Genome sequences of Chlamydia trachomatis MoPn and Chlamydia pneumoniae AR39 (original) (raw)
Abstract
The genome sequences of Chlamydia trachomatis mouse pneumonitis (MoPn) strain Nigg (1 069 412 nt) and Chlamydia pneumoniae strain AR39 (1 229 853 nt) were determined using a random shotgun strategy. The MoPn genome exhibited a general conservation of gene order and content with the previously sequenced C.trachomatis serovar D. Differences between C.trachomatis strains were focused on an ~50 kb ‘plasticity zone’ near the termination origins. In this region MoPn contained three copies of a novel gene encoding a >3000 amino acid toxin homologous to a predicted toxin from Escherichia coli 0157:H7 but had apparently lost the tryptophan biosyntheis genes found in serovar D in this region. The C.pneumoniae AR39 chromosome was >99.9% identical to the previously sequenced C.pneumoniae CWL029 genome, however, comparative analysis identified an invertible DNA segment upstream of the uridine kinase gene which was in different orientations in the two genomes. AR39 also contained a novel 4524 nt circular single-stranded (ss)DNA bacteriophage, the first time a virus has been reported infecting C.pneumoniae. Although the chlamydial genomes were highly conserved, there were intriguing differences in key nucleotide salvage pathways: C.pneumoniae has a uridine kinase gene for dUTP production, MoPn has a uracil phosphororibosyl transferase, while C.trachomatis serovar D contains neither gene. Chromosomal comparison revealed that there had been multiple large inversion events since the species divergence of C.trachomatis and C.pneumoniae, apparently oriented around the axis of the origin of replication and the termination region. The striking synteny of the Chlamydia genomes and prevalence of tandemly duplicated genes are evidence of minimal chromosome rearrangement and foreign gene uptake, presumably owing to the ecological isolation of the obligate intracellular parasites. In the absence of genetic analysis, comparative genomics will continue to provide insight into the virulence mechanisms of these important human pathogens.
INTRODUCTION
Chlamydiae are obligate eubacterial parasites classed into four species, two of which, Chlamydia trachomatis and Chlamydia pneumoniae, are pathogenic for humans. All chlamydiae share a common biology (1). The organisms grow only within a specialized vacuole in the post-Golgi exocytic vesicular compartment of the eukaryotic cell. They undergo a distinct developmental cycle that alternates between an extracellular transmission cell, termed the elementary body (EB), and an intracellular replicating cell, termed the reticulate body (RB). As parasitic bacteria they have extremely streamlined genomes and are auxotrophic for most nucleotides and amino acids (2). The organisms are capable of persisting in cells of an immune host presumably due to evolved capabilities for immune evasion. Infection causes host cells to produce a variety of pro-inflammatory cytokines which likely contribute to disease pathogenesis (3).
Recently, the genomes of two chlamydiae species have been published. The C.trachomatis serovar D genome contains 1 042 519 nt and an estimated 894 protein coding genes (4). The C.pneumoniae genome contains 1 230 230 nt and an estimated 1052 protein coding genes (5). Chlamydia trachomatis also contains an extrachromosomal plasmid genome of 7493 nt whereas C.pneumoniae has no identified extrachromosomal elements. The compact genomes for these organisms make them particularly suitable for rapid genomic sequence analysis. The major new findings from these two genome studies from the viewpoint of pathogenesis included the identification of a new multigene family of sequence-variant putative outer membrane proteins and the complete components for a type III secretion system. Both genomes contain homologs for these two virulence attributes.
Despite extraordinary similarity in biology, chlamydiae display extreme diversity in tissue trophism and disease expression for which genome analysis has not yet provided sufficient explanation. For instance, C.trachomatis serovars A–C cause the ocular disease trachoma while serovars D–K cause a variety of sexually transmitted disease syndromes. All these serovars are classified as the trachoma biovar and produce infection limited to the mucosal surfaces of the host. On the other hand, C.trachomatis serovars L1–L3 are classified as the lympho-granuloma venereum biovar and produce systemic infection mainly of the lymphatic tissue. Chlamydia pneumoniae similarly displays a remarkable range in disease expression. Chlamydia pneumoniae infects the mucosal surfaces of the respiratory tract causing pharyngitis, bronchitis and pneumonitis. Recent epidemiological data also suggest that C.pneumoniae may disseminate from the respiratory tract to produce vascular infection and contribute to atherogenesis (6).
The genetic basis for the diversity of disease expression and tissue trophism remains a major unanswered question in Chlamydia biology. Knowledge in this area may contribute to elucidating the fundamental mechanisms of chlamydial disease pathogenesis and to the identification of new targets for vaccine and drug design. We therefore undertook to sequence two additional chlamydial genomes to, in part, explore these issues. We chose the mouse trophic strain or biovar (Nigg) of C.trachomatis (designated in this report MoPn) because of its apparent wide separation from the human biovars of C.trachomatis and a strain of C.pneumoniae (AR39) isolated from a human case of respiratory tract infection that is epidemiologically distinct from the initial sequenced strain of C.pneumoniae (CWL029).
MATERIALS AND METHODS
Library preparation and random sequencing of C.trachomatis
Chlamydia trachomatis mouse pneumonitis strain Nigg (MoPn) was the kind gift of Dr J. Schachter. The organism was propagated in HeLa 229 cells. EBs were harvested and purified by step gradient density centrifugation. Purified EBs were lysed with 10% SDS and proteinase K. The DNA was extracted twice with buffered phenol and once with 25:24:1 phenol:chloroform:isoamyl alcohol and precipitated with alcohol.
Cloning, sequencing and assembly were as described previously for genomes sequenced by TIGR (7–10). One small-insert plasmid library (1.5–2.5 kb) was generated by random mechanical shearing of genomic DNA. One large-insert λ library was generated by partial _Tsp_5091 digestion and ligation to the λ-DASHII/_Eco_RI vector (Stratagene). In the initial random sequencing phase, ~7-fold sequence coverage was achieved with 19 754 sequences from 11 869 plasmid clones (average read length 530 bases). The plasmid and λ sequences were jointly assembled using TIGR Assembler. Sequences from both ends of 368 λ clones served as a genome scaffold, verifying the orientation, order and integrity of the contigs. Sequence gaps were closed by editing the ends of sequence traces and/or primer walking on plasmid clones. Physical gaps were closed by direct sequencing of genomic DNA or combinatorial PCR followed by sequencing of the PCR product. The final genome sequence is based on 18 889 sequences.
