Benchmarking the CATMA Microarray. A Novel Tool forArabidopsis Transcriptome Analysis (original) (raw)
Abstract
Transcript profiling is crucial to study biological systems, and various platforms have been implemented to survey mRNAs at the genome scale. We have assessed the performance of the CATMA microarray designed for Arabidopsis (Arabidopsis thaliana) transcriptome analysis and compared it with the Agilent and Affymetrix commercial platforms. The CATMA array consists of gene-specific sequence tags of 150 to 500 bp, the Agilent (Arabidopsis 2) array of 60mer oligonucleotides, and the Affymetrix gene chip (ATH1) of 25mer oligonucleotide sets. We have matched each probe repertoire with the Arabidopsis genome annotation (The Institute for Genomic Research release 5.0) and determined the correspondence between them. Array performance was analyzed by hybridization with labeled targets derived from eight RNA samples made of shoot total RNA spiked with a calibrated series of 14 control transcripts. CATMA arrays showed the largest dynamic range extending over three to four logs. Agilent and Affymetrix arrays displayed a narrower range, presumably because signal saturation occurred for transcripts at concentrations beyond 1,000 copies per cell. Sensitivity was comparable for all three platforms. For Affymetrix GeneChip data, the RMA software package outperformed Microarray Suite 5.0 for all investigated criteria, confirming that the information provided by the mismatch oligonucleotides has no added value. In addition, taking advantage of replicates in our dataset, we conducted a robust statistical analysis of the platform propensity to yield false positive and false negative differentially expressed genes, and all gave satisfactory results. The results establish the CATMA array as a mature alternative to the Affymetrix and Agilent platforms.
At the turn of the century, the completion of the Arabidopsis (Arabidopsis thaliana) genome sequencing project (The Arabidopsis Genome Initiative, 2000) marked the emergence of the next challenge: the assignment of function to each gene identified in the chromosome sequence. This daunting task depends largely on the availability of a diverse collection of functional genomics resources to the research community (Hilson et al., 2003). It is clear that in addition to the established gene-by-gene research approach, novel ways of working will be essential. Indeed, some 25 years of Arabidopsis molecular biology and genetics have now yielded experimental proof of function for some 3,500 genes (Berardini et al., 2004). It is generally believed that to increase the rate of functional annotation of genes significantly, it will be crucial to adopt computational approaches for mining and modeling information from complex molecular phenotypes. Such phenotypes include transcriptome, proteome, and metabolome data. Already well established, clustering of microarray experiment data can yield valuable clues on the function of poorly annotated genes, dubbed the guilt-by-association approach (for review, see Quackenbush, 2003). Furthermore, initial studies now demonstrate that modeling based on large microarray data compendia shows potential for the reverse engineering of genetic networks (D'Haeseleer et al., 2000; Hughes et al., 2000; Maki et al., 2001; Lee et al., 2002). Thus, the value of microarray-based transcript analysis will most probably continue to increase.
In this context, we have built a novel microarray platform for the systematic analysis of the Arabidopsis transcriptome: the Complete Arabidopsis Transcriptome MicroArray, or CATMA, array. It is the result of a collaborative project joining the efforts and resources of laboratories in eight European countries. The CATMA consortium aims to produce, through PCR and from genomic DNA, gene-specific sequence tags (GSTs) for most annotated Arabidopsis genes (Hilson et al., 2004). A sizeable repertoire of sequence tags is already available today, and all GST information is accessible through the CATMA database (Crowe et al., 2003; http://www.catma.org; also relayed by other Arabidopsis Web sites). The GSTs are designed to have minimal homology with any other sequence in the Arabidopsis genome and consist of 150 to 500 bp fragments (Thareau et al., 2003). The GST amplicons as well as arrays printed with the CATMA GSTs are available from the Nottingham Arabidopsis Stock Centre (NASC; http://nasc.nott.ac.uk/). The GST amplicons can easily be reamplified, and subsets can be picked as preferred to print dedicated arrays. Furthermore, the GSTs can be cloned and used for other functional studies, including gene silencing (Hilson et al., 2004). The CATMA array is being used to build a compendium of Arabidopsis gene expression profiles. The main objective of the European Framework project titled Compendium of Arabidopsis Gene Expression (http://www.psb.ugent.be/CAGE) is to demonstrate that a collective of research laboratories can produce a compendium of high-quality microarray data that supports future microarray experimentation and forms the core of a growing repository of molecular phenotype data.
The Arabidopsis research community has been blessed with multiple independent resources for transcript profiling, both from commercial sources and academic core facilities. However, today, microarrays that do not carry probes for the majority of transcription units identified in the genome, prominently cDNA arrays, are quickly becoming obsolete. Therefore, this is an opportune moment to introduce the CATMA array as an alternative to limited coverage cDNA or commercial oligonucleotide arrays. The aim of our work was to describe in detail the performance of the CATMA array in comparison with the oligonucleotide-based platforms commercialized by Agilent (Arabidopsis 2 oligo array; Palo Alto, CA) and Affymetrix (ATH1 GeneChip probe array; Santa Clara, CA; Redman et al., 2004), and to present these results as a reference to the Arabidopsis research community.
Several studies have already described microarray platform comparison and quality assessment based on various approaches (Chudin et al., 2002; Kuo et al., 2002; Yuen et al., 2002; Lee et al., 2003; Nimgaonkar et al., 2003; Tan et al., 2003). A common method for platform comparison is to determine the concordance of differential expression measurements between contrasted biological samples. Such studies either pointed to platform-specific expression differences (Kuo et al., 2002; Moreau et al., 2003; Tan et al., 2003) or illustrated a broad concordance between different platforms (Barczak et al., 2003). We have chosen not to focus on gene-for-gene comparison of ratio reports between platforms, but rather on the comparative analysis of RNA samples designed specifically to test the hybridization characteristics of the platforms. We have spiked aliquots of a single biological sample with a range of calibrated quantities of in vitro synthesized poly(A) RNAs. These series of synthetic RNAs provided detailed information about the dynamic response of the microarrays, in the context of an invariant base sample transcript profile. Aliquots of a single set of spiked RNA targets were used for all platforms. Our results indicate that CATMA arrays perform equally well as Agilent or Affymetrix arrays in terms of sensitivity, specificity, and the ability to prevent detection of false negative and false positive genes in differential expression studies. However, both the long and short oligonucleotide platforms suffer from signal saturation at high target concentrations, whereas the CATMA array does not. The solid performance of the CATMA array makes it a valid platform for functional genomics studies, and a well-managed core facility may be able to offer CATMA array service at a cost highly competitive with commercial alternatives.
RESULTS
In Silico Coverage
Several genome-scale microarrays are now available for Arabidopsis transcript profiling, and choosing a particular platform will depend on various criteria including genome coverage, data quality, dynamic range, and sensitivity, as well as more practical factors such as availability, price, and logistics. We present here a detailed analysis of the main technical characteristics of the CATMA array, and compare them with the Agilent Arabidopsis 2 oligo array (Agilent array) and the Affymetrix ATH1 genome array (Affymetrix array). Together, these arrays cover the three probe types now used in genome-scale microarrays: PCR amplicons (150–500 bp, CATMA), long oligonucleotides (60mer, Agilent), and short oligonucleotide sets (25mer, Affymetrix).
First, to determine which genes are represented in each of the compared arrays, the sequences of their respective DNA features, or probes, were analyzed with BLAST against all the transcription units described in the Arabidopsis genome annotation provided in January 2004 by The Institute for Genome Research (TIGR; release 5.0). The total number of array probes or probe sets was 18,981 (CATMA v1), 22,072 (CATMA v2), 21,500 (Agilent), and 22,763 (Affymetrix). Note that the CATMA GSTs have been produced in two successive rounds and that this in silico analysis presents both the data on CATMA v1 and CATMA v2 (http://www.ebi.ac.uk/arrayexpress). All hybridization data presented below were obtained with arrays printed with the initial version of the repertoire, CATMA v1 (Hilson et al., 2004; see “Materials and Methods”). Also, approximately 1,000 of the probe sets on the Affymetrix arrays permit cross-hybridization to 1 or more other closely related genes, thus allowing transcript detection of up to 24,000 genes. The TIGR 5.0 genome annotation contains a total of 26,207 protein-coding genes. In addition, it describes genomic regions with homology to open reading frames of transposable elements (2,355) and pseudogenes (1,652), accounting for an additional 3,786 annotations. The coverage is summarized in Table I. The probe design for all platforms was done with genome annotations predating TIGR 5.0. With the continued refinements in the gene prediction algorithms and the increased availability of experimental full-length cDNA sequences, some of these gene models have become obsolete. As a result, all platforms contain probes designed according to previous TIGR gene models that do not appear anymore in the latest release. The table shows that the CATMA array also contains probes for gene models uniquely predicted by the EuGène gene finder software (Schiex et al., 2001) and that Affymetrix is the only platform containing probes for mitochondrial and chloroplast genes (for further details on in silico coverage, see Supplemental Fig. 1 and Supplemental Table I).
