Regulation of the Resident Chromosomal Copy of c-myc by c-Myb Is Involved in Myeloid Leukemogenesis (original) (raw)
Abstract
c-myb is a frequent target of retroviral insertional mutagenesis in murine leukemia virus-induced myeloid leukemia. Induction of the leukemogenic phenotype is generally associated with inappropriate expression of this transcriptional regulator. Despite intensive investigations, the target genes of c-myb that are specifically involved in development of these myeloid lineage neoplasms are still unknown. In vitro assays have indicated that c-myc may be a target gene of c-Myb; however, regulation of the resident chromosomal gene has not yet been demonstrated. To address this question further, we analyzed the expression of c-myc in a myeloblastic cell line, M1, expressing a conditionally active c-Myb–estrogen receptor fusion protein (MybER). Activation of MybER both prevented the growth arrest induced by interleukin-6 (IL-6) and rapidly restored c-myc expression in nearly terminal differentiated cells that had been exposed to IL-6 for 3 days. Restoration occurred in the presence of a protein synthesis inhibitor but not after a transcriptional block, indicating that c-myc is a direct, transcriptionally regulated target of c-Myb. c-myc is a major target that transduces Myb's proliferative signal, as shown by the ability of a c-Myc–estrogen receptor fusion protein alone to also reverse growth arrest in this system. To investigate the possibility that this regulatory connection contributes to Myb's oncogenicity, we expressed a dominant negative Myb in the myeloid leukemic cell line RI-4-11. In this cell line, c-myb is activated by insertional mutagenesis and cannot be effectively down regulated by cytokine. Myb's ability to regulate c-myc's expression was also demonstrated in these cells, showing a mechanism through which the proto-oncogene c-myb can exert its oncogenic potential in myeloid lineage hematopoietic cells.
myb was originally identified as an oncogene following its transduction into the avian retroviruses avian myeloblastosis virus and E26 (16, 32), which cause acute myeloblastic leukemia or erythroblastosis. Its oncogenic potential was further demonstrated in animal models, where the cellular proto-oncogene c-myb was shown to be a target of retroviral insertional mutagenesis, causing T- and B-cell lymphomas in chickens (28, 52, 53, 55) and myeloid leukemias in mice (21, 46, 60, 61, 72). Despite many reports over the last several years that described potential target genes of the transcription factors v-Myb and its cellular counterpart c-Myb, little is known about the regulatory pathways through which c-myb exerts its oncogenic potential.
The product of the c-myb gene is a highly conserved transcription factor of 75 kDa (4, 37). In hematopoietic cells of all types, it is expressed in the immature phase (29, 62) and is down regulated during terminal differentiation (15, 22, 69). Functions assigned to this gene regulator are apoptosis, differentiation, and proliferation (reviewed in references 70 and 71). Its role in apoptosis was impressively demonstrated by inactivation of its function in transgenic mice using a dominant interfering Myb, targeted specifically to T cells; thymocytes from these mice died from apoptosis at a higher rate than normal. It was shown that the antiapoptotic role of c-Myb in both T cells and myeloid cells is connected to Myb's ability to regulate expression of bcl-2 (19, 65). Its function in differentiation is evident from studies which have identified, as c-Myb targets, genes like mim-1, TCRδ, CD4, and the neutrophil elastase gene (reviewed in reference 48), all of which are markers of mature cell phenotypes. Furthermore, the detection of the homeobox gene GBX2 as a direct target gene of c-Myb demonstrates how Myb can be responsible, in a more global way, for lineage commitment in differentiation (31).
Myb's involvement in maintaining proliferation is thought to be an important function in regard to its oncogenic potential. This function also has been clearly demonstrated for normal cell development. For example, homozygous knockout mice were shown to have an embryonic lethal phenotype due to a severe reduction in the number of progenitor cells (44). In addition, in vitro experiments in both erythroid and myeloid cells have demonstrated that constitutive expression of exogenous c-myb blocks growth arrest associated with terminal differentiation (7, 12, 42, 58). Genes implicated as targets of c-Myb and involved in its role in proliferation are the cell cycle regulator gene cdc2 (31), the DNA polymerase α gene (64), c-kit (27, 54, 68), and c-myc (13, 17, 73). These genes were implicated primarily in studies using in vitro transactivation assays, where reporter gene constructs containing the given promoters were transfected into different cell types. More conclusive data were obtained recently for a role of c-kit in transducing c-Myb's signal to proliferate in fetal liver-derived primitive myeloid cells when it was shown that the endogenous chromosomal copy of the gene was regulated directly by c-Myb (27). In contrast, in the case of c-myc, recent studies have raised doubts as to whether it is regulated by c-Myb, because analysis of its expression in lymphoid, erythroid, or primitive myeloid cells, in response to conditional expression of an active c-Myb or a dominant negative Myb, failed to show effects on the resident chromosomal gene (27, 35, 65).
Despite the fact that recent studies have raised doubts concerning whether c-myc is a gene regulated by c-Myb, experimental observations from leukemogenesis studies have led us to further investigate this topic in cells of the myeloid lineage. Myeloid leukemias resulting from inoculation of retroviruses into young adult mice fall into two categories. One type, monocyte-macrophage leukemias induced by a retrovirus expressing c-myc, do not express c-myb (2, 3, 72). In contrast, promonocytic leukemias, which have been induced following retroviral insertion of the c-Myb locus and have inappropriate expression of c-myb, have constitutively up regulated c-myc (72; L. Wolff, unpublished data). These leukemias are called MML, for murine leukemia virus-induced myeloid leukemia (71). Interestingly, expression of c-myc is not observed in normal cells of this late stage of differentiation. These examples suggested the intriguing possibility that c-myc is a downstream target of c-Myb in myeloid leukemogenesis.
MATERIALS AND METHODS
Cells and viruses.
The murine myeloid cell line M1 (33) was maintained in RPMI 1640 medium supplemented with 10% heat-inactivated horse serum. The murine leukemia cell line RI-4-11 (45) was maintained in Dulbecco modified Eagle medium containing 10% fetal bovine serum, as were the murine ecotropic GP + E-86 (38) and murine amphotropic _env_AM12 (39) packaging cell lines. For activation of the estrogen receptor (ER) fusion proteins, 4-hydroxytamoxifen (4-OHT; Sigma) was added to the medium at a concentration of 1 to 2 nM; for induction of the metallothionein (MT) promoter, ZnCl2 was used at a concentration of 50 to 85 μM.
Retroviruses were prepared as previously described (7) by transfecting LXSN-based (43) recombinant retrovirus DNA into the amphotropic packaging cell line GP + _env_AM12 by the calcium phosphate method. The ecotropic packaging cell line GP + E-86 was infected 48 h later, in the presence of Polybrene (4 μg/ml; Sigma), with conditioned, virus-containing supernatant of the transfected GP + _env_AM12 culture. Infected cells were selected in medium containing G418 (400 μg/ml; GIBCO-BRL). Selected cells producing recombinant retroviruses were treated with mitomycin C (10 ng/ml; Sigma) for 3 h and used to infect M1 or RI-4-11 cells by cocultivation. The infected M1 or RI-4-11 cells were subsequently selected for neomycin resistance with 400 or 800 μg respectively, of G418 per ml.
Electroporation was used for transfection of M1 cells. Briefly, 5 × 106 cells in 500 μl of phosphate-buffered saline (PBS) were electroporated with a Bio-Rad Gene Pulser II (400 V, 50 μF). After transfection, M1 cells containing recombinant vectors were selected in medium containing G418 (400 μg/ml) or puromycin (2 μg/ml).
For M1 cell differentiation, cells were seeded at a concentration of 1 × 105 to 2 × 105/ml in medium containing interleukin-6 (IL-6). Stocks of secreted murine recombinant IL-6 were prepared from SF9 cells infected with PVL 6A8 mouse IL-6 baculovirus (a kind gift of J. Van Snick, Ludwig Institute for Cancer Research, Brussels, Belgium). SF9 cells grown in Grace's insect medium with 10% fetal calf serum were infected for 2 days for the preparation of secreted IL-6. One-liter stocks of IL-6 were prepared by Cell Trends, Inc. (Middletown, Md.) and used at a concentration of 1:500 after testing for the ability to induce differentiation.