Polymorphisms were noted in C.trachomatis MoPn at positions 58882 (T or G) and 58904 (T or G), with a small deletion between 469219 and 469238.
Library preparation and random sequencing of C.pneumoniae AR39
Chlamydia pneumoniae strain AR39 was purchased from the Washington Research Foundation courtesy of Dr C.C. Kuo. The organism was propagated in 6-well plates in HL cells. EBs and DNA were purified as described for C.trachomatis MoPn. The C.pneumoniae genome was completed using 26 754 sequence reads (average length 521 nt) from 16 224 clones, including PCR walks off the ends of inserts in 288 bacteriophage λ clones. The final chromosome and phage sequences comprised data from 19 903 sequence reads.
ORF prediction and gene family identification
An initial set of ORFs likely to encode proteins was identified by GLIMMER9 (11) and those shorter than 30 codons eliminated. ORFs that overlapped were visually inspected and, in some cases, removed. ORFs were searched against a non-redundant protein database as previously described. Frameshifts and point mutations were detected and corrected where appropriate as described previously. Remaining frameshifts and point mutations are considered authentic and corresponding regions were annotated as ‘authentic frameshift’ or ‘authentic point mutation’, respectively. Annotation was completed using the methodology described previously (10). Two sets of hidden Markov models (HMMs) were used to determine ORF membership in families and superfamilies. These included 527 HMMS from pfam v2.0 and 199 HMMS from the TIGR ortholog resource. TopPred46 was used to identify membrane-spanning domains (MSD) in proteins.
Comparative genomics
The Chlamydia genomes were rotated based on the results of GC skew analysis (12) so that the first base was near the hemB genes. All genes and predicted proteins from each Chlamydia genome, as well as from all other completed genomes, were compared using Fasta3. For determination of the presence and absence of particular genes in each Chlamydia genome, protein comparisons were used to better detect distantly related homologs. A gene was considered to be absent from a genome if there was no match to that gene with a P value <10–8. For comparisons of chromosome organization between two genomes, gene (i.e. DNA) comparisons were used. Each gene in species 1 was paired with its most similar gene (as measured by P value) in species 2. Frameshifts and small unique ORFs (<30 amino acids) were excluded from the analysis. For the identification of recent gene duplications all genes from C.pneumoniae and C.trachomatis were compared to each other. A gene was considered to be recently duplicated if its most similar gene (as measured by P value) was another gene within the same genome (relative to genes from the two other genomes).
Database submission
The nucleotide sequences of the whole genomes of C.trachomatis MoPn and C.pneumoniae AR39 were submitted to GenBank under accession nos AE002160 and AE002161, respectively.
RESULTS AND DISCUSSION
Chlamydia genome architecture
The two C.trachomatis and the two C.pneumoniae genomes sequenced to date are highly conserved in gene content and order (Fig. 1 and Table 1). Scatter plots based on the results of Fasta3 searches are presented in Figure 2A and B. The C.trachomatis MoPn and serovar D plots were almost linear (Fig. 2A), indicating that despite evolutionary separation that has allowed an average difference in orthologous genes of ~10%, there have been no major rearrangements in the chromosomes. The exception to the overall synteny is in an area of ~50 kb near to the predicted termination origin, which appears to be a ‘plasticity zone’ (13). The C.pneumoniae AR39 and CWL029 chromosomes were essentially identical, with only a few small deletions and ~300 single nucleotide polymorphisms (SNPs) distinguishing the two strains, although the AR39 sequence included a novel infecting bacteriophage (described later). Only when the C.trachomatis genome was compared to the C.pneumoniae genome (Fig. 2B) was there evidence of chromosomal rearrangements. It appears that there have been several large DNA inversions (inverted diagonals on the scatter plot) in the period since the two species had diverged from their common ancestor. The C.pneumoniae chromosome also has a plasticity zone near its termination origin where there has been a higher rate of DNA reorganization, although this region is more extended in the C.pneumoniae genome than in the C.trachomatis genome (~160 versus ~50 kb). From Figure 1 it is notable that many of the divergent genes (red or blue ticks) in the chlamydiae are clustered, suggesting that they are in units involved in a similar cellular function. The significance of these groups of divergent genes is that they might represent determinants of strain-specific functions, for instance host tropism or specific virulence activity.
Figure 1.

Comparison of the C.pneumoniae and C.trachomatis genomes. The DNA genomes of C.trachomatis MoPn, C.trachomatis serovar D and C.pneumoniae (CWL029 and AR39 are effectively identical at this level of resolution) are represented as circles. Genomes are scaled in 100 000 nt increments. The three outer rings of each genome represent assignment of genes sharing identity in a Fasta3 comparison with a score of <0.00000001. Each tick indicates the location of the 5′-end of a gene. In the first, outer ring the green ticks represent genes encoding proteins conserved in all Chlamydia genomes, the purple ticks are genes conserved in C.pneumoniae and one C.trachomatis genome and are hence assumed to be deleted from the other C.trachomatis genome. The second circle (red) shows species-specific genes. The third circle (blue) illustrates genes encoding proteins not similar at a score of <10–8 to any other chlamydial protein. The fourth circle shows the location of the tRNAs and the fifth the position of the rRNA operons. The inner, sixth circle shows the results of GC skew analysis using a 1000 nt window size (12). Windows with a positive skew value are shown as cyan ticks, with a negative skew as yellow. The origin of replication and the termination region are defined as the points of inflection from positive to negative and negative to positive skew, respectively. The approximate positions of the plasticity zones near the origins of replication are indicated by the letters PZ. The genomes have been branched in a phylogenetic tree based on the average identity of homologous genes. Branch lengths (bracketed value) are the average percent difference in the homologous genes.
