Poliovirus Proteins Induce Membrane Association of GTPase ADP-Ribosylation Factor (original) (raw)
Abstract
Poliovirus infection results in the disintegration of intracellular membrane structures and formation of specific vesicles that serve as sites for replication of viral RNA. The mechanism of membrane rearrangement has not been clearly defined. Replication of poliovirus is sensitive to brefeldin A (BFA), a fungal metabolite known to prevent normal function of the ADP-ribosylation factor (ARF) family of small GTPases. During normal membrane trafficking in uninfected cells, ARFs are involved in vesicle formation from different intracellular sites through interaction with numerous regulatory and coat proteins as well as in regulation of phospholipase D activity and cytoskeleton modifications. We demonstrate here that ARFs 3 and 5, but not ARF6, are translocated to membranes in HeLa cell extracts that are engaged in translation of poliovirus RNA. The accumulation of ARFs on membranes correlates with active replication of poliovirus RNA in vitro, whereas ARF translocation to membranes does not occur in the presence of BFA. ARF translocation can be induced independently by synthesis of poliovirus 3A or 3CD proteins, and we describe mutations that abolished this activity. In infected HeLa cells, an ARF1-enhanced green fluorescent protein fusion redistributes from Golgi stacks to the perinuclear region, where poliovirus RNA replication occurs. Taken together, the data suggest an involvement of ARF in poliovirus RNA replication.
Poliovirus, the best-characterized member of the family Picornaviridae, is a small nonenveloped virus containing a single-stranded RNA genome of positive polarity. The virion RNA of about 7,500 nucleotides includes 5′ and 3′ nontranslated regions as well as a single long open reading frame and a poly(A) tail. The translated polyprotein is cleaved in cis and in trans by viral proteases to produce about 10 final products as well as a number of cleavage intermediates, many of which have been shown to possess unique activities necessary for virus propagation distinct from those of their cleavage products. Poliovirus proteins initiate a major remodeling of intracellular membranes, so that the cytoplasm of the infected cell becomes filled with tightly associated vesicles serving as sites for viral RNA replication (8-10). The reactions leading to this process are poorly understood, although COPII-mediated vesicle budding from the endoplasmic reticulum (ER) has been suggested to occur during formation of the replication vesicles at the beginning of infection (60), and data implicating the cellular autophagic pathway have been presented as well (64, 69).
It is well documented that poliovirus infection is sensitive to the fungal metabolite brefeldin A (BFA) (24, 34, 40). BFA is known to inhibit the activation and function of the small GTPases that comprise the ADP-ribosylation factor (ARF) family but has no reported effect on COPII-dependent vesicle budding (44, 55, 62, 63) or autophagy (56). The ARF family consists of six members, which are divided into three classes based on their primary structures. The proteins participate in formation of coated membranous vesicles originating from different organelles and plasma membrane as well as in cytoskeleton remodeling and regulation of phospholipase D activity. ARFs cycle between GTP- and GDP-bound states, with ARF-GTP required to interact with different membrane proteins and initiate membrane remodeling (46, 48). ARF that is associated with membranes is often referred to as activated. BFA prevents regeneration of ARF-GTP from ARF-GDP by interacting with guanine exchange factors, the proteins that accelerate replacement of bound GDP by GTP (44, 55, 62, 63). Data suggesting involvement of ARF in poliovirus replication have been presented previously by Cuconati and coworkers (20). They studied replication of poliovirus RNA in an in vitro translation-replication system (6, 42) that has greatly facilitated investigation of different aspects of the poliovirus life cycle. The system allows not only translation and proper processing of poliovirus proteins but also RNA replication and assembly of infectious virions. Addition of peptides corresponding to the N terminus of ARF1 to in vitro translation-replication reactions had deleterious effects on poliovirus RNA replication. The interpretation of these data may be uncertain, however, as those peptides may nonspecifically inhibit ARF-dependent as well as ARF-independent pathways, presumably due to their potential membrane-disrupting properties (29). A requirement for intact membranes to support viral RNA synthesis in the in vitro system was demonstrated by Fogg et al. (30). They showed that compounds such as nonionic detergents and cerulenin (a membrane-altering inhibitor of fatty acid synthesis), as well as BFA, prevented efficient VPg uridylylation and poliovirus RNA replication, although the morphology of the membranes supporting viral RNA synthesis in vitro had little in common with the highly organized vesicular structures found in infected cells.
