with ICIs but not in patients receiving chemotherapy or surgery. Among them, POLE mutations provide an important proofreading function during DNA replication, leading to a distinct hypermutated but MSS phenotype with improved ICI efficacy. In NSCLCs, mutations in XPO1 and ERBB4 are positively associated with TMB-H and high PD-L1 expression, which resulted in higher response rates to ICIs17,18. MAP2K1, MAP2K4, RAF1, and PTPN11 mutations can lead to the activation of the MAPK pathway which we have previously shown to increase benefit from ICI treatment in patients with gastroesophageal adenocarcinomas19. The inactivation of EZH2 can sensitize tumors to ICIs by improving effector functions of CD8+ T cells, promoting IFN-γ production and cytotoxicity20. JAK2 loss-of-function mutations, such as nonsense mutations, was considered as a predictive marker for hyperprogression; however, emerging evidence has shown that JAK2 missense mutations, especially p.V617F, could confer sensitivity to ICI treatment21. In our study, we did not include JAK2 nonsense mutations in the mTMB signature. The association between various mutation sites of JAK2 and ICI efficacy should be explored further. The inactivation of PP2A (encoded by PPP2R1A and PPP2R2A) could convert “cold” MSS into MSI tumors via triggering neo-antigen production, cytotoxic T cell infiltration and ICI sensitization22. STAG2 is associated with genomic stability. STAG2 deficiency may induce interferon response and PD-L1 expression, leading to the increased response to ICI in melanoma23. Mutations in PIK3R1 may lead to the activation of PI3K/AKT pathway, which was reported as a primary resistance mechanism to ICI in MSI-H GI cancers24. In contrast, in MSS colorectal cancer, PIK3CA mutations are associated with increased cytotoxic T cell infiltration, higher PD-L1 expression, and greater clinical benefit from ICI25, highlighting that the regulation of tumor immunity by PI3K-AKT pathway is context-dependent. No studies about the association between CTNNA1, CDC73, RUNX1 and ICI efficacy were reported. Based on their biology, we speculated that CDC73 and RUNX1 were involved in RNA polymerase II transcription26,27; thus, mutations in CDC73 and RUNX1 may increase the transcriptional stress and genomic instability. However, not all the gene mutations in mTMB signature are equal to induce efficient neoantigens presented by their MHC. More in depth research efforts about the optimization of predictive models for ICI treatment are warranted.
Copy number alteration burden is negatively associated with ICI efficacy. Patients with high TMB and low copy number alteration cancer can be an optimal subgroup for ICI therapy in GI cancers28. Patients with 11q13 amplification may have inferior efficacy to ICI treatment in esophageal squamous cell cancer29. Mechanically, 11q13 amplifications might impair the antitumor activity, indicated by the decrease in CD8+ T cell, NKT and B cell infiltration30. In our study, we found copy number amplification was more prevalent in MSS tumors, especially in Chr11q13 (CCND1, FGF3, FGF4, FGF19), strengthening the rationale for combination therapies with agents targeting the CCND1 and FGFs with ICIs. In addition, our data also showed that MSS GI tumors with TMB-H has a higher frequency of HER2 amplification compared to dMMR/MSI-H and MSS&TMB-L GI tumors, which may provide one of the theoretical explanations for the enhanced efficacy of the combined ICIs with anti-HER2 treatment.
Limitations of this work need to be mentioned, including its retrospective nature and the heterogeneity of cancer types between different cohorts. Due to the population of the MSS&TMB-H subgroup, we had to analyze all the cancer types together. It is worthy to explore the genomic signatures related to TMB-H and the association with ICI efficacy in each cancer type separately. Second, due to the small sample size and the lake of clinical information (e.g. T cell infiltration density, CRP levels, antibiotic treatment, etc.) of ICI cohorts in our study which may influence ICI efficacy, prospective randomized studies with a larger sample size of GI cancers are warranted to further to confirm the predictive value of mTMB signature for ICIs and its interaction with TIME by integrated analyses of multiple omics.