De novo protein synthesis was inhibited by cultivating the cells in the presence of cycloheximide (Sigma) at a concentration of 10 μg/ml for 3 to 6 h. Under these experimental conditions, 35S incorporation was found to be reduced to more than 90% in treated cell samples.
De novo transcription was blocked by cultivating the cells in the presence of actinomycin D (Sigma) at a concentration of 10 μg/ml for 3 to 6 h.
Plasmids and construction of retroviral vectors.
The vector pLMybERSN was constructed by cloning the c-Myb–ER (MybER) gene of pJ4Δ4R/MER as _Sac_I/_Cla_I fragment into the _Hpa_I site of LXSN (43). pJ4Δ4R/MER encodes a C-terminally deleted c-Myb (amino acids 1 to 489) fused to the mutant mouse ER and was constructed by cloning a _Sal_I/_Sac_I c-myb fragment from plasmid pJ4ΔR into _Sal_I/_Bam_HI-digested pJ4MER. MybEnER (a fusion protein consisting of the Myb DNA binding region, the Drosophila Engrailed [En] transrepressor, and a modified ER hormone binding domain) and EnER (36) were cut out of pJ4/Myb/En/ER or pJ4En/ER as _Bam_HI/_Cla_I fragments that were inserted into the _Hpa_I site of pLXSN to create pLMybEnERSN or pLEnERSN, respectively. pMTCB6+MybEn and pMTCB6+c-Myb were cloned as a _Bam_HI fragment from the MEnT (1) expression vector pUHIT/MEnT or the c-Myb expression vector pLFLmybSN (42) by insertion into the _Bam_HI site of pMTCB6+ (9, 11). MycER (34) expressing pBabe Puro MycER was transfected directly.
Preparation of RNA and Northern blot analysis.
Total RNA was prepared using a RNAeasy Mini kit (Qiagen). Five-microgram samples of RNA were electrophoresed in a Tris-acetate-EDTA buffer on a 1.0% agarose gel containing 20 mM guanidine thiocyanate as described in reference 20. After blotting to a nylon membrane, the RNA was cross-linked to the membrane by using a Stratalinker 1800 (Stratagene). Probes used for hybridization were labeled using a random priming kit (GIBCO-BRL). The following DNA fragments were used as probes: EnER, _Bam_HI/_Cla_I fragment (36); MybER, _Sac_I/_Cla_I fragment; MybEn, _Bam_HI fragment (1); c-myc, 1.4-kb _Sst_I/_Hin_dIII fragment (63); MycER, 2.2-kb _Eco_RI fragment; c-myb, 2.0-kb _Nco_I fragment (4); rat GAPDH cDNA (18); β-actin cDNA (25); cdc2 cDNA (66); ornithine decarboxylase gene (ODC), 0.7-kb _Pst_I fragment (14); v-kit, 0.7-kb _Sac_I/_Sal_I fragment (5); cyclinD1 cDNA (41).
Analysis of apoptosis.
Early-stage apoptosis was detected using the TACS annexin V apoptosis detection kit (Trevigen, Inc.) according to the manufacturer's instructions. For detection of late-stage apoptosis, DNA was analyzed for fragmentation as described recently (8). Briefly, 2 × 106 cells were lysed in a buffer containing 0.5% sodium dodecyl sulfate (SDS), 0.1 M NaCl, 1 mM EDTA, and 50 mM Tris-HCl (pH 8.0) and incubated for 4 h at 50°C in the presence of proteinase K (0.1 mg/ml; GIBCO-BRL). The samples were then extracted with chloroform-isoamyl alcohol, precipitated with ethanol, dissolved in 10 mM Tris–1 mM EDTA (pH 7.4), and treated with RNase A for 1 h at 37°C. Five micrograms of DNA was electrophoresed on a 2% agarose gel containing 1 mg of ethidium bromide per ml and visualized by UV fluorescence.
Cell cycle analysis.
Cells were prepared for cell cycle analysis as described in reference 50. Cells were harvested and fixed in 70% ethanol for a minimum of 18 h. For cell cycle analysis, the cells were collected by centrifugation and stained in a solution of phosphate-buffered saline (PBS), propidium iodide (50 μg/ml), RNase A (100 U/ml), and glucose (1 g/liter) and measured in a FACScan apparatus (Becton Dickinson). The data were analyzed for cell cycle distribution using the ModfitLT V2.0 program.
Western blot analysis.
Cells were lysed in 10 mM Tris (pH 7.4)–0.15 M NaCl–1% NP-40, 1% sodium deoxycholate–0.1% SDS with 10 mM iodoacetamide, 1 mM phenylmethylsulfonyl fluoride and Complete protease inhibitor cocktail as instructed by the manufacturer (Boehringer Mannheim). Samples of 100 μg of protein were separated by SDS-polyacrylamide gel electrophoresis using 8% gels and a discontinuous Tricine buffer system (57). Western blot analysis was carried out by standard procedures. Briefly, proteins were electrophoretically transferred to nitrocellulose membranes, then blocked for 2 to 24 h in PBS with 10% nonfat dry milk, and washed twice in PBS with 0.5% Tween 20 for 5 to 10 min. Incubation with a monoclonal antibody to c-Myc (N-262; Santa Cruz Biotechnology) or c-Myb (a kind gift from Eric Westin and Tim Bender) was carried out at room temperature in PBS with 5% dry milk. Protein was detected using an ECL kit (Amersham Life Sciences Inc.).
RESULTS
A conditionally active Myb reverses IL-6-induced growth arrest in M1 cells and directly activates c-myc.
M1 cells cultivated in the presence of IL-6 undergo terminal differentiation to a mature macrophage phenotype over the course of 3 to 4 days. This effect is accompanied by growth arrest 2 to 3 days after treatment and by cell death through apoptosis 5 to 7 days poststimulation (6, 58). c-myb RNA is reduced to nondetectable levels in the differentiating cells by as early as 3 h after stimulation with IL-6, while c-myc RNA, after a transient increase for 3 h, gradually decreases until it is no longer detectable at around 24 h (6, 58). To determine if c-Myb can positively regulate c-myc, we investigated whether a conditionally active c-Myb could induce proliferation and reactivate c-myc expression in M1 cells. For this study, we used a conditionally active MybER fusion protein (see Materials and Methods) (Fig. 1A). It has been demonstrated that in the absence of hormone stimulation, constitutively expressed chimeric proteins such as this one are inactive but can change to an active conformation upon binding of the estrogen analog 4-OHT to the ER part of the protein (10, 35, 51). A plasmid containing MybER was transfected into M1 cells. Western blot analysis of M1 cells stably transfected with MybER (M1/MybER cells) confirmed expression of the chimeric MybER protein (Fig. 1B).
FIG. 1.

Effects of MybER on cell growth in IL-6-treated M1 myeloblastic cells. (A) Structure of the retroviral vector LXSN with sequences encoding MybER inserted downstream of the virus long terminal repeat (LTR). SV, simian virus 40 promoter; neo, neomycin resistance gene; TA, transactivation domain; LZ, leucine zipper; aa, amino acids. (B) Western blot analysis showing expression of MybER. The protein was detected using a monoclonal c-Myb antibody. (C) Growth curve of M1/MybER cells cultivated in the presence of IL-6 for 3 days prior to treatment with 4-OHT; control cultures were incubated with IL-6 alone or 4-OHT alone. Shown is the mean of three independent results with a standard deviation smaller than 15%.
The effect of MybER protein on cell growth was examined first. As shown in Fig. 1C, M1/MybER cells that had been cultivated in the presence of IL-6 for 3 days and had undergone growth arrest were able to proliferate again following stimulation with 4-OHT (Fig. 1C). In fact, the doubling time for this IL-6 and 4-OHT-treated cell population (30 h) was almost identical to that of the control cells grown only in the presence of 4-OHT (29 h). Therefore, the chimeric MybER protein was shown to have a dramatic influence on M1 cell growth.