Table 1. Chlamydia genomes comparison.
| Features | C.trachomatis (MoPn) | C.trachomatis (serovar D)a | C.pneumoniae (AR39) | C.pneumoniae (CWL029)a |
|---|---|---|---|---|
| GC (%) | 40.3 | 41.3 | 40.6 | 40.6 |
| Size (nt) | 1 069 412 | 1 042 519 | 1 229 853 | 1 230 230 |
| ORFs | 924 | 894 | 1052 | 1052 |
| tRNAs | 37 | 37 | 38 | 38 |
| Extrachromosomal elements | Circular dsDNA plasmid (7501 nt) | Circular dsDNA plasmid (7493 nt) | Circular ssDNA bacteriophage (4524 nt) | None |
| Replication originb | Near hemB gene | Near hemB gene | Near hemB gene | Near hemB gene |
Figure 2.


Dot plots of gene similarities between (A) C.trachomatis MoPn and C.trachomatis serovar D and (B) C.trachomatis MoPn and C.pneumoniae AR39. The criterion for match was a Fasta3 score of <10–8. The genomes have been rotated to better show inversion around the origins. Axes are marked with 200 kb gradations. R and T are the locations of the origin of replication and termination region, respectively.
It is interesting to note that the multiple large inversions in the Chlamydia chromosomes occur around the axes of the origins of replication and termination (Fig. 2B). Recombination across the origins has been seen in other eubacteria (14–16) but the chlamydiae provide one of the clearest illustrations of how this phenomenon affects the architecture of the genome. Another significant feature of the chlamydiae genomes is tandemly repeated genes. Figure 3A and B charts the positions of the duplicated genes of C.trachomatis and C.pneumoniae and those that have the nearest sequence match to another gene in the same chromosome. Mostly, these genes are situated next to each other, indicating a recent recombination event.
Figure 3.

Gene duplications in (A) C.pneumoniae AR39 and (B) C.trachomatis MoPn. Duplicated genes in each chromosome were identified as having a score in a Fasta3 comparison of <10–8. The location of each gene and its duplicate on the chromosome were plotted.
The origin-linked inversions and tandem duplication events are more clearly observable in Chlamydia than in other bacteria because of the apparent paucity of intra-genomic DNA rearrangement and the rarity of sequences from extraneous sources. There are no IS elements or other dispersed repeated sequences in the genomes to promote intramolecular rearrangements and disrupt the synteny of the genetic organization. Judging from a lack of variation in the ratio of GC to AT nucleotides across the genomes (data not shown) there are no regions from any of the four sequenced Chlamydia genomes that have recently been transferred from an evolutionarily diverged organism.
The C.trachomatis plasticity zones
Considering that Chlamydia are isolated from genetic exchange with other bacteria owing to their obligate intracellular ecological niche, it is not surprising that there is a great deal of overall synteny between the C.trachomatis genomes. For the same reasons, it is significant that there is one segment of each genome, the plasticity zone (Fig. 1), that has undergone genetic reorganization to a much higher degree than the rest of the chromosome. Outside the single plasticity zone, syntenic differences between the C.trachomatis strains is limited to three novel genes together with rare gene duplications in the MoPn genome. Two of the novel MoPn genes encode DNA helicases, while the other specifies uracil phosphoribosyl transferase (upp).
The C.trachomatis plasticity zone extends between the conserved orthologs dbsB (disulphide bond oxidoreductase) and ycfV, encoding an ABC-transporter of unknown substrate specificity. The distance between the 3′-ends of these genes is 22 922 nt in serovar D and 50 624 nt in MoPn. This size difference in the plasticity zones (27 702 nt) is of the same order as the total difference between the two genomes (26 893 nt; Table 1). The genetic composition of the C.trachomatis plasticity zones are outlined in Figure 4. There are several differences in the plasticity zone between the human and mouse trophic genomes that suggest an influence on Chlamydia pathogenesis. Chlamydia trachomatis MoPn, in common with C.pneumoniae, contains guaAB and adenosine deaminase (add) apparently arranged as a single operon. In the same location relative to the 5′-end of the opp gene, C.trachomatis serovar D has the trpRBA trypophan biosynthesis cluster. This arrangement suggests strongly that in the human C.trachomatis strain trpRBA has replaced the guaAB and adenosine deaminase genes.
Figure 4.

Gene map of C.trachomatis MoPn and serovar D plasticity zones. Schematic diagram showing the gene content of the plasticity zones of the two C.trachomatis strains between the conserved dbsB and ycrfV loci. The line across the two homologs of the large toxin gene of MoPn TC0439 indicates a frameshift mutation. The ‘toxin’ genes of serovar D are homologous to regions of the larger TC0439 protein.
Another striking difference between the two C.trachomatis plasticity zones is the presence of a 9675 nt gene, ORF TC0439, encoding a putative toxin protein of predicted molecular weight 364 kDa. The protein bears an overall similarity of 53% to a 3192 amino acid putative toxin encoded by the Escherichia coli 0157:H7 virulence plasmid (17,18). Both the MoPn and 0157:H7 toxins have similarity at their N-terminus to the N-terminus of large clostridial toxins (LCTs; 19). This portion of LCT molecules has been shown to interfere with eukaryotic cell chemistry by glycosylating GTP-binding proteins of the Ras superfamily. A conserved motif in LCTs and yeast glycosyltransferases (LxxxGGxYxDxD) (17) was found at the N-terminus of the MoPn and E.coli toxins, suggesting similar activity by the latter proteins. In addition to the catalytic region, LCTs contain domains for recognizing cell surface receptors and translocation through the outer membrane. The C-terminal ligand-recognizing portions typically contain multiple repeated motifs. Hydrophobicity plots of the Chlamydia and E.coli toxin (data not shown) indicate potential MSD in the center of the molecules but the C-termini are not repetitive in the manner of LCTs. Adjacent to the toxin are two other very similar large toxin-encoding genes but these ORFs contain multiple frameshift mutations (Fig. 4). The serovar D strain also contains what appears to have been an entire toxin gene that has accumulated numerous frameshift mutations, arguing that there has been selection against expression of the entire toxin in the human trophic strain but not in the mouse trophic strain.