Table I.
Overview of in silico coverage
*, Approximately 1,000 genes were not taken into account because their cognate transcripts were detected by overlapping probe sets.
| CATMA v1 | CATMA v2 | Agilent Arabidopsis 2 | Affymetrix ATH1 | |
|---|---|---|---|---|
| Probes/probe sets | 18,852 | 22,072 | 21,500 | 22,763 |
| Transposable elements plus pseudogenes | 363 | 575 | 572 | 946 |
| TIGR 5.0 | 18,122 | 21,019 | 20,921 | 22,348* |
| On TIGR annotation prior to 5.0 | 46 | 57 | 579 | 260 |
| EuGène annotation | 684 | 996 | – | – |
| Organelle genomes | – | – | – | 155 |
Calibrated RNA Samples and Hybridization Series
We chose to evaluate the performance of the three array types by performing the same standardized experiment on each of these platforms, with target labeling, hybridization, and data extraction protocols commonly considered to be optimal for the platform. The targets were derived from the same series of RNA samples that contain known concentrations of calibrated transcripts. The base for all samples was a single batch of total RNA extracted from whole shoots of Arabidopsis ecotype Columbia (Col) harvested at the developmental stage 1.04 (Boyes et al., 2001), also known as TAIR 0000399 (http://www.arabidopsis.org/index.jsp). The different RNA samples were assembled by the addition of in vitro synthesized polyadenylated RNA species (from now on referred to as spike RNAs) to the shoot total RNA. The genes corresponding to the spike RNAs fulfilled the following criteria: (1) they were not transcribed at a detectable level in Col shoots, as shown in prior experiments with either cDNA or Affymetrix arrays; (2) they preferably had to be represented on all three arrays; and (3) plasmids with a cognate polyadenylated cDNA sequence flanked by the T7 promoter, convenient for in vitro transcription, had to be available in an in-house collection of 6,000 clones used for the production of spotted cDNA arrays.
A total of 14 cDNA clones were thus selected (Supplemental Table II) and used as templates to synthesize bona fide polyadenylated spike RNAs. We assumed that 14 spikes would allow an in-depth cross-platform comparison while still constituting a number that could practically be handled. Each spike RNA was calibrated and mixed in equal amount with one of the other spike RNAs to obtain seven pairs at equal concentration (labeled a–g in Fig. 1). These seven spike RNA pairs were then combined systematically to construct seven complex spike mixes in a design similar to an ordered Latin square, each mix containing six of the seven spike pairs in staggered concentrations covering five logs (Table II). As a result, all spike mixes contained equal quantities [amounting to approximately 7.4% of the endogenous cellular poly(A) RNA content] of in vitro synthesized poly(A) RNA. To prevent loss of spike RNA through adsorption to the plasticware, the spike mixes were prepared in 0.5 _μ_g _μ_L−1 shoot total RNA, resulting in a range of concentration from 0.1 to 10,000 copies per cellular equivalent (cpc), assuming that the total RNA contained 1% poly(A) mRNA and that a cell contained on average 300,000 transcripts. To convert the spike hybridization signals to ratios, an eighth sample was prepared, called the reference sample, consisting of the base shoot total RNA completed with all spike RNAs at a concentration of 100 cpc. Thereby, the comparison of any of the seven RNA samples to the reference sample should theoretically yield signal ratios ranging from 100-fold to 0.001-fold across the gene subset corresponding to the spike RNAs and a signal ratio of 1 for all other genes.
Figure 1.

Schematic representation of the experimental design. Each graph represents the content of spike RNA in the RNA sample(s) hybridized to a single array. A, Two-channel arrays (CATMA and Agilent). In series 1 to 7, the RNA samples containing the spike RNAs in staggered concentration were used as template to synthesize the Cy5-labeled targets, whereas the reference sample was used for the Cy3-labeled target. The inverse configuration applied to the 1′ to 7′ series. Cy3 and Cy5 were cohybridized. B, One-color arrays (Affymetrix). The seven RNA samples containing the spike RNAs in staggered concentrations and the eighth reference sample were each used as template for hybridization on a single array.
Table II.
Concentration (copies per cell) of the 14 spike RNAs for the seven different spike mixes and the reference mixa
Each spike RNA was calibrated and mixed in equal amount with one of the other spike RNAs to obtain seven pairs at equal concentration (labeled a–g).
| Spike No. | Spike Mix 1 | Spike Mix 2 | Spike Mix 3 | Spike Mix 4 | Spike Mix 5 | Spike Mix 6 | Spike Mix 7 | Reference Mix |
|---|---|---|---|---|---|---|---|---|
| 1, 8 (a) | 10,000 | 0 | 0.1 | 1 | 10 | 100 | 1,000 | 100 |
| 2, 9 (b) | 1,000 | 10,000 | 0 | 0.1 | 1 | 10 | 100 | 100 |
| 3, 10 (c) | 100 | 1,000 | 10,000 | 0 | 0.1 | 1 | 10 | 100 |
| 4, 11 (d) | 10 | 100 | 1,000 | 10,000 | 0 | 0.1 | 1 | 100 |
| 5, 12 (e) | 1 | 10 | 100 | 1,000 | 10,000 | 0 | 0.1 | 100 |
| 6, 13b (f) | 0.1 | 1 | 10 | 100 | 1,000 | 10,000 | 0 | 100 |
| 7, 14 (g) | 0 | 0.1 | 1 | 10 | 100 | 1,000 | 10,000 | 100 |
Hybridization series were set up to perform all possible combinations with the available RNA samples. For two-color arrays (Cy3/Cy5; CATMA and Agilent), each individual RNA sample was compared directly to the reference sample, and both dye swaps were analyzed, resulting in 14 slides for each platform (Fig. 1A). For the one-color arrays (Affymetrix), each sample, including the reference, was hybridized to one slide, resulting in eight slides (Fig. 1B). Although the total number of hybridizations on Affymetrix arrays was only one-half that of the two-color arrays, this fact actually reflects the practical application of the different platforms for a single observation: two-color arrays, one probe per gene in a dye swap; one-color array, multiprobe set per gene in a single hybridization.
Dose-Response Curves
Microarray data typically provide information about the level of transcripts relative to a common reference. Therefore, it is critical to investigate the dynamic range of the different platforms, i.e. whether they display a linear dose-response relationship between transcript abundance and hybridization signal, and to determine the span of this dynamic range. In our experimental design, all target nucleic acids were synthesized from the same series of eight RNA samples, and the spike RNA concentration range covered all biologically relevant transcript levels. This experimental set up allowed a straightforward comparison of the three systems.
The technical specifications for all hybridization and raw data collection protocols are provided in “Materials and Methods.” For each hybridization series, the raw signals were preprocessed according to statistical methods generally accepted as standard by the microarray data community to produce ratio measurements. A debatable topic remains the issue of background subtraction. Because it is still customary to include background subtraction, we used it as default, although for some of the particular analyses described below we applied approaches both with and without background subtraction. To determine the dynamic range, CATMA array data were normalized after subtracting for each feature the median background intensity from the mean foreground intensity. The background-subtracted data were then normalized using the standard locally weighted scatter plot smoothing (LOESS) fit, for each print-tip separately. For each feature, the LOESS normalized log2 ratios were averaged over the two dye swaps, and the final ratio was computed as the exponential base 2 of that average. Similarly, the log10 ratios calculated from Agilent array hybridizations as supplied by the service provider were averaged over the two dye swaps, and the final ratio was also expressed as the exponential (Agilent, 2003). The raw Affymetrix data were preprocessed alternatively with two software packages: the Affymetrix Microarray Suite (MAS) 5.0 (Affymetrix, 2001) and RMA (Irizarry et al., 2003). Because the Affymetrix platform does not allow direct, within GeneChip, comparisons of two samples, ratios were calculated for the seven samples with staggered spike concentrations relative to the eighth reference sample.