Northern blot analysis of c-myc expression showed that in M1/MybER cells incubated with IL-6 alone, c-myc was down regulated 24 h following treatment with the cytokine. M1/MybER cells with the inactive fusion protein showed, therefore, a response to treatment with IL-6 comparable to that reported for M1 wild-type cells by Bies et al. (6) and Selvakumaran et al. (58). In sharp contrast was the continued expression of c-myc in the same cells 24 h following treatment with IL-6 in the presence of 4-OHT (Fig. 2A and B). Significantly, c-myc expression, in addition to being maintained at high levels when 4-OHT was added at the same time as IL-6, could be restored when 4-OHT was added to cells that had been differentiating in the presence of IL-6 for up to 3 days, the longest period tested (Fig. 2B). 4-OHT itself had no effect on c-myc expression as shown in Fig. 2C. To determine if the effect of the modified c-Myb was direct or indirect, we treated the cells with cycloheximide to inhibit protein synthesis. As shown for parental M1 cells, cycloheximide itself had no effect on c-myc mRNA levels during the incubation period used for these experiments (Fig. 2C). Since c-myc expression was restored in M1/MybER cells in the presence of cycloheximide, as demonstrated in Fig. 2C, we conclude that MybER directly up regulates c-myc in these myeloid cells and is likely responsible for reversing growth arrest. Furthermore, we have evidence that the effect of c-Myb on c-myc expression is through regulation at the transcriptional level, because c-myc expression could not be restored in cultures that were treated with an inhibitor of transcription, actinomycin D (Fig. 2D).
FIG. 2.

Northern blot analysis showing regulation of c-myc by c-Myb in M1/MybER cells differentiating in the presence of IL-6. (A) Down regulation of c-myc RNA in M1 cells stimulated with IL-6. Total RNA was separated on a agarose gel, blotted on nitrocellulose, and hybridized with a c-myc probe. The same blot was rehybridized with a probe for GAPDH to control for sample loading. (B) Analyses of IL-6-stimulated cells showing continued expression of c-myc or reactivation of c-myc due to activation of MybER by 4-OHT. Cells were cultivated in the presence of IL-6. 4-OHT was added at either 0 or 72 h following IL-6 treatment, as shown on the left, and RNA was analyzed for c-myc expression at 0, 3, 12, 24, and 48 h after activation of MybER. (C) Reactivation of c-myc expression in IL-6-treated M1/MybER cells in the presence or absence of the protein synthesis inhibitor cycloheximide. As a control, parental M1 cells were tested for effects of 4-OHT or cycloheximide on c-myc RNA expression. Cells were incubated with IL-6 for 72 h prior to addition of 4-OHT and/or cycloheximide. RNA was analyzed for c-myc expression at 0, 3, and 6 h after treatment with 4-OHT and/or cycloheximide. (D) Reactivation of c-myc expression in IL-6-treated M1/MybER cells is blocked in the presence of the transcription inhibitor actinomycin D. Cells were incubated with IL-6 for 72 h prior to addition of 4-OHT and actinomycin D. RNA was analyzed for c-myc expression at 0, 3, and 6 h after treatment with 4-OHT and actinomycin D.
To rule out the possibility that the ER part of the MybER fusion protein is responsible for the observed effect on c-myc expression, we switched to a conditionally active system that allows us to express the full-length, native c-Myb protein. For this purpose, we cloned full-length c-myb under control of the metallothionein (MT) promoter (MTCB6+c-Myb) and transfected the plasmid into M1 cells. Northern blot analysis of the resulting single-cell clones showed expression of c-myb following stimulation of the MT promoter with ZnCl2. An example is shown in Fig. 3A. Cultivation of these cells in the presence of IL-6 and ZnCl2 resulted in inhibition of the down regulation of c-myc expression. This is in contrast to the observed down regulation in control cells, expressing the empty MTCB6+ vector (Fig. 3B). Furthermore, c-myc expression could be restored by c-Myb in cells that were already differentiating in the presence of IL-6 for 24 h prior to activation of the MT promoter with ZnCl2 (Fig. 3B). The up regulation was not as dramatic or as long lasting as that observed for MybER and may be explained by the fact that the overall activation of the MT promoter was significantly lower and started dropping a few hours following stimulation with ZnCl2 (Fig. 3A). Use of both conditionally active systems led us to the same result, that activation of c-Myb in M1 cells during differentiation can not only inhibit the IL-6-induced down regulation of c-myc but reinduce its expression as well.
FIG. 3.

Conditional expression of c-Myb using the MT promoter also results in up regulation of c-myc RNA in M1/MTCB6+c-Myb cells differentiating in the presence of IL-6. (A) M1/MTCB6+c-Myb cells were first incubated with IL-6 for 72 h to down regulate endogenous c-Myb and then treated with ZnCl2. Total RNA was examined for c-Myb expression at the indicated time points by Northern blot analysis using c-Myb as a probe. Depicted here is the result for the single-cell clone used in the following experiments. (B) Analyses of IL-6-stimulated cells showing continued expression of c-myc or reactivation of c-myc due to activation of exogenous c-Myb in M1/MTCB6+c-Myb cells. c-myc is down regulated in M1/MTCB6+ control cultures. Cells were cultivated in the presence of IL-6. ZnCl2 was added at either 0 or 24 h following IL-6 treatment, as shown on the left, and RNA was analyzed for c-myc expression at 0, 3, 12, 24, and 48 h or at 0, 3, 7, 14, and 24 h after activation of c-Myb.
Dominant negative Myb influences growth of an MML cell line with c-Myb activated by insertional mutagenesis.
A goal of our study was to determine if the regulation of c-myc by c-Myb in M1 cells also functions in leukemias in which c-Myb is proposed to be the direct cause for the development of the disease. For this purpose, we studied both c-Myb's functional roles and its regulatory properties in the leukemic cell line RI-4-11 (45). This cell line is one of many leukemias derived in vivo by inoculation of a murine retrovirus in pristane-treated mice. It, like others derived in this model system (61, 72), has undergone insertional mutagenesis of the c-myb locus and demonstrates constitutive expression of c-Myb. In these cells, unlike in M1 cells, c-myb cannot easily be down regulated by treatment with differentiation inducers such as IL-6. Therefore, to determine whether constitutive expression of c-myb may contribute to the continuous growth behavior through regulation of c-myc, we took a different approach by inhibiting Myb's function using a dominant negative Myb (MybEnER) (Fig. 4A) (36). RI-4-11 and M1 cells were infected with a retrovirus expressing either MybEnER or EnER as a control. To obtain cells expressing the dominant negative protein at high levels, infected RI-4-11 and M1 cells were selected in G418, cloned, and analyzed for MybEnER or EnER expression by Northern blot analysis. Figure 4B shows examples of the clones chosen for the studies presented here. Protein levels, determined by Western blot analysis, correlated with the detected RNA levels (data not shown).
FIG. 4.

Analysis of M1 or RI-4-11 cells with blocked Myb function due to expression of a dominant negative Myb (MybEnER). (A) Structure of the fusion genes MybEnER and the control EnER, inserted downstream of the virus long terminal repeat in LXSN (Fig. 1A). aa, amino acids. (B) Total RNA from individual cell clones of transfected M1 or RI-4-11 cells were analyzed for MybEnER or EnER expression. A EnER probe was used for hybridization. (C) Effect of the dominant negative Myb on the number of viable cells. Viable counts were determined using trypan blue dye exclusion in clonal populations of M1/MybEnER cells, RI-4-11/MybEnER cells, or their respective controls expressing EnER, following activation of the fusion protein. Data shown are calculated as the mean of three independent experiments with a standard deviation smaller than 15% and plotted as ratio of the number of viable cells in the stimulated population versus the number of cells in the unstimulated population and expressed as percentage.
RI-4-11/MybEnER and M1/MybEnER cells were first characterized for changes in the ability to proliferate after stimulation with 4-OHT. Activation of the dominant negative Myb caused a substantial decrease in the number of viable cells. There was a slight inhibitory effect on all cells due to 4-OHT alone; however, the inhibitory effect of MybEnER on proliferation in the tumor cell line RI-4-11 was 5- to 10-fold stronger than that observed in EnER cultures (Fig. 4C). A less dramatic but still reproducible effect was observed in M1 cells, where proliferation was two- to threefold less in MybEnER-expressing clones than in the EnER-expressing clone.