The plasticity zone is also the location of an unusual family of genes encoding phospholipase D–endonuclease (PLD) superfamily proteins previously reported by Kalman et al. (5). These proteins have little overall similarity to other PLD enzymes and lack a type II secretion signal sequence but contain conserved duplicated HKD motifs typical of this family (20). Chlamydia trachomatis serovar D contains four PLD paralogs between ycfV and the toxin genes arranged in an operon. MoPn contains five paralogs in this location and two on the other side of the toxin genes in the opposite orientation (Fig. 4). PLD genes on the same genome are generally more closely related to each other than to paralogs from the other strain, indicating that frequent intragenomic duplication and deletion has occurred in this gene family.
Overall, the C.trachomatis plasticity zones are the location of several genes suspected to be involved in pathogenesis, such as the trp genes, the large toxin and the unusual PLD-like enzymes, suggesting that these regions might be sites for horizontal gene exchange. The GC content and codon adaptation values (21) of genes in this area are in line with other chlamydial loci, providing no evidence of recent horizontal movement of genes from outside the genus. More likely, genetic rearrangements in this area are endogenous, with deletions or duplications of existing loci (the latter case explaining the expansion of the numerous PLD paralogs). Chlamydia pneumoniae has either deleted the toxin and PLD genes from this region at an earlier point in its evolution or C.trachomatis acquired these genes after the speciation event. The mechanisms driving rearrangements at the plasticity locus are not clear. There are no features reminiscent of ‘pathogenicity islands’ as seen in other Gram-negative bacteria, such as long flanking repeats and associated transposase or recombinase genes. It is likely important that the plasticity zone is close to the predicted termination origin. Perhaps genomic rearrangement is facilitated at this site by stalled replication forks caused by lack of processivity of the chlamydial DNA polymerase enzyme at the termination origin.
Why is MoPn a mouse pathogen and serovar D a human pathogen?
The extraordinary similarity in gene content and order in the C.trachomatis MoPn and serovar D genomes is surprising given prior reports which suggested that the two biovars exhibited only low to moderate homology by DNA:DNA hybridization and amplified fragment length polymorphism (AFLP) studies (22). On the one hand, the extraordinary conservation offers encouragement for investigators using the MoPn biovar to model disease caused by human biovars of C.trachomatis. On the other hand, there are no macroscopic features deduced from comparative genomic analysis to explain the observed differences in host range and pathogenicity between the two C.trachomatis biovars. Instead, host species tropism might be attributable to a few genes on the chromosomes that influence the ecology of infection within a species. The finding that serovar D contains tryptophan biosynthesis genes not present in MoPn has potential significance. One of the primary host defences against chlamydial infection is the pro-inflammatory cytokine interferon-γ (IFN-γ). IFN-γ modulates the depletion of intracellular tryptophan through induction of indoleamine 2,3-dioxygenase (23). The trp genes of serovar D might allow for increased survival inside a tryptophan-depleted human cell, thus producing persistent infection more readily than MoPn.
Serovar D may require persistent infection in order to achieve successful transmission from human to human through density-independent sexual contact, whereas acute high level respiratory infection with MoPn may facilitate aerosol transmission under the density-dependent conditions of a rodent colony (24). In this regard, the large toxin encoded by the MoPn genome may be an important virulence determinant that promotes acute high level infection and might be the reason why MoPn replicates more readily in vivo and in vitro compared to human C.trachomatis isolates. It is notable that serovar D appears to have accumulated mutations in its copy of the toxin gene that prevent expression of the entire molecule, suggesting that the toxin could be an example of a virulence determinant important in infection of one host (mice) but unnecessary or disadvantageous for pathogenesis in a second host (human). In addition, several of the few genes specific to MoPn but not found in serovar D (guaAB, adenine deaminase, upp) are involved in scavenging of nucleotides. This differential capacity for nucleotide metabolism could also contribute to defining the host range of tissues each organism is capable of infecting (discussed later).
Comparison of the C.pneumoniae AR39 and CWL029 genomes
The C.pneumoniae strain sequenced by TIGR (AR39) and the strain (CWL029) reported previously (5) are the most similar published genomes to date. Comparison of the 1.23 Mb chromosomes by the MUMmer suffix tree analysis method (25) revealed only 296 SNPs and 21 single base frameshift mutations. There are two small insertions in the AR39 strain (25 and 85 bp) and five insertions in the CWL029 isolate (5, 5, 7, 89 and 305 bp). Previous studies based on AFLP (22) highlighted close similarities of C.pneumoniae isolates but suggested that AR39 might be a phylogenetically separated isolate with a sequence difference of 6% from the main set of strains. However, the genome data on these two C.pneumoniae isolates show that they are close enough to have diverged within recent human history.
The C.pneumoniae AR39 genome data contain 304 polymorphisms (areas where there is a mixture of two variant sequences). Most of these variants are either SNPs or variations in the number of repeated nucleotides. The largest polymorphism by far is the deletion of one unit of a tandem 1649 bp repeat containing a tyrosine transport protein gene and partial ORFs of a tyrosine permease and glucosamine-fructose 6-phosphate aminotransferase. In comparison, the C.trachomatis MoPn sequence has only three polymorphic areas (see Materials and Methods). The reason for the differences in numbers of polymorphisms between the two chlamydiae is unknown.
Having whole genome sequence data for two strains as closely related as the C.pneumoniae strains provides a unique opportunity to observe the process of mutagenic change. Many of the mutations (including polymorphisms in the AR39 sequence) occur in intergenic regions of the chromosome, suggesting a predominantly neutral phenotypic effect. Comparing the AR39 and the CWL029 genomes, only 161 of 1165 proteins are not identical. By far the majority of mutation events are purine–purine or pyrimidine–pyrimidine transitions (90%), in line with other studies of C.trachomatis omp1 gene polymorphisms (26). There was no indication of clustering of the SNPs at any particular genomic location.
Given the high degree of similarity between the C.pneumoniae chromosomes, the small differences that are observed become important, as they offer potential targets for strain differentiation assays and for ideas about gene function. One notable change is the apparent loss of a 393 bp iterated segment in AR39 from the large polymorphic outer membrane protein pmp6, showing how cell surface variability could be generated in otherwise very similar bacteria.