The ratio measurements for 13 spike RNAs (one spike RNA turned out to be faulty; see “Materials and Methods”) and all platforms are shown in Figure 2. The graph in each panel is the summary of a complete hybridization series (14 arrays for CATMA and Agilent; 8 arrays for Affymetrix) where each curve represents the signal ratios associated with 1 of the 13 spike RNAs and is plotted left to right from the highest to the lowest concentration. The panels provide a concise overview of the hybridization dynamic range. In all of them and as expected, ratios calculated for samples at 100 cpc were close to 1 because the reference sample contains all spike RNAs at that same concentration. CATMA arrays displayed a near-perfect dynamic range over three logs (10,000–10 cpc), whereas Agilent and Affymetrix arrays had a somewhat wider spread of the curves with dynamic range seldom beyond two logs (1,000–10 cpc), depending on the spike RNA and on the preprocessing method for Affymetrix. For CATMA, dose-response curves obtained with background subtraction were significantly better than without (Supplemental Fig. 3).
Figure 2.

Normalized intensity ratios. The abscissa indicates the cell copy number equivalent in spike mixes 1 to 7. The ordinate shows the resulting ratios relative to the reference mix (all at concentration of 100 cpc) for the different platforms. A, CATMA. B, Agilent. C, Affymetrix with MAS 5.0 preprocessed data. D, Affymetrix with RMA preprocessed data. Figure 2A has been previously shown in Hilson et al. (2004; p. 2180) and is reprinted with permission from Cold Spring Harbor Laboratory Press.
The leftmost portion of the curves provides information about the high concentration spikes (ratios superior to 1), in particular concerning saturation effects. Clearly, only the CATMA platform reported accurately ratios for spike RNAs at the highest concentration (10,000 cpc; 100-fold ratio; Fig. 2A), whereas both the Affymetrix and Agilent platforms showed a marked collapse (Fig. 2, B, C, and D). Interestingly, probes that showed signal saturation were automatically flagged in the Agilent data output. Out of the 12 probes corresponding to spike RNAs and included on the Agilent array (see “Materials and Methods”), 10 were flagged as saturated in both channels when hybridized with spikes at 10,000 cpc. None were flagged at lower concentrations. Notably, 27 additional Agilent probes, sharing no homology with the spike RNAs, were also flagged for saturation (see Supplemental Table III). Most of them represent nuclear genes involved in chloroplast function.
The rightmost portion of the curves (ratios below 1) provides information on the sensitivity of each platform, as it shows how signals of the lower target concentrations get confounded with background noise. Overall, for all three platforms, linearity of the dynamic range ends around 10 cpc and the signal reaches a bottom plateau marking the limit of sensitivity around 1 cpc. Although the positions of the plateaus for some spikes may in fact reflect a low level of transcription for the spike RNA cognate genes, they most probably indicate nonspecific background hybridization because the curves are not ranked in any conserved order across the platforms. Together, these observations suggest that the three platforms have similar sensitivity.
In Vivo Coverage
The percentage of the probes on an array that report a hybridization signal can also be interpreted as a measure of platform sensitivity. However, the comparative analysis of this parameter across the platforms is difficult because it depends on many factors, including scanner characteristics, data extraction software, and, subject to many different interpretations, the decision rule to declare that a signal is above background hybridization level. Aware of these caveats, we present a summary of the results as they were exported by the particular data extraction software specific to each platform (see “Materials and Methods”) to emphasize that they each rely on a different method to distinguish detectable genes and that these methods may yield strikingly distinct results. Only genes transcribed in the base Col shoot sample were considered in this analysis, based on the three hybridization series (Fig. 1). All spike probes and the various controls were omitted.
For CATMA data, a signal was considered “above background” if it fulfilled the following criterion for both channels:
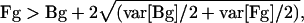 |
(1) |
|---|
i.e. a signal or foreground intensity (Fg) is called significant if it is larger than the background intensity (Bg) plus 2 times the sd of background and foreground, computed as the square root of the average of their variances (var). The fraction of CATMA probe signals above this threshold ranged between 40.4% and 54.3% (average 50.6%). Separate experiments with leaf and shoot RNAs conducted with CATMA arrays also routinely showed that more than 50% of the probes yielded signal significantly above background according to the same criteria (data not shown).
For Agilent, the information was extracted from the features “gIsWellAboveBG” and “rIsWellAboveBG” (Agilent, 2003) that were provided in the raw data files in which the vast majority of probes were labeled with signal above background in both channels: between 93.6% and 99.6% (average 96.9%). Because it is highly unlikely that more than 95% of the Arabidopsis genes are actually transcribed in Col shoots, we investigated the background and foreground values for control features in the complete Agilent dataset. As expected, an average of 99.1% of the positive controls displayed signal above background, but oddly some 74% of the negative controls were also flagged as such. When we changed the feature extraction mode to “spatial detrending” instead of “background subtraction” (Feature Extraction Software version 7.5), we observed some improvement. With these settings, the percentage of flagged negative controls decreased from 74% to 25.9%, but on average still 91.9% of all Arabidopsis probes gave a “significant” signal. We have not tried other alternative procedures for feature extraction, and we used the data obtained following standard background subtraction for all subsequent analyses presented below. Our observations, however, suggest that the raw data features gIsWellAboveBG and rIsWell-AboveBG about signal significance have no absolute biological relevance. Applying the same decision rule (Eq. 1) as for the CATMA dataset in subsequent data preprocessing resulted in an even larger percentage of probes with signal above threshold, above 99.85% for all hybridizations. By setting an alternative threshold defined as the median signal of the negative controls plus 2 sds of the median signals, 63.1% of the Arabidopsis probes scored positive. Also note that 0.15% to 1.5% of all probes were assigned surrogate values by the Agilent software to minimize artifacts resulting from division by 0 in ratio calculation (Agilent, 2003).
For Affymetrix data, we simply took the number of probe sets labeled as “present” by the Detection Call function in the MAS 5.0 software. Between 50.5% and 57.0% of all probe sets were assigned “present” calls (average of 53.9%).
For CATMA and Affymetrix, we made a more detailed in vivo coverage comparison. Based on Arabidopsis Genome Initiative codes, 14,844 genes had matching probes both on the CATMA v1 array and the Affymetrix chips. The overlap between the “present” and “absent” calls was computed for this gene set, considering that “present” meant the signal was above the platform-specific threshold for a particular gene in at least one-half of the hybridizations. The results are shown in Supplemental Table IV. The in vivo coverage estimated for CATMA and Affymetrix is similar, again suggesting that the sensitivity of the two platforms is comparable. This is further substantiated by the observation that 83.7% of the genes detected on CATMA arrays are also detected by Affymetrix, and 79.4% vice versa (Supplemental Table IV). Because the Agilent data were inherently incoherent with the built-in controls, we were unable to include that platform in the comparison.
Specificity
Probe specificity was assessed by looking for cross-hybridization of spike RNAs to probes other than the true cognate. For each of the spike RNAs, we focused on the three highest concentrations among the labeled targets and checked probes that most closely matched the spike cDNA sequences in BLAST searches. We could not detect hybridization patterns associated with any of these sets of spike RNAs, for any of the spike RNAs tested, in any of the microarray types. This is remarkable considering that a spike RNA at 10,000 cpc represents an estimated 3.3% of the total mRNA pool. For a more detailed description of the results, see Supplemental Table V.
Signal Reproducibility
Because the majority of the labeled target consisted of a single Col shoot RNA, transcript level measurements should theoretically be invariant across all hybridizations for all genes, except for those corresponding to spike RNAs. Therefore, the different hybridization series essentially consisted of 8 or 14 repetitions (Fig. 1) that are valuable to assess the platforms using robust statistical methods.
In particular, our dataset was used to investigate whether the relationship between signal reproducibility and intensity depends on the platform across the transcript level range. Because the array signal is defined as platform-specific intensity, the log2 intensity values were first converted to a unique scale by Z-score transformation so that the signal value distribution had a mean equal to 0 and a sd equal to 1 (see “Materials and Methods”; Tan et al., 2003). Furthermore, to compare similarly sized datasets, we calculated and plotted the Z-score curves for specific subsets of the data. We took the converted values from the seven Affymetrix hybridizations with RNA samples containing the spike RNAs in staggered concentration (1–7 in Fig. 1B, excluding the reference sample). For two-color arrays, we used the seven pair-wise averages of the Cy5 and Cy3 intensities corresponding to the same RNA samples in the reciprocal dye swaps (Cy5 from 1–7 and Cy3 from 1′–7′ in Fig. 1A, excluding the reference channels). In doing so, we used a 7-slide data equivalent for all three platforms (2-color datasets typically include a dye-swap hybridization) and compared the Affymetrix 11-probe set design (which actually measures each transcript 11 times, exporting an average) with the dye-swap design. Furthermore, only the set of 13,036 genes with cognate probes on all three arrays were considered, omitting, however, those matching the spike RNAs.