To determine if apoptosis was responsible for the cell death observed, clonal cell populations were examined by the annexin V assay. Stimulation with 4-OHT led to increases in the number of apoptotic cells (Fig. 5A). In RI-4-11 cells with activated MybEnER, the first evidence that the cells were undergoing apoptosis was found at 24 h poststimulation, while in M1 cells apoptosis became evident at 36 to 48 h postinduction. The maximum increases in apoptotic cells were 8.2-fold in RI-4-11 cells and 5.4-fold in M1 cells compared to nonstimulated cells. Consistent with these results, we detected a strong increase in DNA fragmentation after stimulation of the MybEnER clones with 4-OHT (Fig. 5B). Because the antiapoptotic gene bcl-2 has been demonstrated to be a target of c-Myb in some cell systems, we examined its expression by Northern blot analysis in hormone-stimulated cells expressing MybEnER. Although we were unable to show large changes in bcl-2 expression after 4-OHT treatment in the dominant negative system, activation of MybER in M1/MybER cells caused a clear up regulation of bcl-2 expression several hours following stimulation, suggesting that c-Myb is also able to influence the antiapoptotic pathway in this cellular system (data not shown).
FIG. 5.


Activation of MybEnER in clonal populations of M1 and RI-4-11 cells leads to apoptosis and cell cycle arrest. (A) Increase in apoptotic cells as determined by the TACS annexin V apoptosis assay. The y axis depicts the fold increase in the amount of apoptotic cells, and the x axis depicts the hours after stimulation with 4-OHT. Data shown are the mean of three independent results with a standard deviation smaller than 15%. (B) A DNA fragmentation assay used to detect late-stage programmed cell death. DNA laddering is prominent in MybEnER clones following activation of the fusion protein. (C) Flow microfluorometric analysis of the cell cycle distribution in RI-4-11/MybEnER or RI-4-11/EnER cells stimulated with 4-OHT for 24 h. Cell cycle distribution was examined using a Becton Dickinson FACScan and analyzed with the ModFitLT V2.0 program. The proportions of cells in G0-G1, G2-M, and S are shown in boxes in each graph as percentages of the total population.
Although it could be demonstrated, as described above, that apoptosis accounts, at least in part, for reduced cell growth in MybEnER cells, we wanted to determine if alterations in the cell cycle could also be responsible for this effect. MybEnER cells treated with 4-OHT for 24 h, a period corresponding to one doubling time, were shown to have an increase in the proportion of cells remaining in G1 compared to unstimulated cells (Fig. 5C). Cell cycle arrest was observed in both RI-4-11 (clone 16 [cl16] and cl38) and M1 (cl9 and cl16) cells, with increases in the G1 populations of 1.4-, 1.3-, 1.3-, and 1.6-fold, respectively, compared with the negative control EnER. We concluded that the reduced cell numbers in MybEnER cells, stimulated with the synthetic steroid, was due to G1 arrest as well as increased apoptosis. We also concluded that deregulated c-Myb is responsible for the uncontrolled growth of the leukemia cells.
Use of a dominant negative Myb to substantiate the regulatory connection between c-myc and c-Myb in an MML induced by disregulated c-myb.
As shown in Fig. 2, in partially differentiated and growth-arrested M1 cells, induction of active MybER led to a reactivation of c-myc expression. We wanted to determine if c-myc is also responsible for Myb-dependent proliferation in the RI-4-11 leukemia cell line with constitutively activated c-myb. Following stimulation with 4-OHT, RI-4-11/MybEnER and M1/MybEnER cells were examined for c-myc expression by Northern blot analysis. c-myc was down regulated in RI-4-11 cell clones expressing the dominant negative Myb, 3 to 7 h after stimulation with the synthetic steroid (Fig. 6A). Similar results were obtained for M1 cells (Fig. 7A). In contrast, no change in c-myc RNA levels in individual clones expressing EnER was observed. Western blot analysis using a monoclonal c-Myc antibody confirmed the down regulation at the protein level (Fig. 6B and 7B).
FIG. 6.

Down regulation of c-myc in RI-4-11/MybEnER cells stimulated with 4-OHT. (A) Clonal populations of RI-4-11 cells expressing MybEnER or EnER were treated with 4-OHT and analyzed at the indicated times poststimulation for expression of c-myc RNA by Northern blot analysis. β-actin was used to probe the same blots as a control for loading. (B) c-Myc protein expression was determined by Western blot analysis in RI-4-11/MybEnER or EnER cells at 0 and 24 h following stimulation with 4-OHT.
FIG. 7.

Down regulation of c-myc in M1/MybEnER cells stimulated with 4-OHT. (A) Clonal populations of M1 cells expressing MybEnER or EnER were treated with 4-OHT and analyzed at the indicated times poststimulation for expression of c-myc RNA by Northern blot analysis. β-actin was used to probe the same blots as a control for loading. (B) c-Myc protein expression was determined by Western blot analysis in M1/MybEnER or EnER cells at 0 and 24 h following stimulation with 4-OHT.
To rule out the possibility that the ER part of the dominant negative protein was responsible for the observed effects, we placed the MybEn part of the fusion gene under the control of the inducible MT promoter in the pMTCB6+. In M1 cells stably transfected with MTC6+MybEn, MybEn could be detected within 14 h when stimulated with 50 μM ZnCl2 (Fig. 8A). Consistent with our previous results, c-myc expression was found to be significantly down regulated by MybEn, while there was no change in c-myc mRNA levels in cells transfected with the empty plasmid pMT-CB6+ (Fig. 8B). The reduction in c-myc first became visible after 7 h after stimulation with ZnCl2 in M1/MybEn cells. Interestingly, this is 4 h later than the response observed with the fusion protein MybEnER and can be explained by the fact that expression of MybEn must be induced at the RNA level, while MybEnER is constitutively expressed. Therefore, we concluded that in both inducible systems, Myb controls expression of c-myc in leukemic cells.
FIG. 8.

Conditional expression of dominant negative Myb in M1 cells using the MT promoter also results in down regulation of c-myc RNA. (A) M1/MTC6+MybEn cells were treated with ZnCl2 and analyzed for MybEn RNA expression at the indicated times. Induction of MybEn RNA expression was examined by Northern blot analysis using MybEn as a probe. (B) c-myc expression was examined in the same cells by Northern blot analysis at the indicated times after addition of ZnCl2. Cells transfected with the empty pMTC6+ vector were used as a control. GAPDH was used as a probe to control for loading.
We noticed that the effect on c-myc was transient in both conditional systems used to activate the dominant negative Myb. Analysis of the MybEnER protein itself revealed no differences in the levels of expression during the stimulation period (data not shown). In addition, restimulation of M1/MybEnER cells 48 h after the first induction had no influence on prolonging the inhibitory effect of the dominant negative protein. Since this observation cannot be explained by inactivation of the drug or a feedback mechanism affecting the receptor level, we suggest that the effect is due to a negative selection process against cells in which c-myc and other genes are down regulated. As activation of the dominant negative Myb leads to cell cycle arrest and apoptosis around 24 h following induction, RNA isolated at later time points comes from an increasing percentage of cells that are not affected by the treatment.
Expression of other putative c-Myb target genes in M1/MybEnER cells.
In addition to c-myc, several putative target genes of c-Myb have been described which may be involved in c-Myb's function in regulating proliferation, such as cdc2, c-kit, and c-myb itself (48, 71). By examining the endogenous expression of c-myb in activated M1/MybEnER cells by Northern blot analysis, we determined that c-myb was down regulated 3 to 7 h after stimulation with 4-OHT, in a pattern analogous to what we had observed for c-myc (Fig. 9A). This result indicated that the mouse c-Myb protein is able to influence its own expression in myeloid cells. This observation was not completely unexpected, as it has been shown before that the human c-Myb protein is able to autoregulate its own promoter in T cells and fibroblasts (24, 49).
FIG. 9.

Expression of putative c-Myb target genes in M1/MybEnER cells treated with 4-OHT. (A) MybEnER cl38 cells were treated with 4-OHT; at the indicated times, expression of endogenous c-myb RNA was examined by Northern blot analysis. GAPDH was used as a probe to control for loading. (B) RNA analysis was carried out as for panel A, using probes for cdc2, cyclinD1, and ODC. β-actin was used to probe the same blots as a control for loading.