In conclusion, our data suggest that not all the mutations related to TMB-H can improve anti-tumor immune response, and specific gene mutations, such as SMAD2, MTOR, KEAP1, NFE2L2, RB1, TERT and RASA1 should also be taken into consideration. The combination of TMB and gene mutations positively regulating the anti-tumor immunity (such as mTMB) may be a promising tool for patient selection for ICI treatment. Our data also provide novel insights for the combination of targeting CCND1/FGFs and ICI treatment.
Supplementary Material
Supplementary Appendix
Research in context:
Evidence before this study
We searched PubMed for peer-reviewed, original studies (with no start date and up to January 10, 2022), using the search terms “Tumor mutation burden”, “immune checkpoint inhibitor”, “microsatellite stable”, and “cancer”. We also reviewed congress abstracts in the fields of immunotherapy in microsatellite stable (MSS) cancers. Although pembrolizumab is recommended for patients with high tumor mutation burden (TMB-H) solid tumors based on KEYNOTE-158 trial, studies in MSS gastrointestinal (GI) cancers have shown mixed results for TMB-H as a predictive biomarker for immune checkpoint inhibitors (ICIs). Increasing evidence revealed that quality of neoantigens, which could be affected by genetic mutations, may predict ICI efficacy more accurately than the quantity. Several gene mutations, such as POLE, are associated with TMB, independent of microsatellite instability-high (MSI-H). However, no systematic analysis for molecular features of MSS GI tumors with TMB-H was reported.
Added value of this study
This is the largest study to explore the molecular features of MSS GI tumors with TMB-H, providing novel insights for patient selection, the biology of the tumor-immune interaction, and the development of rational immunotherapy combinations in the future. Mutations in SMAD2, MTOR, NFE2L2, RB1, KEAP1, TERT, and RASA1 were associated with suppressed tumor immune environment, despite high TMB. Gene mutations in 16 genes (CDC73, CTNNA1, ERBB4, EZH2, JAK2, MAP2K1, MAP2K4, PIK3R1, POLE, PPP2R1A, PPP2R2A, PTPN11, RAF1, RUNX1, STAG2, XPO1) were related to TMB-H with improved anti-tumor immune response independent of dMMR/MSI-H, constructing a predictive model (modified TMB, mTMB) for ICI efficacy. Copy number amplification in Chr11q13 (CCND1, FGFs) was more prevalent in the MSS&TMB-H tumors.
Implications of all the available evidence
Our study indicated that not all the genes associated with TMB-H can improve anti-tumor immune response. Mutations in SMAD2, MTOR, NFE2L2, RB1, KEAP1, TERT, and RASA1, should be taken into consideration, when patients with TMB-H are considered for ICI treatment. In addition to TMB, more composite biomarkers should be developed (such as mTMB signature) for more effective patient selection for ICIs. Our data also provide novel insights for the combination of targeting CCND1/FGFs and ICIs.
Funding
This work was partly supported by National Cancer Institute (grant number P30CA014089), Gloria Borges WunderGlo Foundation, Dhont Family Foundation, Gene Gregg Pancreas Research Fund, San Pedro Peninsula Cancer Guild, Daniel Butler Research Fund, Victoria and Philip Wilson Research Fund, Fong research project, Ming Hsieh research fund, Shanghai Sailing Program (22YF1407000), China National Postdoctoral Program for Innovative Talents (BX20220084), China Postdoctoral Science Foundation (2022M710768), National Natural Science Foundation of China (82202892). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute of the National Institutes of Health.
Declaration of interests
H.-J.L. reports receiving honoraria from consultant/advisory board membership for Merck Serono, Bayer, and Genentech. J.X, A. F, B.Y, M.O, D.S, and W.M.K. are employers of Caris Life Sciences. A.F.S reports funding for research, travel, and the speakers bureau from Caris Life Sciences. B.A.W reports receiving honoraria from Bayer, Sirtex, Lilly, Taiho, and HalioDx. Caris and the university of Southern California have not licensed any intellectual property on mTMB at this time. All remaining authors have declared no conflicts of interest.