Figure 3A shows the corresponding Z-score frequency plots. Because these plots illustrate the distribution of the normalized data within and across platform, they allow a direct comparison of the hybridization characteristics of the different systems. The Z-score distributions of the individual arrays in any given group were all very similar, indicating that hybridizations were very reproducible. The frequency distributions of CATMA, Agilent, and Affymetrix RMA values had profiles suggestive of a Gaussian distribution, but sometimes with quite distinct shoulders. For instance, the CATMA data displayed a significant broadening of the peak, and the Affymetrix MAS 5.0 values even showed a distinct bimodal distribution with an additional smaller peak at lower intensity. Affymetrix data analyzed with RMA had a Z-score distribution very similar to the distribution of CATMA data. The difference between MAS 5.0 and RMA indicates that at least part of the bimodality of the distributions resulted from data preprocessing. To visualize the signal reproducibility in function of intensity, we plotted the Z-score sd against the Z-score mean for each gene (Supplemental Fig. 2). The LOESS lines representing the overall trend for each system are shown collectively in Figure 3B. CATMA values for background-subtracted data (CATMA BGS) showed variability independent of signal for high to medium intensity but gradually increasing for low signal. By contrast, CATMA non-background-subtracted data (CATMA non-BGS) resulted in a flatter LOESS showing a somewhat decreased variability at low intensity. Agilent had overall higher variability increasing at both ends of the intensity spectrum. We presume that the variability at low intensity results from background subtraction, whereas higher intensity values may reflect saturation. Finally, MAS 5.0 variability was low for high to medium signal, but with a sharp increase followed by a conspicuous drop for the lower intensity values. This profile was strikingly different for RMA-processed Affymetrix data, where the variability was overall very low and independent of intensity. This behavior is consistent with the statistical strategy behind RMA, which aims at reducing signal variance. Signal intensity was also used to assess the correlation between intensity values across platforms. We restricted the analysis to the genes that were present on all three platforms and that displayed a significant signal on both the CATMA and Affymetrix arrays (see above). The resulting plots indicate that there is significant correlation between the individual signal values and, hence, the hybridization characteristics of the probe elements (Supplemental Fig. 4). This is particularly satisfactory considering that the strategy for probe design was quite different for the three array types. The correlation coefficients for pair-wise comparisons are listed in Table III. Not surprisingly, the highest correlation was measured between the MAS 5.0 and RMA expression values, both obtained from the same Affymetrix chips. Furthermore, there was a fair agreement of signal intensities when Affymetrix was compared to either CATMA or Agilent. The comparison of CATMA to Agilent yielded the lowest correlation.
Figure 3.

Relationship between signal intensity and variability. A, Comparison of the distribution of Z-score values. B, Visualization of the signal reproducibility in function of intensity. The LOESS lines represent for each dataset the overall trend of the Z-score sd as a function of the Z-score mean for each gene.
Table III.
Correlation between the platforms
Correlation between the platforms was calculated for the log2 intensity signals of genes with probes on all three platforms. Only those genes were compared that were given a present call by the Affymetrix MAS 5.0 software in at least four of the eight hybridizations and scored above background for at least seven out of the 14 hybridizations on CATMA arrays.
| Platforms | Correlation |
|---|---|
| r2 | |
| CATMA/Agilent | 0.5833 |
| CATMA/Affymetrix RMA | 0.6619 |
| CATMA/Affymetrix MAS 5.0 | 0.6681 |
| Agilent/Affymetrix RMA | 0.7157 |
| Agilent/Affymetrix MAS 5.0 | 0.7292 |
| Affymetrix MAS 5.0/Affymetrix RMA | 0.9728 |
False Positive and False Discovery Rate
One of the most important issues in microarray analysis is the reliability in the measurement of gene expression differences. On the one hand, poorly chosen boundaries to define meaningful fold changes may include too many false positives or false negatives. On the other hand, microarray statistics must cope with genome-wide datasets and minimize the number of false positives that may result from the multiple-testing problem (Benjamini and Hochberg, 1995; Storey and Tibshirani, 2003). However, it is now generally accepted that the Bonferroni correction, also referred to as the “panic approach” by Y. Benjamini (personal communication), is much too restrictive. We have investigated systematically the accuracy of the platforms in calling differentials using various statistical tools. Although our experimental design does not address the reliability of small fold changes (our lowest actual real fold change is 10), it is useful because we have ample a priori knowledge about fold changes equal to 1. Again, we benefit from the fact that all hybridizations rely on a single batch of Col shoot RNA; therefore, the hybridization series essentially consist of eight or 14 repetitions (Fig. 1) and are valuable to assess in depth the robustness of the platforms. Taking advantage of our datasets and excluding the spike controls, we estimated the fraction of genes that are erroneously called differentially expressed using a statistical tool called LIMMA (Smyth, 2004). LIMMA uses a moderated t statistic in which an empirical Bayes method is used to estimate the sd of the log-fold changes (Smyth, 2004). The moderated t statistic follows a t distribution with augmented degrees of freedom, which makes the test more powerful than the conventional t test, especially in experiments with few arrays. The LIMMA package is part of the Bioconductor statistical analysis software (http://www.bioconductor.org; Gentleman et al., 2004). A gene was called differentially expressed if the moderated t test had a P value, corrected to control for the false discovery rate (FDR), smaller than 0.05 (Benjamini and Hochberg, 1995). To simulate a biological sample comparison for each platform, data from eight hybridizations were randomly assembled in two groups of four hybridizations. For Affymetrix, the expression measurements of these two subgroups were compared as two different samples each hybridized four times. For the two-channel arrays, one subgroup was used to calculate log ratios of a two-sample comparison, whereas the second group was used to obtain dye-swap ratios. We next used LIMMA to identify genes that appeared to be differentially expressed, based on these eight log ratios. To get an average estimate of this false positive fraction, the procedure was repeated for all 70 possible different permutations of two sets of four arrays from the eight Affymetrix hybridizations, and for 70 different random assemblies of the 2-channel platform array sets. The results are shown in Table IV. Because identical samples were compared, all differential genes constituted false positive observations. For each platform, the minimum, average, and maximum false positive rates are shown. The average false positive fraction was 2.16% for CATMA BGS, whereas it was 3.43% and 8.62% for Agilent and Affymetrix (MAS 5.0), respectively. The RMA-processed Affymetrix data yielded a smaller fraction of 7.71%, whereas the CATMA non-BGS gave 0.73% false positives. These percentages would result in significant numbers of falsely identified differentially expressed genes, as indicated in the last column of Table IV. Interestingly, CATMA BGS gave the lowest range in the false positive fractions calculated in the 70 iterations, with a sd of 0.189. These results have to be treated with some caution, as they not only reflect platform characteristics but also how well the LIMMA model fits the different datasets.
Table IV.
Detection of false positives
For each platform, we selected all gene probes, omitting the spikes. For each platform and data preprocessing method the percentages and numbers are given of genes flagged by the LIMMA procedure as differentially expressed. The results reflect 70 iterations of the LIMMA procedure, as described in the text.
| Platforms | Low | High | Mean | sd | Mean False Positives | Total Gene No. |
|---|---|---|---|---|---|---|
| % | % | % | % | |||
| CATMA BGS | 1.60 | 2.77 | 2.16 | 0.19 | 410 | 18,967 |
| CATMA non-BGS | 0.30 | 1.98 | 0.73 | 0.33 | 138 | 18,967 |
| Agilent | 0.56 | 14.59 | 3.43 | 2.60 | 559 | 21,487 |
| Affymetrix MAS 5.0 | 5.58 | 19.69 | 8.62 | 3.59 | 1,959 | 22,732 |
| Affymetrix RMA | 1.72 | 36.11 | 7.71 | 7.52 | 1,753 | 22,732 |
The results are also presented as Volcano plots (Fig. 4). In such a graph, the fold changes (on log2 scale) between the two samples are plotted against the log-odds ratio. The horizontal axis displays the extent of the observed differential expression, and the vertical axis the confidence associated with that observation. The resulting dot plots allow an intuitive assessment of both the extent of fold changes and the corresponding significance of these observations. For each platform, we used a representative sample comparison, i.e. a comparison that gave a percentage of differentially expressed genes closest to the average obtained from the iterative procedure. The representation of fold changes dramatically changed when CATMA BGS was compared to CATMA non-BGS results and Affymetrix MAS 5.0 with RMA processed data. Assessment of differential expression based on the moderated t test resulted in markedly lower numbers of false positives for CATMA non-BGS (Table IV). It is evident from the Volcano plots that a more detailed assessment of the results can be achieved when we weigh both significance and fold-change measurements to call cases of differential gene expression. For example, the plots for CATMA non-BGS, Agilent, and Affymetrix RMA show considerable numbers of differentially expressed genes, but predominantly associated with relatively small fold changes (often much lower than 2-fold). Interestingly, the CATMA non-BGS, Agilent, and Affymetrix RMA results had a fold-change spread sufficiently narrow to eliminate most false positives with a fold-change threshold much lower than 2.