One of the most intriguing differences between the two C.pneumoniae chromosomes is in the area upstream of the uridine kinase gene (Fig. 5) where there is a 23 nt sequence in AR39 that is in an inverted orientation relative to the CWL029 genome. Phase variable expression of key virulence determinants involving inversion of promoter DNA mediated by site-specific recombinases is a common feature of Gram-negative bacteria, for example the hin and piv systems of Salmonella and Moraxella bovis, respectively (27,28). The likelihood of a recombinase-mediated DNA inversion in the C.pneumoniae genome is indicated by the fact that the 23 nt flipped segment is flanked by a 15 nt inverted repeat sequence (Fig. 5). The 23 nt inverted segment contains a reasonable consensus –10 RNA polymerase binding site (TATAGT; Fig. 5), therefore, it is possible that inversion of this piece of DNA may result in switching on or off of expression/transcription of the uridine kinase gene. In CWL029 the putative –10 site is orientated for transcription of the gene; in AR39 it is in the opposite orientation. As uridine kinase is apparently a key enzyme in nucleoside metabolism in C.pneumoniae (see below), a phase variation system that appears to result in potential lack of expression of the protein is a fascinating observation. Chlamydia pneumoniae contains two genes specifying homologs to integrase/recombinase enzymes although neither are situated near the uridine kinase gene and both are conserved in the other Chlamydia genomes. Neither the inverted repeats nor the inverted 23 nt are found anywhere else in any of the Chlamydia genomes.
Figure 5.
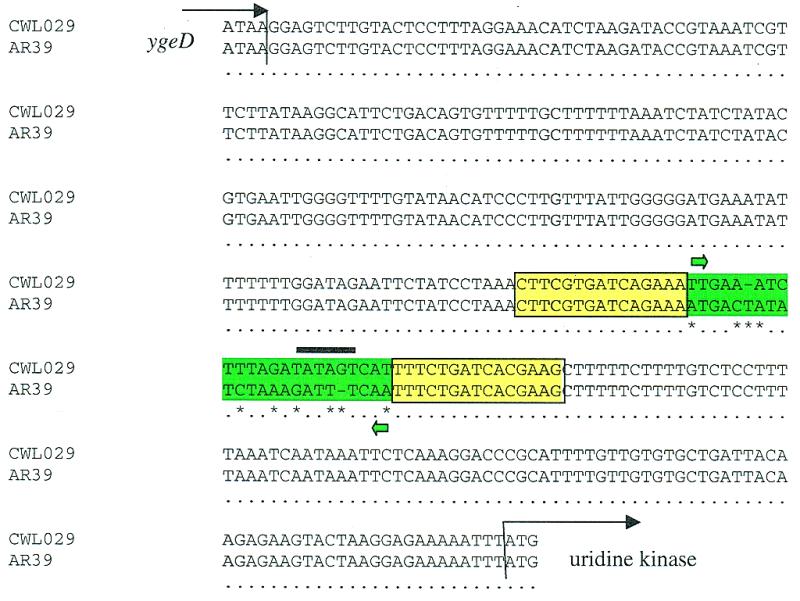
Regions upstream of the uridine kinase gene in C.pneumoniae CWL029 and AR39. A comparison of the sequence between ygeD and uridine kinase of the CWL029 and AR39 strains is shown. Differences between the sequences (asterisks) are limited to the 23 bp inverted sequence (green highlight). This inverted segment contains a putative –10 sequence (black bar) TATAGT in the same orientation as the uridine kinase gene in CWL029. Flanking the inverted sequence are 15 kb inverted repeats (boxed yellow).
The C.pneumoniae AR39 bacteriophage
The C.pneumoniae AR39 genome includes a 4524 nt circular molecule homologous to members of the single-stranded (ss)DNA microviridae class of bacteriophages previously reported in Chlamydia, Spiroplasma and E.coli (29–31). The phage genome reported here bears 49% nucleotide sequence identity to the Chp1 phage from an avian strain of Chlamydia psittaci. We therefore believe that we have sequenced the dsDNA circular intracellular replicative form of an ssDNA C.pneumoniae bacteriophage present in the sample lysate.
The discovery of a small ssDNA bacteriophage genome during sequencing of the C.pneumoniae AR39 genome was unexpected. The phage was not found in the otherwise almost identical C.pneumoniae CWL029 strain sequenced by Kalman et al. (5). A map of the phage genome is shown in Figure 6. Three genes encode products homologous to microviridae structural proteins VP1–VP3. The C.pneumoniae phage also contains sequence homologous to the φX174 gene A nicking/closing protein ORF4. Interestingly, the C.pneumoniae chromosome contains a truncated version of this gene with 73% identity to the truncated phage gene 4 product, suggesting that at some time in the past the C.pneumoniae phage was integrated into the chromosome. This gene was seen in both the AR39 and CWL029 genomes. This is the first _C.pneumoniae_-infecting bacteriophage described and while it has fundamental organizational similarities with other microviridae, the level of sequence divergence from its nearest neighbour, Chp1, indicates that it is a novel branch of this virus family. The presence of extra phage bands is possibly the reason for the figure of 6% divergence of the AR39 isolate from the main group of C.pneumoniae strains reported by Meijer et al. (22) using AFLP.
Figure 6.

Comparison of C.pneumoniae AR39 phage with other sequences. The figure details putative ORFs of the 4524 nt ssDNA phage sequence (inner circle) with similarities to previously sequenced proteins. Chp1, C.psittaci Chp1 bacteriophage; Spv, Spiroplasma virus 4.
The presence of a bacteriophage in an obligate intracellular pathogen raises interesting biological questions. For instance, does the phage infect the RB or EB of Chlamydia? How does the phage transfer between and co-infect new host organisms? Pioneering work on the C.psittaci virus by Richmond et al. (32) suggested that the RB is the target for phage replication, with the phage identified as multiple electron-dense particles in the cytoplasmic compartment. While nothing like the crystalline structures noted by Richmond have been described in C.pneumoniae, it is interesting to speculate that the intracellular and periplasmic particles termed ‘minibodies’ (33,34) observed in C.pneumoniae strains AR39 and TW183 could actually be associated with the virus.