Data sharing
Study protocol and statistical analysis plan are available in the paper. Other data (including the summary of clinical and genomic data.) will be made available upon reasonable request with the permission of caris life science. Individual participant data of CARIS cohort from this study, are not available for sharing.
Reference:
- 1.Marabelle A, Fakih M, Lopez J, et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. The Lancet Oncology 2020; 21(10): 1353–65. [DOI] [PubMed] [Google Scholar]
- 2.Wang F, Wei XL, Wang FH, et al. Safety, efficacy and tumor mutational burden as a biomarker of overall survival benefit in chemo-refractory gastric cancer treated with toripalimab, a PD-1 antibody in phase Ib/II clinical trialNCT02915432. Ann Oncol 2019; 30(9): 1479–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Valero C, Lee M, Hoen D, et al. Response Rates to Anti-PD-1 Immunotherapy in Microsatellite-Stable Solid Tumors With 10 or More Mutations per Megabase. JAMA Oncol 2021; 7(5): 739–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim ST, Cristescu R, Bass AJ, et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat Med 2018; 24(9): 1449–58. [DOI] [PubMed] [Google Scholar]
- 5.Fukuoka S, Hara H, Takahashi N, et al. Regorafenib Plus Nivolumab in Patients With Advanced Gastric or Colorectal Cancer: An Open-Label, Dose-Escalation, and Dose-Expansion Phase Ib Trial (REGONIVO, EPOC1603). J Clin Oncol 2020; 38(18): 2053–61. [DOI] [PubMed] [Google Scholar]
- 6.Jardim DL, Goodman A, de Melo Gagliato D, Kurzrock R. The Challenges of Tumor Mutational Burden as an Immunotherapy Biomarker. Cancer Cell 2021; 39(2): 154–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thorsson V, Gibbs DL, Brown SD, et al. The Immune Landscape of Cancer. Immunity 2018; 48(4): 812–30.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Samstein RM, Lee CH, Shoushtari AN, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nature genetics 2019; 51(2): 202–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiao X, Wei X, Li S, et al. A genomic mutation signature predicts the clinical outcomes of immunotherapy and characterizes immunophenotypes in gastrointestinal cancer. npj Precision Oncology 2021; 5(1): 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Merino DM, McShane LM, Fabrizio D, et al. Establishing guidelines to harmonize tumor mutational burden (TMB): in silico assessment of variation in TMB quantification across diagnostic platforms: phase I of the Friends of Cancer Research TMB Harmonization Project. J Immunother Cancer 2020; 8(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frigola J, Carbonell C, Irazno P, et al. High levels of chromosomal aberrations negatively associate with benefit to checkpoint inhibition in NSCLC. Journal for immunotherapy of cancer 2022; 10(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goodman AM, Sokol ES, Frampton GM, Lippman SM, Kurzrock R. Microsatellite-Stable Tumors with High Mutational Burden Benefit from Immunotherapy. Cancer immunology research 2019; 7(10): 1570–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bai X, Yi M, Jiao Y, Chu Q, Wu K. Blocking TGF-beta Signaling To Enhance The Efficacy Of Immune Checkpoint Inhibitor. Onco Targets Ther 2019; 12: 9527–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Knudsen ES, Pruitt SC, Hershberger PA, Witkiewicz AK, Goodrich DW. Cell Cycle and Beyond: Exploiting New RB1 Controlled Mechanisms for Cancer Therapy. Trends Cancer 2019; 5(5): 308–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu H, Xie D, Yu Y, et al. KEAP1/NFE2L2 Mutations of Liquid Biopsy as Prognostic Biomarkers in Patients With Advanced Non-Small Cell Lung Cancer: Results From Two Multicenter, Randomized Clinical Trials. Frontiers in oncology 2021; 11: 659200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dizman N, Lyou Y, Salgia N, et al. Correlates of clinical benefit