Figure 4.

Volcano plots. The log2 ratio is plotted versus the log odds. Log odds is the loge of the probability that a gene is differentially expressed over the probability that it is not. The lower the log odds, the more likely it is that a gene is not differentially expressed. A, CATMA BGS. B, CATMA non-BGS. C, Agilent. D, Affymetrix with MAS 5.0 preprocessed data. E, Affymetrix with RMA preprocessed data. Horizontal lines mark log odds thresholds of 10,000 to 1; vertical lines mark 2-fold log2-ratio boundaries.
False Negatives
Finally, we compared the accuracy of the platforms based on their ability to avoid false negative observations. Instead of investigating intensity values for invariant genes, we now focused on those corresponding to the 13 spike RNAs and determined whether the data supported the correct statistical identification of 10-fold concentration increases. For that purpose, the LIMMA procedure was used to test whether spike genes were detected as differentially expressed when comparing consecutive spike mixes (1 versus 2, 2 versus 3, etc.; Table II). The P values obtained from the moderated t test were corrected to control the FDR, according to the method of Benjamini and Hochberg (1995), with a significance threshold P < 0.05. The results of the consecutive concentration comparisons are given in Table V. For both CATMA and Agilent data, LIMMA failed to distinguish correctly between a transcript absent and present at 0.1 cpc or between 0.1 and 1 cpc, confirming that the sensitivity threshold was between 1 and 10 cpc. In the CATMA dataset, this difference was correctly detected for 10 out of 13 cases, and for 6 out of 12 in the Agilent data. Additionally, for Agilent, four of the spikes were not accurately differentiated between 1,000 and 10,000 cpc, which can be explained by the saturation effect already observed in the dose-response curves (Fig. 2). The number of false negatives from the Affymetrix data could not be estimated because of the insufficient numbers of replicates.
Table V.
Detection of false negatives
The LIMMA procedure was used to compare consecutive sets of concentrations (0.1 cpc against 0 cpc, 1 cpc against 0.1 cpc, etc.). “−1” and “+1” indicate that the gene is flagged by LIMMA as down-regulated or up-regulated, respectively, whereas “0” is used for genes that do not appear to be differentially expressed. All pair-wise comparisons should theoretically be assigned “−1”.
| Spike RNA | 0 vs 0.1 | 0.1 vs 1 | 1 vs 10 | 10 vs 100 | 100 vs 1000 | 1000 vs 10,000 |
|---|---|---|---|---|---|---|
| CATMA | CATMA | CATMA | CATMA | CATMA | CATMA | |
| 1 | 0 | 0 | −1 | −1 | −1 | −1 |
| 2 | 0 | 0 | 0 | −1 | −1 | −1 |
| 3 | −1 | +1 | −1 | −1 | −1 | −1 |
| 4 | 0 | 0 | −1 | −1 | −1 | −1 |
| 5 | −1 | 0 | −1 | −1 | −1 | −1 |
| 6 | +1 | −1 | −1 | −1 | −1 | −1 |
| 7 | 0 | 0 | −1 | −1 | −1 | −1 |
| 8 | 0 | +1 | −1 | −1 | −1 | −1 |
| 9 | 0 | +1 | −1 | −1 | −1 | −1 |
| 10 | 0 | 0 | −1 | −1 | −1 | −1 |
| 11 | 0 | 0 | 0 | −1 | −1 | −1 |
| 12 | 0 | 0 | −1 | −1 | −1 | −1 |
| 14 | 0 | 0 | 0 | −1 | −1 | −1 |
| Agilent | Agilent | Agilent | Agilent | Agilent | Agilent | |
| 1 | 0 | 0 | −1 | −1 | −1 | 0 |
| 2 | 0 | 0 | −1 | −1 | −1 | −1 |
| 3 | 0 | 0 | 0 | −1 | −1 | −1 |
| 4 | 0 | 0 | 0 | −1 | −1 | −1 |
| 5 | 0 | 0 | −1 | −1 | −1 | 0 |
| 7 | 0 | 0 | 0 | −1 | −1 | −1 |
| 8 | −1 | 0 | −1 | −1 | −1 | −1 |
| 9 | 0 | +1 | 0 | 0 | −1 | −1 |
| 10 | 0 | −1 | 0 | −1 | −1 | 0 |
| 11 | −1 | 0 | −1 | −1 | −1 | −1 |
| 12 | 0 | 0 | −1 | −1 | −1 | 0 |
| 14 | 0 | 0 | 0 | −1 | −1 | −1 |
DISCUSSION
Two technologies have dominated the microarray field: cDNA and oligonucleotide arrays. The main advantage of cDNA microarrays has been their relatively low cost. Affymetrix oligonucleotide arrays, however, take advantage of the available genome sequence and are considered to offer higher reproducibility, albeit at a higher cost. More recently, long oligonucleotide platforms (60–80mers) have emerged as a competing technology. Whereas the cost of these oligonucleotide-based technologies is slowly decreasing, multiple problems have eroded the attractiveness of the cDNA-based arrays: difficulty in obtaining full-genome coverage (because of the limited depth of expressed sequence tag libraries), lack of standardization among laboratories (hampering data integration), higher levels of noise, and cross-hybridization between homologous transcripts. Here, we present the CATMA array for Arabidopsis that addresses these shortcomings. It is based on a standardized genome-scale PCR amplicon library, with minimal crosshybridization and high quality control. The library is available at low cost for the production of spotted arrays.
To assess the quality of the data obtained with CATMA arrays, we included Affymetrix and Agilent arrays in our performance study. In each case, RNA labeling, hybridization, scanning, and data extraction were performed by a laboratory offering routine microarray services with that particular platform, and following its standard protocols and processes: VIB-MAF microarray facility to process the CATMA arrays, ServiceXS (a service facility in The Netherlands) for Agilent, and the Nottingham Arabidopsis Stock Center for Affymetrix (GARNet program, GARNet application no. SM03b/006). Thus, all datasets were produced independently by laboratories best positioned to provide service with their particular platform. The differences observed resulted from a combination of factors: the arrays themselves but also all the equipment necessary for their processing, including the hybridization and washing station, the slide scanner, and the software application producing the raw microarray data file. In all three cases, the platforms were equipped with the standard suite of hardware and software commercially distributed by the Amersham BioSciences (Little Chalfont, UK), Agilent, and Affymetrix companies, respectively.
The comparison was based on a single, large shoot RNA sample spiked with synthetic poly(A) RNAs in various quantities. These were added to evaluate signal detection over a range of biologically meaningful abundance classes. The spike concentrations spanned a wide range of subsequent 10-fold dilutions, covering both the high, intermediate, and scarce abundance classes, allowing us to establish the detection dynamic range. We chose to use a significant number of spikes (14) to guarantee the robustness of the study and to attempt to address more extensively than most studies the potential for illegitimate hybridization. Because we used spikes that resembled bona fide transcripts, our approach tested the entire data production process, and not only the hybridization and data extraction part. Except for the faulty Spike 13, all spike RNAs showed extremely similar hybridization characteristics, and the hybridization results, combining the spike genes and the genes transcribed in Arabidopsis shoots, constituted an extensive dataset for a detailed comparison of the different platforms. The CATMA array performed very well when compared to the commercial oligonucleotide systems. Even at the highest concentrations (10,000 copies per cell), it showed no sign of saturation or signal decrease, whereas Agilent and Affymetrix arrays conspicuously lacked signal linearity in that range. For Affymetrix, RMA-processed data were slightly less saturated compared to MAS 5.0. In the Agilent data output file, some of the spike probes at the highest concentrations were flagged as saturated, together with 27 other probes, almost all corresponding to nuclear genes with chloroplast function (see Supplemental Table III), suggesting they still represented biologically relevant transcript levels. Although we could have performed multiple scans at different laser powers or detector gains, we chose to use a single setting because that is how microarray data are produced routinely by service providers. Also, integration of data resulting from multiple scans is cumbersome. Our results indicated that for abundant mRNAs, the CATMA array performed substantially better than both the short and long oligonucleotide arrays and will yield more accurate ratio-fold changes for such transcripts.