It is possible that the C.pneumoniae phage may play a role in pathogenesis. Lysis of intracellular C.pneumoniae could cause the release of cell-activating proteins such as Chlamydia heat shock protein 60 (35) or of multiple immunogenic epitopes which could result in an enhanced inflammatory response to pathogenic epitopes such as the MAxxxST motif (36). Alternatively, lytic phage may reduce the antigenic mass and promote the persistence of C.pneumoniae by preventing the accumulation of a strong anti-chlamydial immune response, thereby preventing immune-mediated clearance.
As well as its potential importance in C.pneumoniae pathogenesis, the phage has exciting promise as a genetic vector for a bacterium where genetic analysis has so far proved difficult. The finding of a phage gene apparently inserted into the chromosome is encouraging as it suggests that the virus might co-integrate at some frequency. It is also significant that the C.pneumoniae chromosomally located partial ORF4 is found within the plasticity zone; another indication that plasticity zones might have increased susceptibility to uptake of foreign DNA than the rest of the genome.
Comparative genomics of C.trachomatis and C.pneumoniae
Kalman et al. (5) noted that ~80% of the C.pneumoniae and C.trachomatis serovar D predicted coding sequences were orthologs. Unsurprisingly, given the synteny of the C.trachomatis genomes, the number of shared orthologs between MoPn and C.pneumoniae is of the same order (854/924). Despite the number of orthologous proteins and their relatively high similarity, as well as the overall homology in genome organization (Fig. 1), there is only a relatively low level similarity in the nucleotide sequence of orthologous genes between C.pneumoniae and C.trachomatis (81.5%). This argues for conservation for the basic functions necessary for intracellular growth in the chlamydiae despite a long separation of the C.pneumoniae and C.trachomatis species.
An overall comparison between the C.pneumoniae and C.trachomatis genomes is shown in Figure 1. The C.pneumoniae genome is ~0.15 Mb larger than that of C.trachomatis and contains ~200 genes not found in C.trachomatis. As detailed by Kalman et al. (5), most of the ‘extra’ genes found in C.pneumoniae are either expansions of paralogous families (for instance, there are 21 pmp outer membrane protein genes in C.pneumoniae but only nine in C.trachomatis) or encode hypothetical proteins without current database matches. Many of the additional C.pneumoniae genes are located in the plasticity zone portion of the genome (Fig. 1). Proteins with homologs of known function encoded by C.pneumoniae but by neither C.trachomatis MoPn nor serovar D include tryptophan hydroxylase, genes involved in biotin synthesis and uridine monophosphate synthase and uridine kinase. It is interesting that both the MoPn and C.pneumoniae genomes contained guaAB and adenosine deaminase homologs whereas these genes are not present in the serovar D genome.
The few C.trachomatis genes without homologs in C.pneumoniae are restricted to the plasticity zone, with the exception of the three apparently inserted genes of MoPn: two DNA helicases and uracil phosphoribosyltransferase (upp). Plasticity zone genes unique to C.trachomatis include those encoding the large toxins, the family of PLD-like proteins lacking signal sequence, the tryptophan biosynthesis cluster of serovar D and several proteins without homologs in other species.
An important result of comparative genome sequencing is the identification of proteins conserved within bacterial species. Table 2 lists the Chlamydia orthologous proteins that have >90% sequence identity over >90% of their length. Presumably sequence conservation at this level when the general level of similarity of orthologs between C.pneumoniae and C.trachomatis is ~65% reflects strong conservative selection on the protein. Most of the proteins in Table 2 are conserved across all bacteria: ribosomal proteins, σ factors and transcriptional elongation factors, for example. Some of the conserved proteins elaborate highly specific structures important in the chlamydial lifestyle, such as SctN and SctV, type III secretion transporters. One of the conserved proteins, encoded by TC0313, is a hypothetical protein without homologs in another organism. Possibly this protein fulfills a unique role in chlamydial virulence or intracellular survival and may be an important subject for studies on pathogenesis and molecular typing.
Table 2. Highly conserved chlamydial proteinsa.
| Ribosomal proteins | S19, L19, L14, L11, S12, S10, S21, L33, S1, S15, L35, L36 |
|---|---|
| Other transcription/translation | σ70, Rho, FusA, TufA, TufB, RpoA, RpoB, IhfB |
| Type III secretion structural proteins | SctN, SctV |
| Other known function | MreB, ClpP, GroEL |
| Unknown functions | TC0687 and TC068 (conserved hypothetical proteins), TC0313 (_Chlamydia_-specific hypothetical protein) |
It was recently reported (36) that the chlamydial 60 kDa cysteine-rich outer membrane protein (omp2) contains a conserved MAxxxST motif that can induce autoimmune inflammatory heart disease in mice through molecular mimicry with heart muscle α-myosin proteins. Comparative genomic analysis revealed another chlamydial protein with the conserved MAxxxST structure: a homolog of the E.coli cell division protein FtsH. In common with FtsH found in other bacteria, the chlamydial protein contains ATPase and zinc metalloprotease motifs. However, the FtsH homologs of the Chlamydiae contain a 400 amino acid N-terminal domain with multiple transmembrane helices not seen in any other organism. It is possible that FtsH plays a unique role in the outer envelope of Chlamydia and might be recognized by the immune system.
All four Chlamydia genomes contain highly conserved determinants for a complete type III secretion system spread over three chromosomal regions. The high level of similarity between the proteins in dispersed locations is a further argument for the key role of these systems in survival of the bacterium in the intracellular vacuole. The chlamydial type III systems have homologs to other type III structural, targeting and regulation proteins and chaperones in Yersinia, Shigella, E.coli and Gram-negative plant pathogens (37) but there are no obvious matches to known type III secreted effector proteins. This situation is common with other type III systems and reflects the versatility and adaptability of these important pathogenesis mechanisms. Identification of the effectors is therefore a critical focus for research. Potential effectors revealed by genomic analysis include MoPn TC0044, which has a conserved serine/threonone kinase motif and is located within type III gene cluster 2. This molecule, when injected into the infected cell via type III secretion, might interfere with intracellular signaling in a manner beneficial to the parasitic Chlamydia. Other possible effectors are encoded by TC0042, TC0867 and TC0868, which are also situated close to the type III gene clusters and have low level similarity to other proteins such as E.coli EspB, Salmonella typhimurium SspB and SspC and the plant pathogen Pseudomona aeruginosa Harpin HrpO (37).