Ku et al. demonstrated that c-Myb is able to transactivate the human cdc2 promoter and suggested that c-Myb may regulate cell cycle progression by activating expression of the cdc2 gene (31). We detected no changes in cdc2 expression after stimulating M1/MybEnER cl38 cells with 4-OHT for 96 h (Fig. 9B). A connection between c-Myb and CDC2, therefore, could not be confirmed in mouse myeloid cells using the dominant negative Myb. Another regulator of the cell cycle, c-Kit, a tyrosine kinase receptor that plays an important role in hematopoietic cell growth, has recently been suggested to be regulated by c-Myb and to be important for proliferation of very early myeloid cells derived from fetal liver (27, 54). However, we were unable to study the potential effects of c-Myb on c-kit in the system used here, because expression of this gene could not be detected in M1 or RI-4-11 cells (data not shown). The stem cell factor is normally expressed only in very early progenitor cells.
Interestingly, we found an inhibitory effect on cyclinD1 expression after stimulation of the dominant negative Myb protein in M1/MybEnER cl38 cells (Fig. 9B). Cyclin D1 is known to be an important factor for cell cycle progression by driving cells from the quiescent G0 phase to G1 phase, and it was shown to be regulated by c-Myc (26). As expected, the effects on cyclinD1 occurred later than those on c-myc. To further evaluate downstream effects of deregulated c-myc expression due to activation of MybEnER, we examined the influence of the dominant negative protein on another target gene of c-Myc, ODC. ODC is involved in polyamine biosynthesis and has been shown to be required for entry and progression through the cell cycle (23, 26). In M1/MybEnER cl38 cells, stimulation with 4-OHT caused a reduction of ODC several hours later than the down regulation of c-myc with a time course similar to that seen for cyclinD1 (Fig. 9B). This experiment demonstrated that the blocking of c-Myb function not only influences expression of direct target genes like c-myc but also has an effect on genes further down in this regulatory pathway of proliferation.
A conditionally active Myc reverses IL-6-induced growth arrest in M1 cells comparably to MybER.
Next we wanted to determine whether c-Myb might control proliferation in myeloid cells solely through c-myc. In the first experiment, we attempted to reverse the growth arrest, induced in cells with the dominant negative Myb, by constitutive over expression of c-myc. However, treatment of the double transfectants with tamoxifen led to a dramatic increase in the number of apoptotic cells and was responsible for inconsistent cell cycle results. Induction of apoptosis in these cells was probably due to an acquired increased sensitivity to drugs, since this has been reported recently for cells which overexpress c-myc (47). Because of this, we went back to the M1 differentiation model to establish a role for c-Myc as the major transducer of Myb's proliferation signal. In this model, we compared the effects of MycER with those of MybER on cell cycle distribution. M1 cells transfected with a MycER fusion gene were checked for expression by Northern blot analysis (Fig. 10; Table 1). Significantly, activation of MycER in cells that were cultivated with IL-6 for 3 days, and therefore growth arrested and nearly terminally differentiated, reversed growth arrest to almost the same degree as activation of MybER (Table 1) (40.6% to 70.5% reversal, versus 70.6% to 107.0%). Although we cannot rule out the possibility that an additional c-Myb target gene(s) may be involved in proliferation in myeloid cells, this experiment demonstrates that c-myc plays a very dominant role in this process.
FIG. 10.
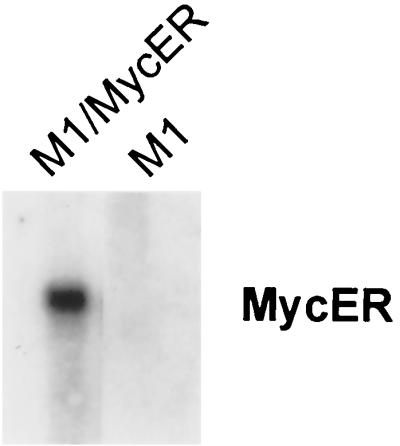
Northern blot analysis showing expression of MycER in transfected M1 cells. Total RNA from M1 cells transfected with the c-myc expression vector pBabe Puro MycER was monitored for MycER expression, using MycER as the probe.
TABLE 1.
Activation of both MybER and MycER reverses growth arrest in IL-6-treated M1 cells
| Expta | Treatment | % cells in G1c | |||
|---|---|---|---|---|---|
| M1/MybER | M1/MycER | ||||
| 24 h | 48 h | 24 h | 48 h | ||
| 1 | IL-6 | 54.7 | 71.6 | 54.9 | 69.6 |
| 4-OHT | 41.3 | 33.4 | 38.8 | 40.1 | |
| IL-6 + 4-OHT | 44.8 | 42.4 | 49.3 | 52.8 | |
| % Inhibitionb | 73.8 | 76.4 | 34.8 | 56.9 | |
| 2 | IL-6 | 59.0 | 60.6 | 63.9 | 66.3 |
| 4-OHT | 35.7 | 31.6 | 34.4 | 41.8 | |
| IL-6 + 4-OHT | 34.1 | 40.1 | 51.9 | 49.0 | |
| % Reversal | 107.0 | 70.7 | 40.7 | 70.6 |
DISCUSSION
The data presented here show for the first time that the expression of endogenous chromosomal c-myc, a gene well known to be involved in cell growth, can be directly regulated by c-Myb. By demonstrating that inhibition of c-Myb simultaneously leads to growth arrest and down regulation of c-myc, we are able to suggest a mechanism by which the proto-oncogene c-Myb exerts its oncogenic activity. Additionally, this study provides an explanation for the leukemogenic phenotype observed in an MML-derived cell lines where c-myb has been activated by retroviral insertional mutagenesis.
Evidence for the regulation of c-myc by c-Myb came from four conditional expression systems. Two of these systems exploited the M1 cell differentiation model, in which IL-6 treatment causes growth arrest and down regulation of both c-myb and c-myc. In this model, it was possible by using MybER to restore both growth and c-myc expression even after the cells were nearly terminally differentiated. To eliminate potential regulatory effects of the ER portion of the fusion protein, the coding sequences for full-length c-Myb were placed under control of the MT promoter. With this construct, it was possible not only to confirm Myb's ability to inhibit the down regulation of c-myc during differentiation but also to confirm its ability to reinduce expression of c-myc in cells that were already differentiating. Two additional systems utilizing conditional expression of dominant negative Myb proteins were initiated to study the function of c-Myb in the leukemia cell line RI-4-11, in which c-myb cannot be easily down regulated by cytokines. When activated in RI-4-11 cells, the dominant negative Myb led to apoptosis and cell cycle arrest, effects which are likely to be caused, at least in part, by the observed major reduction in the level of c-Myc.
Expression of downstream targets of c-Myc were also affected by MybEnER subsequent to its effect on c-myc. While expression of c-myc was down regulated 3 to 7 h following activation of the dominant negative protein, expression of two proposed c-Myc target genes, cyclinD1 and ODC (26, 56, 67), was reduced several (7 to 14) hours later. Repression of indirect target genes demonstrates how Myb transduces proliferation signals down a pathway to other important regulators of the cell cycle.
c-Myb's ability to regulate the expression of c-myc may be tissue specific, because studies in lymphoid and erythroid cells, using conditionally active Myb or a conditionally active dominant negative Myb, did not support this connection. For example, in a murine erythroleukemia cell line, a conditionally active MybER blocked differentiation but had no effect on proliferation or c-myc expression (35). Similarly, dominant negative Myb caused apoptosis in T cells, but this effect was not accompanied by growth arrest or deregulation of c-myc (65). The fact that regulation of the resident c-myc by c-Myb was observed in myeloid cells and not lymphoid or erythroid cells is consistent with reporter gene experiments which showed that transactivation of the c-myc promoter by c-Myb is significantly higher in myeloid cells than in all other cell types examined (13).
The c-myc promoter has been shown to have binding sites for many transcription factors, in addition to those for c-Myb, and it is presumed that the transactivation of this promoter by c-Myb would require additional transcription factors as well as cofactors. Known cofactors that bind to the DNA binding and transactivation domains of c-Myb are p100 and p300/CBP, respectively (48). Presumably, additional known factors, as well as perhaps unknown factors, could be involved in the activation of c-myc. As c-Myb was able to induce c-myc in nearly terminally differentiated cells during IL-6-induced differentiation, it must be assumed that if such transcription factors or coactivators are necessary, they have remained at sufficiently high levels during the differentiation process.