In order to identify the BFA-sensitive step(s) in poliovirus RNA replication and to clarify the proposed involvement of ARF proteins, we initiated an investigation of ARF in the cell-free, translation-replication system. In this study we present direct evidence for specific translocation of different members of the ARF family to membranes in response to synthesis of poliovirus proteins and show that two individual virus proteins, 3A and 3CD, can induce such translocation independently.
MATERIALS AND METHODS
Plasmids and RNA synthesis.
Plasmid pARF1-EGFP has been described elsewhere (78). All constructs used for expression of poliovirus proteins are based on the ribozyme-containing plasmid pXpA (33). To facilitate cloning, two unique sites were introduced by PCR-based mutagenesis before and immediately after the poliovirus coding sequence: SalI in position 724 to 729 and HpaI in position 7372 to 7377. All other constructs were made by inserting PCR fragments obtained from the pXpA template with 5′ primers containing a SalI site and ATG initiator codon before the codon for the first amino acid of the corresponding protein sequence and 3′ primers containing an HpaI site following a TAG stop codon into the modified pXpA vector. All constructs were verified by sequencing. PCR fragments containing a mutated 3CD sequence with substitution of active site His for Ala (amino acid 40 in the 3C sequence) and active site Cys for Ala (amino acid 147 in the 3C sequence) were obtained from plasmid pPVΔP1 3C*, kindly provided by N. Teterina (National Institute of Allergy and Infectious Diseases, National Institutes of Health [NIH]). A plasmid encoding the 3A-2 mutant (7) was created by mutagenesis of our plasmid for 3A expression with direct and reverse oligonucleotides coding for insertion of an additional serine codon (AGT) after the 15th codon of the 3A sequence with Stratagene's QuikChange XL mutagenesis kit. RNAs were synthesized using the T7 MegaScript kit from Ambion (Austin, TX) with plasmids linearized at the EcoRI site. For use in translation-replication reactions, RNAs were subjected to two phenol-chloroform extractions followed by one extraction with chloroform, ethanol precipitation, and further purification on BD Biosciences' Chroma-Spin 100 DEPC H2O columns.
In vitro translation-replication.
HeLa S10 extracts for translation-replication reactions were prepared and used essentially as described elsewhere (30). Translation reaction mixtures of 50 μl included 2.5 μg of viral RNA transcripts or 1.25 μg of purified viral RNA in the presence of 2 mM guanidine-HCl. BFA (Sigma-Aldrich) dissolved in ethanol was added to a final concentration of 80 μg/ml where indicated; control reaction mixtures contained ethanol only. An aliquot of 9 μl from each reaction mixture was mixed with 1 μl of Redivue [35S]methionine (Amersham) and incubated for 3.5 h at 34°C, after which one-fourth of the material was resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) mini-gel for visualization of translation products. The remaining 40 μl was also incubated for 3.5 h at 34°C and then centrifuged for 20 min at 16,000 × g at 4°C. Total pellets were used in replication reactions performed essentially as described previously (30) or assayed in Western blot assays with the ECL Advance system (Amersham) according to the manufacturer's recommendations. Membrane stripping and reprobing were performed with Chemichon's Re-blot Plus mild solution according to their suggested procedure.
Antibodies.
Anti-ARF antibody that recognizes all known ARFs except for ARF4 was purchased from Affinity Bioreagents (Golden, CO). Antibodies against ARF3, -5, and -6 were generous gifts from Martha Vaughan and Joel Moss (National Heart, Lung and Blood Institute, NIH). Poliovirus 2C antiserum was described previously (18). Antibodies against GRP1 and PAPα were kindly provided by Paul Randazzo (National Cancer Institute, NIH).
Microscopy.
HeLa R19 cells grown on coverslips placed in 12-well culture plates were transfected with pARF1-EGFP plasmid with FuGene 6 reagent (Roche Applied Science) according to the manufacturer's recommendations. The next day they were infected with Mahoney strain, type I poliovirus at a multiplicity of 50 PFU/cell. At the indicated times postinfection cells were fixed with 2% paraformaldehyde in phosphate-buffered saline (PBS) for 10 min, washed three times with PBS, and permeabilized for 5 min with 0.1% Triton X-100 in PBS. Primary and secondary antibodies were diluted in 5% fetal bovine serum in PBS. Following three washes in PBS, cells were treated with mouse monoclonal anti-2C antibody solution supplemented with Hoechst 33342 dye (Sigma-Aldrich; 10 ng/ml) for 1 h. After three washes with 5% fetal bovine serum in PBS, secondary goat anti-mouse Cy5-conjugated antibodies were applied for 1 h at room temperature. Cells were washed three times with PBS and mounted on microscope slides using ProLong antifade reagent (Molecular Probes). Micrographs were taken with a Leica DMIRE 2 fluorescent microscope.