Different strategies for nucleotide salvaging among different strains of Chlamydia
One of the interesting insights to emerge from comparative chlamydial genome sequencing is the different pathways used by the four chlamydial strains for acquiring nucleotides. During the course of evolution toward an obligate intracellular lifestyle, Chlamydia spp. appear to have abandoned much of their genome necessary for self-sustaining existence (2). A vital set of genes missing from the chlamydial genomes are those necessary for de novo synthesis and/or salvage of three of the four ribonucleotides, making them dependent on import of nucleotides from the host. Genome sequence analysis indicates that all four Chlamydia contain a CTP synthetase which converts UTP to CTP. All four Chlamydia genomes also encode two proteins known to be dedicated to the transport of nucleotide triphosphates, Tlc1 and Tlc2, both homologs of Tlc, an ATP/ADP translocase from the obligate intracellular parasite Rickettsia prowazakii (38). The Tlc1 protein of C.trachomatis serovar L2 is an ATP/ADP tranlocase, whereas the Tlc2 protein, although sharing a high degree of sequence similarity, is a more general NTP transporter, apparently utilizing an H+ pump to energize the process (39). These differences are likely true for the four completely sequenced chlamydial genomes.
As discussed earlier, four of the genes present in the C.trachomatis MoPn but not serovar D genomes are involved in nucleoside/nucleobase anabolism. The guaAB_–_add operon should allow for conversion of ATP to GTP, while upp facilitates biosynthesis of UTP from uracil. MoPn upp is an interesting gene. It is apparently inserted into the genome between loci that are adjacent in both the serovar D and C.pneumoniae genomes. The protein has most identity (56%) with upp gene products from Gram-positive bacteria but also contains an ~100 amino acid N-terminus without database homology, suggesting that the molecule has a second function. Although C.pneumoniae contains guaAB_–_add, these proteins are probably not expressed due to frameshift mutations. Like MoPn, C.pneumoniae can also synthesize UTP, however, uridine kinase is the salvage enzyme employed in C.pneumoniae, rather than upp, which is employed in MoPn. An interesting finding in the C.pneumoniae genome arises from the observation that uridine kinase may undergo phase variable expression (Fig. 6), hinting that either there is some other novel determinant in the genome that provides for UTP synthesis or that, under certain conditions, the cell can import UTP directly. The human C.trachomatis serovar D does not appear to contain additional nucleotide biosynthesis genes.
Based on these observations regarding nucleotide metabolism, we speculate that the MoPn biovar is the least dependent on its host cell in its requirement for ATP to initiate purine biosynthesis and uracil for pyrimidine biosynthesis. Chlamydia pneumoniae appears dependent only on scavenging uridine for pyrimidine anabolism. Serovar D appears dependent on the host cell for three of the four ribonucleotides. The key difference in the different chlamydial strains could lie in the independent evolution of the substrate specificity of the Tlc2 transporter, possibly a simple ATP transporter in MoPn, an ATP/GTP transporter in C.pneumoniae and a more general NTP importer in serovar D.
With regard to deoxyribonucleotide biosynthesis, all chlamydiae contain a ribonucleotide diphosphate reductase for the generation of dNDPs. Ribonucleotide reductase directly provides dATP, dGTP and dCTP, but not dTTP. Interestingly, none of the chlamydial genomes contain homologs of either thymidylate synthase or thymidine kinase, the only two enzymes known to be capable of dTMP biosynthesis. Previous studies have shown that Chlamydia cannot obtain thymidine nucleotides from the host (40), therefore, it still remains unclear as to how Chlamydia obtain the dTTP required for DNA synthesis.
CONCLUSION
Whole genome analysis has provided unexpected insights into Chlamydia biology and offers a rich set of observations that suggest new lines for experimental analysis. These insights would likely not have been found except through genome analysis. Because of the absence of a facile gene transfer system it is likely that genome sequencing will continue to be an important technique in evaluating the biology of this unusual branch of parasitic bacteria. Genome analysis of representative strains of C.psittaci as well as members from other distant branches in the chlamydiae family tree (41) will likely contribute to advancing our understanding of the pathogenic mechanisms used by these organisms as well as help our understanding of their evolutionary origins.
Acknowledgments
ACKNOWLEDGEMENT
This work was funded by NIH grant AI43359 to C.M.F.