Overall, the three platforms were comparable in sensitivity, although results varied somewhat according to spikes. For some, the signal was still above background level at a concentration of 1 copy per cell, equivalent to scarce RNAs. Because of the numerous replicates in the experimental design, the CATMA and Agilent platform sensitivity could be assessed with the LIMMA algorithm. The discrimination between subsequent spike RNA levels started to deteriorate between 1 and 10 copies per cell (Table V), for which CATMA data yielded a correct call for 10 out of 13 spikes, whereas the Agilent data were accurate for 6 out of 12 spikes. Thus, we conclude that the sensitivity of CATMA arrays was at least equivalent to that of the Agilent arrays. Although the hybridization results of Affymetrix arrays were very good (as judged by the signals from the controls; see “Materials and Methods”), a direct LIMMA comparison with the other platforms was not possible because the Affymetrix experiment lacked sufficient replicates. However, the internal controls used as standard for target preparation (BioB 5′ and BioB 3′ controls; see “Materials and Methods”) showed that Affymetrix arrays also had a detection threshold between 1 and 10 RNA copies per cell in these experiments.
The analysis of CATMA data with background signal correction clearly produced the best doseresponse curves (for comparison, see Supplemental Fig. 3). However, background subtraction introduced a significant level of variance into the data, particularly for low signal. These somewhat contradictory findings illustrate the fact that there is still no single solution for data preprocessing: it remains prudent to test various alternatives even at the preprocessing level to thoroughly mine microarray datasets for information about gene expression levels. This is also evident from the differences observed between the Affymetrix results obtained with the MAS 5.0 or RMA packages. In our comparison, the RMA package outperformed MAS 5.0 for all studied parameters: dynamic range, reproducibility across the range of signal intensity, in particular for low or background signal, and FDR. The better performance of the RMA software clearly demonstrates that the GeneChip mismatch features, not taken into consideration by RMA, are better discarded to measure gene expression. Interestingly, the datasets generated for this study, containing numerous repetitions and including three competing systems, may serve for the comparative evaluation of improved and future algorithms. The choice of preprocessing protocols is especially important to establish coherent repositories of data compendia, as such large databases will hold data from heterogeneous sources. A major challenge will be to effectively integrate data from different platforms for analysis and mining purposes, e.g. by using cross-platform normalization methods (Ferl et al., 2003) or by taking P values, computed from the expression measurements of the different experiments (Rhodes et al., 2002).
CATMA array probes were selected to exclude homology exceeding 70% identity. A similar design strategy was used for the probes of the two oligonucleotide arrays. Therefore, it came as no surprise that cross-hybridization could not be detected for any of the arrays, not even with spike RNAs at 10,000 copies per cell representing up to 3.3% of the poly(A) RNA pool. The ability of the tested platforms to exclude cross-hybridization problems because of sequence homology is a big advantage over cDNA-based arrays.
The coverage of the three arrays was matched against the latest TIGR annotation (release 5.0) of the Arabidopsis genome. The CATMA v2 array is on par with the oligonucleotide arrays. Yet, microarray probe design has a moving target and all platforms will further evolve with advances in genome annotation because experimental transcription data are constantly accruing, gene prediction algorithms are continuously improving, and new genome sequences are becoming available. The ongoing design of CATMA v3 may yield an additional 6,000 probes, taking advantage of both the TIGR 5.0 annotation and the gene models obtained with recent improvements of the EuGène gene finder (http://bioinformatics.psb.ugent.be/genomes_ath_index.php). Likewise, Affymetrix is working on a new version of the ATH array, and Agilent has introduced the Arabidopsis 3 oligonucleotide array with close to 40,000 features. It will take a few more years before the Arabidopsis gene repertoire becomes completely stable, and additional updates of the array feature sets will be necessary.
CATMA arrays are now routinely used by the different CATMA consortium partners (Hilson et al., 2004; Lurin et al., 2004). Furthermore, the CATMA microarray is the platform for the production of a large compendium of Arabidopsis gene expression data, made available through the ArrayExpress database of the European Bioinformatics Institute. Although not exhaustive, this data compendium is meant to provide a reference for analysis, mining, and modeling based on transcript profiles. Its structure is such that additional data can be added easily, either through independently produced CATMA arrays (GSTs and GST arrays are available through NASC) or via microarray service (e.g. provided by CATMA partners). Alternatively, we foresee that data produced by alternative platforms may be integrated into the compendium.
The GST probe resource may constitute an affordable alternative to commercial whole-genome arrays. Of course, the up-front cost to develop the resource has not been trivial. Specific amplicon design software needed to be developed, and the CATMA consortium had to invest in PCR primers and amplification of the GST collection, but thanks to this initiative (Hilson et al., 2004), it is now possible that a well-managed microarray core facility will be able to deliver CATMA arrays for € 100 or less.
CONCLUSION
The CATMA array constitutes a novel platform for transcript profiling. Its sensitivity, specificity, and coverage make it a strong competitor for other microarrays currently available for genome-scale transcript profiling. Because its probes are designed from the complete genome sequence rather than selected from available cDNA or expressed sequence tag collections, it minimizes homologies between probes and maximizes the genome coverage. The up-front investment in the clone library has thus resulted in an ideal low-cost alternative for in-house spotting. As the merits of microarray transcriptome analysis are now firmly established, the novel CATMA array may become an important tool for functional analysis of Arabidopsis genes.
MATERIALS AND METHODS
Plant Material and RNA Extraction
Arabidopsis (Arabidopsis thaliana L.) Heynh. Col-1 seeds were sown, cold stratified (at 4°C for 7 d), and grown at long-day conditions (22°C, 16 h light/8 h dark, with cool-white light [tube code: 840] 65 mE m−2 s−1 photosynthetically active radiation) on agar-solidified culture medium (1× Murashige and Skoog [Duchefa, Haarlem, The Netherlands], 0.5 g L−1 MES, pH 6.0, 1 g L−1 Suc, and 0.6% plant tissue culture agar [LabM, Bury, UK]). Whole shoots were harvested at growth stage 1.04 corresponding to a fourth leaf length of approximately 1 mm (Boyes et al., 2001; developmental stage equivalent to The Arabidopsis Information Resource development term 0000399), 6 h after dawn, and immediately frozen in liquid nitrogen. Total RNA was extracted from pooled plant material using the TRIzol reagent (Invitrogen, Carlsbad, CA).
Preparation of Spiked RNA Samples
Spike poly(A) RNAs were synthesized from selected cDNA clones (Supplemental Table II; EMBL accession nos. AI997299, AI996580, AI998315, AI999518, AI995329, AW004197, AI995484, AI993419, AI994579, AI994777, AI992430, AI995003, AI995254, and AI994049) from a 6K cDNA collection distributed originally by Incyte, now available through Open Biosystems (Huntsville, AL; see http://www.microarray.be/servicemainframe.htm) and constructed by _Not_I-_Sal_I directional cloning in either Lambda ZipLox (Invitrogen) or pSPORT1. All clones were validated for this particular study by sequencing. Plasmid DNA was linearized by _Not_I digestion, the restriction site being positioned immediately after the poly(A) tail sequence; 1 _μ_g of linearized plasmid was used as template for the in vitro synthesis of sense transcripts with the T7 RNA polymerase (AmpliScribe T7 High Yield transcription kit; Epicentre, Madison, WI). Following DNAseI treatment, the transcribed RNAs were purified by ammonium-acetate precipitation and resuspended in diethyl pyrocarbonate-treated water. The quality and quantity of all RNA samples (spikes and Col shoot total RNA) were assessed with the RNA LabChip (Bioanalyzer 2100; Agilent Technologies) and classical spectrophotometry. Despite our efforts to carefully quality control all spike RNAs, we originally overestimated Spike 13 RNA concentration and integrity and could not draw meaningful conclusions from it in the analysis of the hybridization data. We therefore omitted this spike from all subsequent analyses.
A large batch (500 _μ_g) of Arabidopsis (Columbia) shoot RNA was diluted to 1 _μ_g _μ_L−1 and used to prepare 7 test samples at a final concentration of 0.5 _μ_g _μ_L−1, each containing a full range of spike RNAs at concentrations ranging from 0.1 to 10,000 cpc. Care was taken to use water containing total RNA at all dilution steps, to prevent the loss of spike RNAs at low concentrations through adsorption on plastic surfaces. An eighth RNA sample was constructed containing all RNA spikes at a concentration corresponding to 100 cpc. The eight RNA samples were constructed each in a single separate tube, aliquoted, and processed according to the protocols specific to each platform. All RNA samples were again checked for quality and quantity with the RNA LabChip at the end of the dilution procedure.