Recently, it was reported that in fetal liver-derived, c-Myb-transformed primitive myeloid cells, c-Myb was able to regulate proliferation by activating c-kit but had no effect on c-myc (27). We were not able to detect expression of the stem cell factor gene c-kit in M1 or RI-4-11 cells. However, both cell lines seem to be more differentiated than those used in the study mentioned above (27) and would not be expected to express c-kit any longer. It is not inconceivable that during the in vivo development of leukemias such as RI-4-11, c-kit may be an additional important target for c-Myb in establishing myeloid tumors. This would fit with the proposed notion that preleukemic myeloid cells, initiated by provirus integration in the c-myb locus, are early progenitors that later differentiate into mature cells in which c-kit is ultimately no longer expressed (46). Alternatively, it is possible that target cells transformed by infection of fetal liver cells in vitro are even more primitive than those transformed in adult mice in the MML model, which gives rise to leukemias such as RI-4-11 (45). c-kit, as a target gene of c-Myb, would in this case not be important for the development of MML.
Although the function of c-Myc is not yet clear, it is generally accepted that it acts as a ubiquitous regulator of proliferation in cycling cells and normally is not detected in growth-arrested cells. There is a rapid increase in c-Myc levels shortly after cells enter the cell cycle which remain high as long as the cells are proliferating (26, 67). Consistent with this observation, c-Myc has been shown to be important for most cells to proceed from the G1 to the S phase of the cell cycle and also functions in G2 (59). Homozygous deletion of c-myc in a fibroblast cell line has been shown to increase the cell cycle length around threefold (40). Here, we attempted to answer the question of whether c-myc might be the sole target of Myb responsible for proliferation by determining if MycER could effectively reverse IL-6-induced cell cycle arrest. We found that the percentage of cells moving back into the cell cycle, after activation of MycER, was almost as high as after activation of MybER. That MycER was not quite as effective as MybER could be due to the fact that the human c-Myc domain expressed in the fusion gene may not be as effective in promoting growth as the authentic murine c-Myc. It is also possible that when c-Myc is expressed as a fusion protein, dimerization with its partner Max and/or DNA binding may occur with lower affinity than with the native protein. An additional consideration is that activation of MybER results in the up regulation of bcl-2, inhibiting the induction of apoptosis in these cultures, while the overexpression of MycER can induce apoptosis, which may impair cell proliferation. Therefore, we conclude that c-myc may be the only gene transducing Myb-dependent proliferation; however, because of the quantitatively greater effects of MybER than of MycER, we cannot rule out the possibility that Myb promotes cell growth through regulation of another gene(s) as well.
As demonstrated here, activation of the dominant negative Myb has an influence on the expression of endogenous c-myb in myeloid progenitor cells, suggesting that an autoregulatory mechanism that involves the mouse c-myb promoter is functioning in these cells. It has been previously reported for the human c-myb promoter that binding of c-Myb to its binding sites in the 5′ flanking region of the c-myb gene leads to a positive autoregulation in fibroblasts. Negative regulation was detected in T cells, while the Myb binding sites seemed to have no function in a myeloid cell line (24, 49). Because of the general repressive nature of MybEnER, due to the fact that it contains the Drosophila En transrepressor, it is not yet clear if the authentic c-Myb protein has a positive or negative effect on regulation of endogenous c-myb expression. However, comparison of endogenous c-myb mRNA expression in M1/MybER cells with that in parental M1 cells suggests a positive feedback loop, which would contribute to Myb's oncogenic potential (data not shown).
In conclusion, the results presented here indicate that c-Myb's role in leukemias in which c-Myb is activated by insertional mutagenesis is to maintain continued expression of c-myc, which is usually down regulated during myeloid differentiation. c-Myb would therefore influence the proliferation state of these cells by carrying out this function. Although the prevention of growth arrest is important to leukemogenesis, it is not necessarily the only function contributing to the transformed state. Previous data and our data presented here suggest that c-Myb's role in preventing apoptosis through the up regulation of Bcl-2 may also contribute to the neoplastic state. Autoregulation of the c-Myb promoter may contribute to this state as well. The exact role of c-Myb in regulating bcl-2, c-myb, and other genes which are potentially involved in the development of myeloid leukemias will require further investigation.
ACKNOWLEDGMENTS
We thank Jacques Van Snick for providing baculovirus expressing IL-6 and Stuart Rudikoff and Emily Shacter for assistance in preparing IL-6 stocks used in this study. We are grateful to Sandra Ruscetti, Dan Liebermann, and Konrad Huppi for kindly providing probes for v-kit, cdc2, ODC, and β-actin and to Alan Friedman for sending the MT promoter plasmid pMT-C6+ as well as to Eric Westin and Tim Bender for the monoclonal c-Myb antibody. We also thank Doug Lowy for critical reading of the manuscript.
REFERENCES
- 1.Badiani P, Corbella P, Kioussis D, Marvel J, Weston K. Dominant interfering alleles define a role for c-Myb in T-cell development. Genes Dev. 1994;8:770–782. doi: 10.1101/gad.8.7.770. [DOI] [PubMed] [Google Scholar]
- 2.Baumbach W R, Keath E J, Cole M D. A mouse c-myc retrovirus transforms established fibroblast lines in vitro and induces monocyte-macrophage tumors in vivo. J Virol. 1986;59:276–283. doi: 10.1128/jvi.59.2.276-283.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baumbach W R, Stanley E R, Cole M D. Induction of clonal monocyte-macrophage tumors in vivo by a mouse c-myc retrovirus: rearrangement of the CSF-1 gene as a secondary transforming event. Mol Cell Biol. 1987;7:664–671. doi: 10.1128/mcb.7.2.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bender T P, Kuehl W M. Murine myb protooncogene mRNA: cDNA sequence and evidence for 5′ heterogeneity. Proc Natl Acad Sci USA. 1986;83:3204–3208. doi: 10.1073/pnas.83.10.3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Besmer P, Murphy J E, George P C, Qiu F H, Bergold P J, Lederman L, Snyder H W, Jr, Brodeur D, Zuckerman E E, Hardy W D. A new acute transforming feline retrovirus and relationship of its oncogene v-kit with the protein kinase gene family. Nature. 1986;320:415–421. doi: 10.1038/320415a0. [DOI] [PubMed] [Google Scholar]
- 6.Bies J, Hoffman B, Amanullah A, Giese T, Wolff L. B-Myb prevents growth arrest associated with terminal differentiation of monocytic cells. Oncogene. 1996;12:355–363. [PubMed] [Google Scholar]
- 7.Bies J, Mukhopadhyaya R, Pierce J, Wolff L. Only late, nonmitotic stages of granulocyte differentiation in 32Dc13 cells are blocked by ectopic expression of murine c-myb and its truncated forms. Cell Growth Differ. 1995;6:59–68. [PubMed] [Google Scholar]
- 8.Bies J, Wolff L. Acceleration of apoptosis in transforming growth factor beta 1-treated M1 cells ectopically expressing B-myb. Cancer Res. 1995;55:501–504. [PubMed] [Google Scholar]