from immunotherapy and targeted therapy in metastatic renal cell carcinoma: comprehensive genomic and transcriptomic analysis. Journal for immunotherapy of cancer 2020; 8(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li X, Zou B, Wang S, Wang L, Yu J. XPO1-mutant NSCLC without STK11/KEAP1 mutations may predict better survival to immunotherapy. Journal of translational medicine 2021; 19(1): 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu X, Xu H, Xue Q, Wen R, Jiao W, Tian K. The role of ERBB4 mutations in the prognosis of advanced non-small cell lung cancer treated with immune checkpoint inhibitors. Molecular medicine (Cambridge, Mass) 2021; 27(1): 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang JY, Xiu J, Baca Y, et al. Distinct genomic landscapes of gastroesophageal adenocarcinoma depending on PD-L1 expression identify mutations in RAS-MAPK pathway and TP53 as potential predictors of immunotherapy efficacy. Ann Oncol 2021; 32(7): 906–16. [DOI] [PubMed] [Google Scholar]
- 20.Wang X, Brea LT, Yu J. Immune modulatory functions of EZH2 in the tumor microenvironment: implications in cancer immunotherapy. Am J Clin Exp Urol 2019; 7(2): 85–91. [PMC free article] [PubMed] [Google Scholar]
- 21.Li SD, Ma M, Li H, et al. Cancer gene profiling in non-small cell lung cancers reveals activating mutations in JAK2 and JAK3 with therapeutic implications. Genome Med 2017; 9(1): 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yen YT, Chien M, Wu PY, et al. Protein phosphatase 2A inactivation induces microsatellite instability, neoantigen production and immune response. Nature communications 2021; 12(1): 7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chu Z, Gu L, Hu Y, et al. STAG2 regulates interferon signaling in melanoma via enhancer loop reprogramming. Nature communications 2022; 13(1): 1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chida K, Kawazoe A, Kawazu M, et al. A Low Tumor Mutational Burden and PTEN Mutations Are Predictors of a Negative Response to PD-1 Blockade in MSI-H/dMMR Gastrointestinal Tumors. Clin Cancer Res 2021; 27(13): 3714–24. [DOI] [PubMed] [Google Scholar]
- 25.Nusrat M, Roszik J, Katkhuda R, et al. Association of PIK3CA mutations (mut) with immune engagement and clinical benefit from immunotherapy in microsatellite stable (MSS) colorectal cancer (CRC) patients (pts). Journal of Clinical Oncology 2019; 37(15_suppl): 3604-. [Google Scholar]
- 26.Rozenblatt-Rosen O, Hughes CM, Nannepaga SJ, et al. The Parafibromin Tumor Suppressor Protein Is Part of a Human Paf1 Complex. Molecular and Cellular Biology 2005; 25(2): 612–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Singh H. Targeting RNA polymerase II Mediator subunits in cancer therapy. Proceedings of the National Academy of Sciences 2021; 118(11): e2100115118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu Z, Chen H, Li S, et al. Tumor copy-number alterations predict response to immune-checkpoint-blockade in gastrointestinal cancer. Journal for immunotherapy of cancer 2020; 8(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang F, Ren C, Zhao Q, et al. Association of frequent amplification of chromosome 11q13 in esophageal squamous cell cancer with clinical benefit to immune check point blockade. Journal of Clinical Oncology 2019; 37(15_suppl): 4036-. [Google Scholar]
- 30.Zhou R, Zhu X, Peng Y, et al. Clinical Impact of 11q13.3 Amplification on Immune Cell Infiltration and Prognosis in Breast Cancer. Int J Gen Med 2022; 15: 4037–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Appendix
Data Availability Statement
Study protocol and statistical analysis plan are available in the paper. Other data (including the summary of clinical and genomic data.) will be made available upon reasonable request with the permission of caris life science. Individual participant data of CARIS cohort from this study, are not available for sharing.