CATMA GST Microarray
Design and synthesis of primary and secondary GST amplicons were described elsewhere (Thareau et al., 2003; Hilson et al., 2004). As described, the GSTs primarily match (3′) exons or 3′ untranslated region (UTR) sequences and occasionally (2.9%) contain intron sequences. The CATMA v1 array used in this study consisted of 19,992 features, including 18,981 unique GSTs, 768 positive/negative controls (Amersham BioSciences), and 243 blanks. GST PCR products were purified with MinElute UF plates (Qiagen, Hilden, Germany) and arrayed in 50% dimethyl sulfoxide on Type VIIstar reflective slides (Amersham BioSciences) using a Lucidea Array spotter (Amersham BioSciences). The spots had a diameter of approximately 100 microns and were 173 × 173 microns apart. The array design can be accessed via the ArrayExpress database as accession number A-MEXP-10 (http://www.ebi.ac.uk/arrayexpress) or via the VIB MicroArray Facility Web site (http://www.microarrays.be). Prior to hybridization, the slides were washed in 2× saline-sodium phosphate-EDTA buffer, 0.2% SDS for 30 min at 25°C.
RNA was amplified using a modified protocol of in vitro transcription as described previously (Puskás et al., 2002). Briefly, 5 _μ_g of total RNA was reverse transcribed to double-stranded cDNA using an anchored oligo(dT) + T7 promoter [5′-GGCCAGTGAATTGTAATACGACTCACTATAGGGAGGCGG-T24(ACG)-3′ (Eurogentec, Seraing, Belgium)]. From this cDNA, RNA was produced via T7-in vitro transcriptase until an average yield of 10 to 30 _μ_g of amplified RNA. The amplified RNA (5 _μ_g) was labeled with dCTP-Cy3 or Cy5 (Amersham BioSciences), by reverse transcription using random nonamer primers (Genset, Paris). The resulting probes were purified with Qiaquick (Qiagen) and analyzed for amplification yield and incorporation efficiency by measuring the DNA concentration at 280 nm, Cy3 incorporation at 550, and Cy5 incorporation at 650 using a Nanodrop spectrophotometer (NanoDrop Technologies, Rockland, DE). A good target had a labeling efficiency of 1 fluorochrome every 30 to 80 bases. For each target, 40 pmol of incorporated Cy5 or Cy3 were mixed in 210 _μ_L of hybridization solution containing 50% formamide, 1× hybridization buffer (Amersham BioSciences), 0.1% SDS. Each spike mix was hybridized against the reference RNA (spikes at 100 cpc) and repeated with dye swap to make up 14 hybridizations in total (Fig. 1).
Hybridization and posthybridization washing were performed at 45°C with an Automated Slide Processor (Amersham BioSciences). Posthybridization washing was done in 1× sodium chloride/sodium citrate buffer (SSC), 0.1% SDS, followed by 0.1× SSC, 0.1% SDS and 0.1× SSC. Arrays were scanned at 532 nm and 635 nm using a Generation III scanner (Amersham BioSciences). Images were analyzed with ArrayVision (Imaging Research, St. Catharines, Canada).
All protocols are available at the VIB MicroArray Facility Web site (http://www.microarrays.be) and at ArrayExpress under accession numbers P-MEXP-578, P-MEXP-579, P-MEXP-581, P-MEXP-582 for Cy3 labeling, Cy5 labeling, hybridization, and scanning, respectively. The CATMA transcript profiling data have been submitted to ArrayExpress under accession number E-MEXP-30.
Agilent and Affymetrix Microarrays
The protocols used by ServiceXS for Agilent data production were published by Agilent Technologies, in particular the manuals Low RNA Input Fluorescent Linear Amplification Kit (version 1.0, February 2003) and Agilent 60-mer Oligo Microarray Processing Protocol (version 7.0, April 2004). Arrays were scanned with maximum (100%) laser intensity in both channels (default settings) to obtain maximum sensitivity. Lower intensity scanning may correct for saturated features. Features were extracted with background subtraction or with spatial detrending (Feature Extraction Software version 7.5). Spatial detrending estimates the background signal by fitting a surface over the lowest 1% to 2% of the intensities. By subtracting this surface fit, a systematic intensity gradient on the microarray is removed, thereby correcting for a background trend rather than local background measurements that may be biased. Apart from a slight decrease in the percentage of spots above background, spatial detrending gave essentially the same result as the background-subtraction method.
The procedures used for Affymetrix data production are described in the documentation provided by NASC (http://nasc.nott.ac.uk/; Craigon et al., 2004), available together with the data from the ArrayExpress database (http://www.ebi.ac.uk/arrayexpress, accession no. E-NASC-32). For Affymetrix data, the hybridization characteristics of the internal RNA controls were monitored as an additional quality control: (1) the 3′:5′ ratios for GAPDH and _β_-actin ranged from 1.0287 to 1.2408 and from 1.8012 to 2.1705, respectively, and are all indicative of successful hybridizations; (2) the spike controls (BioB, BioC, BioD, BioM, and CreX) were present on all chips, except for BioB 5′ and BioB 3′ called “Marginal” for chips 1 and 3, respectively; (3) when scaled to a target intensity of 100 (using Affymetrix MAS 5.0 software), scaling factors for all arrays were within acceptable limits (ranging between 0.311 and 0.518), as were background and mean intensity values. For all hybridizations, quality and quantity of starting RNA were verified by agarose gel electrophoresis and RNA LabChip analysis. The Agilent and Affymetrix transcript profiling data have been submitted to ArrayExpress under accession numbers E-MEXP-197 and E-NASC-43, respectively.
In Silico Coverage
The coverage of the three platforms was compared by BLAST analysis of their probe sequences against TIGR 5.0 gene models. The sequences of these gene models, including pseudogenes and transposable elements, were extracted from the XML files describing the chromosomes (at ftp://ftp.tigr.org/pub/data/a_thaliana/ath1/PSEUDOCHROMOSOMES). The probes of Affymetrix and Agilent were designed based on TIGR annotation releases 2 and 3, respectively (available in the archives at http://www.tigr.org). The probes of CATMA were designed on gene models predicted by the EuGène software (Schiex et al., 2001), supplemented with gene models uniquely described in the TIGR 3 release. For the analysis of Affymetrix and Agilent probes, we used only exonic sequences to correctly position probes that span exon boundaries. In line with the original design criteria employed for the GSTs, we used complete gene models including 3′ UTRs, to be able to correctly locate probes that were designed to span intron-exon boundaries or exon-3′ UTR boundaries. The set of sequences extracted from the TIGR files for the comparison against Affymetrix and Agilent contained the complete gene structure (exons, introns, and 3′ UTR sequences) of all protein-encoding genes, including their splice variants, and the pseudogenes. For CATMA, we extracted exon and intron sequences of all protein-encoding genes, and the pseudogene sequences. For both databanks, we added either the full 3′ UTR sequence or arbitrarily the 150 bases following the stop codon (when the 3′ UTR was shorter than 150 bases or if no 3′ UTR was available).
The sequences of the Affymetrix probe sets were retrieved from the company's Web site (http://www.affymetrix.com/), the sequences of the Agilent probes were retrieved from the company Web site (http://www.agilent.com; restricted pages requiring transfer agreement for access), and CATMA v2 were derived from the Array Design File accession number A-MEXP-58, publicly available at ArrayExpress (http://www.ebi.ac.uk/arrayexpress/). Perl scripts were used to extract the genes from XML files, to reconstitute exonic gene sequences, to adjust 3′ UTR sequences, and to automate the BLAST and extraction of data from the BLAST output files.
CATMA sequences (150–500 bp) matched TIGR 5.0 when aligned over at least 150 bases allowing for at most two discrepancies (base mismatch or gap); Agilent sequences (60mer) when aligned over the whole probe length allowing at most one base mismatch or gap; and Affymetrix probe sets (11 probes of 25 bases each) when at least eight probes from a set aligned perfectly. Splice variants were merged to allow comparison of CATMA hits (BLAST against gene) with Agilent and Affymetrix hits (BLAST against all possible splice variants). TIGR 5.0 genes represented by features in the different arrays were simply counted based on these criteria.