- 9.Braun B S, Frieden R, Lessnick S L, May W A, Denny T. Identification of target genes for the Ewing's sarcoma EWS/FLI fusion protein by representational difference analysis. Mol Cell Biol. 1995;15:4623–4630. doi: 10.1128/mcb.15.8.4623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burk O, Klempnauer K H. Estrogen-dependent alterations in differentiation state of myeloid cells caused by a v-myb/estrogen receptor fusion protein. EMBO J. 1991;10:3713–3719. doi: 10.1002/j.1460-2075.1991.tb04939.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cao W, Britos-Bray M, Claxton D F, Kelley C A, Speck N A, Liu P P, Friedman A D. CBF beta-SMMHC, expressed in M4Eo AML, reduced CBF DNA-binding and inhibited the G1 to S cell cycle transition at the restriction point in myeloid and lymphoid cells. Oncogene. 1997;15:1315–1327. doi: 10.1038/sj.onc.1201305. [DOI] [PubMed] [Google Scholar]
- 12.Clarke M F, Kukowska-Latallo J F, Westin E, Smith M, Prochownik E V. Constitutive expression of a c-myb cDNA blocks Friend murine erythroleukemia cell differentiation. Mol Cell Biol. 1988;8:884–892. doi: 10.1128/mcb.8.2.884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cogswell J P, Cogswell P C, Kuehl W M, Cuddihy A M, Bender T M, Engelke U, Marcu K B, Ting J P. Mechanism of c-myc regulation by c-Myb in different cell lineages. Mol Cell Biol. 1993;13:2858–2869. doi: 10.1128/mcb.13.5.2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cosenza S C, Carter R, Pena A, Donigan A, Borrelli M, Soprano D R, Soprano K J. Growth-associated gene expression is not constant in cells traversing G-1 after exiting mitosis. J Cell Physiol. 1991;147:231–241. doi: 10.1002/jcp.1041470207. [DOI] [PubMed] [Google Scholar]
- 15.Duprey S P, Boettiger D. Developmental regulation of c-myb in normal myeloid progenitor cells. Proc Natl Acad Sci USA. 1985;82:6937–6941. doi: 10.1073/pnas.82.20.6937. . (Erratum, 83:2281, 1986.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eckert E A, Beard D, Beard J W. Dose-response relations in experimental transmission of avian erythromyeloblastic leukosis. I. Host-response to the virus. J Natl Cancer Inst. 1951;12:447–463. [PubMed] [Google Scholar]
- 17.Evans J L, Moore T L, Kuehl W M, Bender T, Ting J P. Functional analysis of c-Myb protein in T-lymphocytic cell lines shows that it trans-activates the c-myc promoter. Mol Cell Biol. 1990;10:5747–5752. doi: 10.1128/mcb.10.11.5747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fort P, Marty L, Piechaczyk M, el Sabrouty S, Dani C, Jeanteur P, Blanchard J M. Various rat adult tissues express only one major mRNA species from the glyceraldehyde-3-phosphate-dehydrogenase multigenic family. Nucleic Acids Res. 1985;13:1431–1442. doi: 10.1093/nar/13.5.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frampton J, Ramqvist T, Graf T. v-Myb of E26 leukemia virus up-regulates bcl-2 and suppresses apoptosis in myeloid cells. Genes Dev. 1996;10:2720–2731. doi: 10.1101/gad.10.21.2720. [DOI] [PubMed] [Google Scholar]
- 20.Goda S K, Minton N P. A simple procedure for gel electrophoresis and Northern blotting of RNA. Nucleic Acids Res. 1995;23:3357–3358. doi: 10.1093/nar/23.16.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gonda T J, Cory S, Sobieszczuk P, Holtzman D, Adams J M. Generation of altered transcripts by retroviral insertion within the c-myb gene in two murine monocytic leukemias. J Virol. 1987;61:2754–2763. doi: 10.1128/jvi.61.9.2754-2763.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gonda T J, Metcalf D. Expression of myb, myc and fos proto-oncogenes during the differentiation of a murine myeloid leukaemia. Nature. 1984;310:249–251. doi: 10.1038/310249a0. [DOI] [PubMed] [Google Scholar]
- 23.Grandori C, Eisenman R N. Myc target genes. Trends Biochem Sci. 1997;22:177–181. doi: 10.1016/s0968-0004(97)01025-6. [DOI] [PubMed] [Google Scholar]
- 24.Guerra J, Withers D A, Boxer L M. Myb binding sites mediate negative regulation of c-myb expression in T-cell lines. Blood. 1995;86:1873–1880. [PubMed] [Google Scholar]
- 25.Gunning P, Ponte P, Okayama H, Engel J, Blau H, Kedes L. Isolation and characterization of full-length cDNA clones for human alpha-, beta-, and gamma-actin mRNAs: skeletal but not cytoplasmic actins have an amino-terminal cysteine that is subsequently removed. Mol Cell Biol. 1983;3:787–795. doi: 10.1128/mcb.3.5.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Henriksson M, Luscher B. Proteins of the Myc network: essential regulators of cell growth and differentiation. Adv Cancer Res. 1996;68:109–182. doi: 10.1016/s0065-230x(08)60353-x. [DOI] [PubMed] [Google Scholar]
- 27.Hogg A, Schirm S, Nakagoshi H, Bartley P, Ishii S, Bishop J M, Gonda T J. Inactivation of a c-Myb/estrogen receptor fusion protein in transformed primary cells leads to granulocyte/macrophage differentiation and down regulation of c-kit but not c-myc or cdc2. Oncogene. 1997;15:2885–2898. doi: 10.1038/sj.onc.1201472. [DOI] [PubMed] [Google Scholar]
- 28.Kanter M R, Smith R E, Hayward W S. Rapid induction of B-cell lymphomas: insertional activation of c-myb by avian leukosis virus. J Virol. 1988;62:1423–1432. doi: 10.1128/jvi.62.4.1423-1432.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kastan M B, Stone K D, Civin C I. Nuclear oncoprotein expression as a function of lineage, differentiation stage, and proliferative status of normal human hematopoietic cells. Blood. 1989;74:1517–1524. [PubMed] [Google Scholar]
- 30.Kowenz-Leutz E, Herr P, Niss K, Leutz A. The homeobox gene GBX2, a target of the myb oncogene, mediates autocrine growth and monocyte differentiation. Cell. 1997;91:185–195. doi: 10.1016/s0092-8674(00)80401-8. [DOI] [PubMed] [Google Scholar]
- 31.Ku D H, Wen S C, Engelhard A, Nicolaides N C, Lipson K E, Marino T A, Calabretta B. c-myb transactivates cdc2 expression via Myb binding sites in the 5′- flanking region of the human cdc2 gene. J Biol Chem. 1993;268:2255–2259. . (Erratum, 268:13010.) [PubMed] [Google Scholar]
- 32.Leprince D, Gegonne A, Coll J, Taisne C, Schneeberger A, Lagrou C, Stehelin D. A putative second cell-derived oncogene of the avian leukemia retrovirus E26. Nature. 1983;306:395–397. doi: 10.1038/306395a0. [DOI] [PubMed] [Google Scholar]
- 33.Liebermann D A, Hoffman-Liebermann B. Proto-oncogene expression and dissection of the myeloid growth to differentiation developmental cascade. Oncogene. 1989;4:583–592. [PubMed] [Google Scholar]
- 34.Littlewood T D, Hancock D C, Danielian P S, Parker M G, Evan G I. A modified oestrogen receptor ligand-binding domain as an improved switch for the regulation of heterologous proteins. Nucleic Acids Res. 1995;23:1686–1690. doi: 10.1093/nar/23.10.1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lyon J J, Watson R J. Conditional inhibition of erythroid differentiation by c-Myb/oestrogen receptor fusion proteins. Differentiation. 1995;59:171–178. doi: 10.1046/j.1432-0436.1995.5930171.x. [DOI] [PubMed] [Google Scholar]
- 36.Lyon J J, Watson R J. Interference of Myb transactivation activity by a conditional dominant negative protein: functional interference in a cytotoxic T-cell results in G1 arrest. Gene. 1996;182:123–128. doi: 10.1016/s0378-1119(96)00531-8. [DOI] [PubMed] [Google Scholar]
- 37.Majello B, Kenyon L C, Dalla-Favera R. Human c-myb protooncogene: nucleotide sequence of cDNA and organization of the genomic locus. Proc Natl Acad Sci USA. 1986;83:9636–9640. doi: 10.1073/pnas.83.24.9636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Markowitz D, Goff S, Bank A. A safe packaging line for gene transfer: separating viral genes on two different plasmids. J Virol. 1988;62:1120–1124. doi: 10.1128/jvi.62.4.1120-1124.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Markowitz D, Goff S, Bank A. Construction and use of a safe and efficient amphotropic packaging cell line. Virology. 1988;167:400–406. [PubMed] [Google Scholar]