REFERENCES
- 1.Moulder J.W. (1991) Microbiol. Rev., 55, 143–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McClarty G. (1994) Trends Microbiol., 2, 157–164. [DOI] [PubMed] [Google Scholar]
- 3.Rasmussen S.J., Eckmann,L., Quayle,A.J., Shen,L., Zhang,Y.X., Anderson,D.J., Fierer,J., Stephens,R.S. and Kagnoff,M.F. (1997) J. Clin. Invest., 99, 77–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stephens R.S., Kalman,S., Lammel,C., Fan,J., Marathe,R., Aravind,L., Mitchell,W., Olinger,L., Tatusov,R.L., Zhao,Q., Koonin,E.V. and Davis,R.W. (1998) Science, 282, 754–759. [DOI] [PubMed] [Google Scholar]
- 5.Kalman S., Mitchell,W., Marathe,R., Lammel,C., Fan,J., Hyman,R.W., Olinger,L., Grimwood,J., Davis,R.W. and Stephens,R.S. (1999) Nature Genet., 21, 385–389. [DOI] [PubMed] [Google Scholar]
- 6.Kuo C.C., Grayston,J.T., Campbell,L.A., Goo,Y.A., Wissler,R.W. and Benditt,E.P. (1995) Proc. Natl Acad. Sci. USA, 92, 6911–6914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fleischmann R.D., Adams,M.D., White,O., Clayton,R.A., Kirkness,E.F., Kerlavage,A.R., Bult,C.J., Tomb,J.F., Dougherty,B.A., Merrick,J.M. et al. (1995) Science, 269, 496–512. [DOI] [PubMed] [Google Scholar]
- 8.Fraser C.M., Gocayne,J.D., White,O., Adams,M.D., Clayton,R.A., Fleischmann,R.D., Bult,C.J., Kerlavage,A.R., Sutton,G., Kelley,J.M. et al. (1995) Science, 270, 397–403. [DOI] [PubMed] [Google Scholar]
- 9.Fraser C.M., Casjens,S., Huang,W.M., Sutton,G.G., Clayton,R., Lathigra,R., White,O., Ketchum,K.A., Dodson,R., Hickey,E.K. et al. (1997) Nature, 390, 580–586. [DOI] [PubMed] [Google Scholar]
- 10.Nelson K.E., Clayton,R.A., Gill,S.R., Gwinn,M.L., Dodson,R.J., Haft,D.H., Hickey,E.K., Peterson,J.D., Nelson,W.C., Ketchum,K.A. et al. (1999) Nature, 399, 323–329. [DOI] [PubMed] [Google Scholar]
- 11.Salzberg S.L., Delcher,A.L., Kasif,S. and White,O. (1998) Nucleic Acids Res., 26, 544–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lobry J.R. (1996) Mol. Biol. Evol., 13, 660–665. [DOI] [PubMed] [Google Scholar]
- 13.Alm R.A., Ling,L.S., Moir,D.T., King,B.L., Brown,E.D., Doig,P.C., Smith,D.R., Noonan,B., Guild,B.C., deJonge,B.L., Carmel,G., Tummino,P.J., Caruso,A., Uria-Nickelsen,M., Mills,D.M., Ives,C., Gibson,R., Merberg,D., Mills,S.D., Jiang,Q., Taylor,D.E., Vovis,G.F. and Trust,T.J. (1999) Nature, 397, 176–180. [DOI] [PubMed] [Google Scholar]
- 14.Schmid M.B. and Roth,J.R. (1983) Genetics, 105, 539–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Segall A., Mahan,M.J. and Roth,J.R. (1988) Science, 241, 1314–1318. [DOI] [PubMed] [Google Scholar]
- 16.Liu S.L. and Sanderson,K.E. (1995) Proc. Natl Acad. Sci. USA, 92, 1018–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burland V., Shao,Y., Perna,N.T., Plunkett,G., Sofia,H.J. and Blattner,F.R. (1998) Nucleic Acids Res., 26, 4196–4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Makino K., Ishii,K., Yasunaga,T., Hattori,M., Yokoyama,K., Yutsudo,C.H., Kubota,Y., Yamaichi,Y., Iida,T., Yamamoto,K., Honda,T., Han,C.G., Ohtsubo,E., Kasamatsu,M., Hayashi,T., Kuhara,S. and Shinagawa,H. (1998) DNA Res., 5, 1–9. [DOI] [PubMed] [Google Scholar]
- 19.von Eichel-Striber C., Boquet,P., Sauerborn,M. and Thelestam,M. (1996) Trends Microbiol., 4, 375–382. [DOI] [PubMed] [Google Scholar]
- 20.Ponting C.P. and Kerr,I.D. (1996) Protein Sci., 5, 914–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sharp P.M. and Li,W.H. (1987) Nucleic Acids Res., 15, 1281–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meijer A., Morre,S.A., van den Brule,A.J., Savelkoul,P.H. and Ossewaarde,J.M. (1999) J. Bacteriol., 181, 4469–4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dai W. and Gupta,S.L. (1990) J. Biol. Chem., 265, 19871–19877. [PubMed] [Google Scholar]
- 24.Anderson R.M. and May,R.M. (1979) Parasitology, 79, 63–94. [DOI] [PubMed] [Google Scholar]
- 25.Delcher A.L., Kasif,S., Fleischmann,R.D., Peterson,J., White,O. and Salzberg,S.L. (1999) Nucleic Acids Res., 27, 2369–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brunham R., Yang,C., Maclean,I., Kimani,J., Maitha,G. and Plummer,F. (1994) J. Clin. Invest., 94, 458–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scott T.N. and Simon,M.I. (1982) Mol. Gen. Genet., 188, 313–321. [DOI] [PubMed] [Google Scholar]
- 28.Tobiason D.M., Lenich,A.G. and Glasgow,A.C. (1999) J. Biol. Chem., 274, 9698–9706. [DOI] [PubMed] [Google Scholar]
- 29.Renaudin J., Pascarel,M.C. and Bove,J.M. (1987) J. Bacteriol., 169, 4950–4961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Storey C.C., Lusher,M. and Richmond,S.J. (1989) J. Gen. Virol., 70, 3381–3390. [DOI] [PubMed] [Google Scholar]
- 31.Chipman P.R., Agbandje-McKenna,M., Renaudin,J., Baker,T.S. and McKenna,R. (1998) Structure, 6, 135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Richmond S.J., Stirling,P. and Ashley,C.R. (1982) FEMS Microbiol. Lett., 14, 31–36. [Google Scholar]
- 33.Chi E.Y., Kuo,C.C. and Grayston,J.T. (1987) J. Bacteriol., 169, 3757–3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuo C.C., Chi,E.Y. and Grayston,J.T. (1988) Infect. Immun., 56, 1668–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kol A., Bourcier,T., Lichtman,A.H. and Libby,P. (1999) J. Clin. Invest., 103, 571–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bachmaier K., Neu,N., de la Maza,L.M., Pal,S., Hessel,A. and Penninger,J.M. (1999) Science, 283, 1335–1339. [DOI] [PubMed] [Google Scholar]
- 37.Hueck C.J. (1998) Microbiol. Mol. Biol. Rev., 62, 379–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Williamson L.R., Plano,G.V., Winkler,H.H., Krause,D.C. and Wood,D.O. (1989) Gene, 80, 269–278. [DOI] [PubMed] [Google Scholar]
- 39.Tjaden J., Winkler,H.H., Schwoppe,C., Van Der Laan,M., Mohlmann,T. and Neuhaus,H.E. (1999) J. Bacteriol., 181, 1196–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fan H.Z., McClarty,G. and Brunham,R.C. (1991) J. Bacteriol., 173, 6670–6677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Everett K.D., Bush,R.M. and Andersen,A.A. (1999) Int. J. Syst. Bacteriol., 49, 415–440. [DOI] [PubMed] [Google Scholar]