Distribution of Materials
Upon request, all novel materials described in this publication will be made available in a timely manner for noncommercial research purposes, subject to the requisite permission from any third-party owners of all or parts of the material. Obtaining any permission will be the responsibility of the requestor.
Acknowledgments
We thank Jean-Jacques Daudin, Marie-Laure Martin-Magniette, and Stéphane Robin (INA-PG, Paris) and two anonymous reviewers for their helpful comments, and Martine De Cock (VIB–Ghent University, Belgium) for help in preparing the manuscript.
1
This work was supported in part by the 5th European Framework Programme (Compendium of Arabidopsis Gene Expression; grant no. QLK3–CT–2002–02035).
[w]
The online version of this article contains Web-only data.
References
- Affymetrix (2001) Microarray Suite User Guide, Version 5. http://www.affymetrix.com/products/software/specific/mas.affx. (November, 2004)
- Agilent (2003) Feature Extraction Software User Manual, Version 7.1. http://www.chem.agilent.com/Scripts/PDS.asp?lPage=2547. (February, 2004)
- The Arabidopsis Genome Initiative (2000) Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408**:** 796–815 [DOI] [PubMed] [Google Scholar]
- Barczak A, Rodriguez WM, Hanspers K, Koth LL, Chuan Tai Y, Bolstad BM, Speed TP, Erle DJ (2003) Spotted long oligonucleotide arrays for human gene expression analysis. Genome Res 13**:** 1775–1785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B 57**:** 289–300 [Google Scholar]
- Berardini TZ, Mundodi S, Reiser L, Huala E, Garcia-Hernandez M, Zhang P, Mueller LA, Yoon J, Doyle A, Lander G, et al (2004) Functional annotation of the Arabidopsis genome using controlled vocabularies. Plant Physiol 135**:** 745–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyes DC, Zayed AM, Ascenzi R, McCaskill AJ, Hoffman NE, Davis KR, Gorlach J (2001) Growth stage-based phenotypic analysis of Arabidopsis: a model for high throughput functional genomics in plants. Plant Cell 13**:** 1499–1510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chudin E, Walker R, Kosaka A, Wu SX, Rabert D, Chang TK, Kreder DE (2002) Assessment of the relationship between signal intensities and transcript concentration for Affymetrix GeneChip arrays. Genome Biol 3**:** RESEARCH0005 [DOI] [PMC free article] [PubMed]
- Craigon DJ, James N, Okyere J, Higgins J, Jotham J, May S (2004) NASCArrays: a repository for microarray data generated by NASC's transcriptomics service. Nucleic Acids Res 32**:** D575–D577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowe ML, Serizet C, Thareau V, Aubourg S, Rouzé P, Hilson P, Beynon J, Weisbeek P, Van Hummelen P, Reymond P, et al (2003) CATMA: a complete Arabidopsis GST database. Nucleic Acids Res 31**:** 156–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Haeseleer P, Liang S, Somogyi R (2000) Genetic network inference: from co-expression clustering to reverse engineering. Bioinformatics 16**:** 707–726 [DOI] [PubMed] [Google Scholar]
- Ferl GZ, Timmerman JM, Witte ON (2003) Extending the utility of gene profiling data by bridging microarray platforms. Proc Natl Acad Sci USA 100**:** 10585–10587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DJ, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al (2004) Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 5**:** R80.1–R80.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilson P, Allemeersch J, Altmann T, Aubourg S, Avon A, Beynon J, Bhalerao RP, Bitton F, Caboche M, Cannoot B, et al (2004) Versatile gene-specific sequence tags for Arabidopsis functional genomics: transcript profiling and reverse genetics applications. Genome Res 14**:** 2176–2189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilson P, Small I, Kuiper MTR (2003) European consortia building integrated resources for Arabidopsis functional genomics. Curr Opin Plant Biol 6**:** 426–429 [DOI] [PubMed] [Google Scholar]
- Hughes TR, Marton MJ, Jones AR, Roberts CJ, Stoughton R, Armour CD, Bennett HA, Coffey E, Dai H, He YD, et al (2000) Functional discovery via a compendium of expression profiles. Cell 102**:** 109–126 [DOI] [PubMed] [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP (2003) Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4**:** 249–264 [DOI] [PubMed] [Google Scholar]
- Kuo WP, Jenssen TK, Butte AJ, Ohno-Machado L, Kohane IS (2002) Analysis of matched mRNA measurements from two different microarray technologies. Bioinformatics 18**:** 405–412 [DOI] [PubMed] [Google Scholar]
- Lee JK, Bussey KJ, Gwadry FG, Reinhold W, Riddick G, Pelletier SL, Nishizuka S, Szakacs G, Annereau JP, Shankavaram U, et al (2003) Comparing cDNA and oligonucleotide array data: concordance of gene expression across platforms for the NCI-60 cancer cells. Genome Biol 4**:** R82.1–R82.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TI, Rinaldi NJ, Robert F, Odom DT, Bar-Joseph Z, Gerber GK, Hannett NM, Harbison CT, Thompson CM, Simon I, et al (2002) Transcriptional regulatory networks in Saccharomyces cerevisiae. Science 298**:** 799–804 [DOI] [PubMed] [Google Scholar]
- Lurin C, Andres C, Aubourg S, Bellaoui M, Bitton F, Bruyere C, Caboche M, Debast C, Gualberto J, Hoffmann B, et al (2004) Genome-wide analysis of Arabidopsis pentatricopeptide repeat proteins reveals their essential role in organelle biogenesis. Plant Cell 16**:** 2089–2103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maki Y, Tominaga D, Okamoto M, Watanabe S, Eguchi Y (2001) Development of a system for the inference of large scale genetic networks. Pac Symp Biocomput 2001**:** 446–458 [DOI] [PubMed] [Google Scholar]
- Moreau Y, Aerts S, De Moor B, De Strooper B, Dabrowski M (2003) Comparison and meta-analysis of microarray data: from the bench to the computer desk. Trends Genet 19**:** 570–577 [DOI] [PubMed] [Google Scholar]
- Nimgaonkar A, Sanoudou D, Butte AJ, Haslett JN, Kunkel LM, Beggs AH, Kohane IS (2003) Reproducibility of gene expression across generations of Affymetrix microarrays. BMC Bioinformatics 4**:** 27.1–27.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puskás LG, Zvara A, Hackler L Jr, Van Hummelen P (2002) RNA amplification results in reproducible microarray data with slight ratio bias. Biotechniques 32**:** 1330–1340 [DOI] [PubMed] [Google Scholar]
- Quackenbush J (2003) Genomics. Microarrays—guilt by association. Science 302**:** 240–241 [DOI] [PubMed] [Google Scholar]
- Redman JC, Haas BJ, Tanimoto G, Town CD (2004) Development and evaluation of an Arabidopsis whole genome Affymetrix probe array. Plant J 38**:** 545–561 [DOI] [PubMed] [Google Scholar]
- Rhodes DR, Barrette TR, Rubin MA, Ghosh D, Chinnaiyan AM (2002) Meta-analysis of microarrays: interstudy validation of gene expression profiles reveals pathway dysregulation in prostate cancer. Cancer Res 62**:** 4427–4433 [PubMed] [Google Scholar]
- Schiex T, Moisan A, Rouzé P (2001) EuGèNE: an eukaryotic gene finder that combines several sources of evidence. Lect Notes Comput Sci 2066**:** 111–125 [Google Scholar]
- Smyth GK (2004) Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Statistical Applications in Genetics and Molecular Biology, Vol 3, No 1, Article 3. http://www.bepress.com/sagmb/vol3/iss1/art3. (January, 2005) [DOI] [PubMed]
- Storey JD, Tibshirani R (2003) Statistical significance for genomewide studies. Proc Natl Acad Sci USA 100**:** 9440–9445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan PK, Downey TJ, Spitznagel EL Jr, Xu P, Fu D, Dimitrov DS, Lempicki RA, Raaka BM, Cam MC (2003) Evaluation of gene expression measurements from commercial microarray platforms. Nucleic Acids Res 31**:** 5676–5684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thareau V, Déhais P, Serizet C, Hilson P, Rouzé P, Aubourg S (2003) Automatic design of gene-specific sequence tags for genome-wide functional studies. Bioinformatics 19**:** 2191–2198 [DOI] [PubMed] [Google Scholar]
- Yuen T, Wurmbach E, Pfeffer RL, Ebersole BJ, Sealfon SC (2002) Accuracy and calibration of commercial oligonucleotide and custom cDNA microarrays. Nucleic Acids Res 30**:** e48. [DOI] [PMC free article] [PubMed] [Google Scholar]