- 40.Mateyak M K, Obaya A J, Adachi S, Sedivy J M. Phenotypes of c-Myc-deficient rat fibroblasts isolated by targeted homologous recombination. Cell Growth Differ. 1999;8:1039–1048. [PubMed] [Google Scholar]
- 41.Matsushime H, Roussel M F, Ashmun R A, Sherr C J. Colony-stimulating factor 1 regulates novel cyclins during the G1 phase of the cell cycle. Cell. 1991;65:701–713. doi: 10.1016/0092-8674(91)90101-4. [DOI] [PubMed] [Google Scholar]
- 42.McClinton D, Stafford J, Brents L, Bender T P, Kuehl W M. Differentiation of mouse erythroleukemia cells is blocked by late up-regulation of a c-myb transgene. Mol Cell Biol. 1990;10:705–710. doi: 10.1128/mcb.10.2.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miller A D, Rosman G J. Improved retroviral vectors for gene transfer and expression. BioTechniques. 1989;7:980–982. , 984–986, 989–990. [PMC free article] [PubMed] [Google Scholar]
- 44.Mucenski M L, McLain K, Kier A B, Swerdlow S H, Schreiner C M, Miller T A, Pietryga D W, Scott W J, Jr, Potter S S. A functional c-myb gene is required for normal murine fetal hepatic hematopoiesis. Cell. 1991;65:677–689. doi: 10.1016/0092-8674(91)90099-k. [DOI] [PubMed] [Google Scholar]
- 45.Mukhopadhyaya R, Wolff L. New sites of proviral integration associated with murine promonocytic leukemias and evidence for alternate modes of c-myb activation. J Virol. 1992;66:6035–6044. doi: 10.1128/jvi.66.10.6035-6044.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nason-Burchenal K, Wolff L. Activation of c-myb is an early bone-marrow event in a murine model for acute promonocytic leukemia. Proc Natl Acad Sci USA. 1993;90:1619–1623. doi: 10.1073/pnas.90.4.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nesbit C E, Fan S, Zhang H, Prochownik E V. Distinct apoptotic responses imparted by c-myc and max. Blood. 1998;92:1003–1010. [PubMed] [Google Scholar]
- 48.Ness S A. The Myb oncoprotein: regulating a regulator. Biochim Biophys Acta. 1996;1288:F123–F139. doi: 10.1016/s0304-419x(96)00027-3. [DOI] [PubMed] [Google Scholar]
- 49.Nicolaides N C, Gualdi R, Casadevall C, Manzella L, Calabretta B. Positive autoregulation of c-myb expression via Myb binding sites in the 5′ flanking region of the human c-myb gene. Mol Cell Biol. 1991;11:6166–6176. doi: 10.1128/mcb.11.12.6166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Noguchi P D. Use of flow cytometry for DNA analysis. In: Coligan J E, Kruisbeek A M, Margulies D H, Shevach E M, Strober W, editors. Current protocols in immunology. New York, N.Y: John Wiley & Sons, Inc.; 1998. p. 5.7.1-5.7.6. [Google Scholar]
- 51.Picard D, Salser S J, Yamamoto K R. A movable and regulable inactivation function within the steroid binding domain of the glucocorticoid receptor. Cell. 1988;54:1073–1080. doi: 10.1016/0092-8674(88)90122-5. [DOI] [PubMed] [Google Scholar]
- 52.Pizer E, Humphries E H. RAV-1 insertional mutagenesis: disruption of the c-myb locus and development of avian B-cell lymphomas. J Virol. 1989;63:1630–1640. doi: 10.1128/jvi.63.4.1630-1640.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pizer E S, Baba T W, Humphries E H. Activation of the c-myb locus is insufficient for the rapid induction of disseminated avian B-cell lymphoma. J Virol. 1992;66:512–523. doi: 10.1128/jvi.66.1.512-523.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ratajczak M Z, Perrotti D, Melotti P, Powzaniuk M, Calabretta B, Onodera K, Kregenow D A, Machalinski B, Gewirtz A M. Myb and its proteins are candidate regulators of c-kit expression in human hematopoietic cells. Blood. 1998;91:1934–1946. [PubMed] [Google Scholar]
- 55.Rouzic E, Perbal B. Retroviral insertional activation of the c-myb proto-oncogene in a Marek's disease T-lymphoma cell line. J Virol. 1996;70:7414–7423. doi: 10.1128/jvi.70.11.7414-7423.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ryan K M, Birnie G D. Myc oncogenes: the enigmatic family. Biochem J. 1996;314:713–721. doi: 10.1042/bj3140713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schaegger H, von Jagow G. Tricine sodium dodecyl sulfate polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100kD. Anal Biochem. 1987;166:368–379. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
- 58.Selvakumaran M, Liebermann D A, Hoffman-Liebermann B. Deregulated c-myb disrupts interleukin-6- or leukemia inhibitory factor-induced myeloid differentiation prior to c-myc: role in leukemogenesis. Mol Cell Biol. 1992;12:2493–2500. doi: 10.1128/mcb.12.6.2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Seth A, Alvarez E, Gupta S, Davis R J. A phosphorylation site located in the NH2-terminal domain of c-Myc increases transactivation of gene expression. J Biol Chem. 1991;266:23521–23524. [PubMed] [Google Scholar]
- 60.Shen-Ong G L, Morse H C, III, Potter M, Mushinski J F. Two modes of c-myb activation in virus-induced mouse myeloid tumors. Mol Cell Biol. 1986;6:380–392. doi: 10.1128/mcb.6.2.380. . (Erratum, 6:2756.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shen-Ong G L, Wolff L. Moloney murine leukemia virus-induced myeloid tumors in adult BALB/c mice: requirement of c-myb activation but lack of v-abl involvement. J Virol. 1987;61:3721–3725. doi: 10.1128/jvi.61.12.3721-3725.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sitzmann J, Noben-Trauth K, Klempnauer K H. Expression of mouse c-myb during embryonic development. Oncogene. 1995;11:2273–2279. [PubMed] [Google Scholar]
- 63.Stanton L W, Watt R, Marcu K B. Translocation, breakage and truncated transcripts of c-myc oncogene in murine plasmacytomas. Nature. 1983;303:401–406. doi: 10.1038/303401a0. [DOI] [PubMed] [Google Scholar]
- 64.Sudo T, Miyazawa H, Hanaoka F, Ishii S. The c-myb proto-oncogene product binds to but does not activate the promoter of the DNA polymerase alpha gene. Oncogene. 1992;7:1999–2006. [PubMed] [Google Scholar]
- 65.Taylor D, Badiani P, Weston K. A dominant interfering Myb mutant causes apoptosis in T cells. Genes Dev. 1996;10:2732–2744. doi: 10.1101/gad.10.21.2732. [DOI] [PubMed] [Google Scholar]
- 66.Th'ng J P, Wright P S, Hamaguchi J, Lee M G, Norbury C J, Nurse P, Bradbury E M. The FT210 cell line is a mouse G2 phase mutant with a temperature-sensitive CDC2 gene product. Cell. 1990;63:313–324. doi: 10.1016/0092-8674(90)90164-a. [DOI] [PubMed] [Google Scholar]
- 67.Thompson E B. The many roles of c-Myc in apoptosis. Annu Rev Physiol. 1998;60:575–600. doi: 10.1146/annurev.physiol.60.1.575. [DOI] [PubMed] [Google Scholar]
- 68.Vandenbark G R, Chen Y, Friday E, Pavlik K, Anthony B, deCastro C, Kaufman R E. Complex regulation of human c-kit transcription by promoter repressors, activators, and specific myb elements. Cell Growth Differ. 1996;7:1383–1392. [PubMed] [Google Scholar]
- 69.Westin E H, Gallo R C, Arya S K, Eva A, Souza L M, Baluda M A, Aaronson S A, Wong-Staal F. Differential expression of the amv gene in human hematopoietic cells. Proc Natl Acad Sci USA. 1982;79:2194–2198. doi: 10.1073/pnas.79.7.2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Weston K. Myb proteins in life, death and differentiation. Curr Opin Genet Dev. 1998;8:76–81. doi: 10.1016/s0959-437x(98)80065-8. [DOI] [PubMed] [Google Scholar]
- 71.Wolff L. Myb-induced transformation. Crit Rev Oncog. 1996;7:245–260. doi: 10.1615/critrevoncog.v7.i3-4.60. [DOI] [PubMed] [Google Scholar]
- 72.Wolff L, Mushinski J F, Shen-Ong G L, Morse H C., III A chronic inflammatory response. Its role in supporting the development of c-myb and c-myc related promonocytic and monocytic tumors in BALB/c mice. J Immunol. 1988;141:681–689. [PubMed] [Google Scholar]
- 73.Zobel A, Kalkbrenner F, Vorbrueggen G, Moelling K. Transactivation of the human c-myc gene by c-Myb. Biochem Biophys Res Commun. 1992;186:715–722. doi: 10.1016/0006-291x(92)90805-u. [DOI] [PubMed] [Google Scholar]