RNA transfection.
HeLa cell monolayers were transfected with serial dilutions of RNA transcripts in DEAE-dextran solution in PBS (500 μg/ml) and overlaid with agarose as described elsewhere (73).
Polymerase assay.
RNA coding for the wild-type or mutated 3D polymerase was translated overnight in vitro as described above. Polymerase reaction mixtures (30 μl) contained 50 mM HEPES (pH 7.2); 0.5 mM ATP, GTP, and CTP; 0.1 mM UTP; 2 μCi of [α-32P]UTP (specific activity, 3,000 Ci/mmol); 4 mM dithiothreitol; 3 mM Mg(OAc)2; 60 μM ZnCl2; 0.1% NP-40; 0.1 μg/μl poly(A); 0.13 μg/μl oligo(U); and 2 μl of translation reaction mixtures as a source of 3D protein. Samples were incubated on ice for 10 min and then transferred to 30°C for 30 min. Reactions were stopped by 33 mM EDTA. Ten microliters of the reaction mix was spotted onto Whatman DE81 paper, dried, washed three times with 5% Na2HPO4 followed by one wash with ethanol, dried, and counted by scintillation spectroscopy.
RESULTS
Redistribution of ARF1 during poliovirus infection.
ARF1 is one of the best-studied members of the ARF family. Its activity is important for retrograde membrane transport from Golgi to ER and intra-Golgi. It is also involved in membrane transport from the _trans_-Golgi network, endosomal trafficking, and exocytosis of synaptic vesicles and nascent secretory vesicles (48, 58). The role of ARF1 in formation of membranous vesicles and its sensitivity to BFA prompted us to consider whether it might be involved in the assembly of poliovirus replication complexes, structures that appear as clustered vesicles in electron micrographs (9, 10) and whose formation is prevented by BFA. We expressed an ARF1-enhanced green fluorescent protein (EGFP) fusion in HeLa cells and followed its localization during poliovirus infection (Fig. 1). Fusions of ARFs with fluorescent proteins are widely used in cellular biology studies and have been shown to retain the distribution properties and biological activities of native proteins (e.g., references 43 and 78). In noninfected or mock-infected cells, fluorescence is concentrated in distinct areas corresponding to the location of the Golgi apparatus (Fig. 1D). Upon progression of infection, fluorescence is markedly redistributed to perinuclear structures closely resembling those of poliovirus replication complexes (Fig. 1A), as shown by staining with antibodies against poliovirus replication protein 2C (Fig. 1B). This distribution was clearly different from localization of EGFP alone, which was evenly spread throughout the cell with somewhat enhanced accumulation in the nuclear area in both infected and mock-infected cells (not shown). ARF1-EGFP present in virus-infected cells migrated as a single band on SDS-polyacrylamide gels with the same electrophoretic mobility as that from uninfected cells, indicating that it was not degraded and therefore the observed redistribution was not due to alteration of the protein (not shown).
FIG. 1.

Intracellular relocation of ARF-1-EGFP protein following infection of HeLa cells with poliovirus. Cells were transfected with pARF1-EGFP 24 h prior to infection with poliovirus (A, B, and C) or mock infection (D, E, and F). Six hours postinfection, cells were examined for EGFP fluorescence (A and D) or stained with anti-polio 2C antibodies (B and E) and Hoechst 33342 for nuclear DNA (C and F).
ARF is translocated to membranes upon translation of poliovirus RNA.
To investigate the possible involvement of ARF in the replication of poliovirus RNA, we used the two-step in vitro translation-replication system (6, 42). During the first step, poliovirus RNA is translated in HeLa cell S10 extracts in the presence of 2 mM guanidine-HCl, which inhibits poliovirus RNA replication but has no effect on polyprotein synthesis and processing. During this step, poliovirus proteins associate with membranes and form structures ready to initiate RNA synthesis once the guanidine-HCl is removed. After translation, membranes are collected by centrifugation and resuspended in buffer in the absence of guanidine-HCl to initiate RNA replication. We fractionated translation reaction mixtures by centrifugation and analyzed the pellets, containing membranes with associated proteins, and 1/10 of the supernatants by immunoblotting with an anti-ARF monoclonal antibody that recognizes ARF1, -3, -5, and -6 and reacts weakly with ARF4. Translation of poliovirus RNA resulted in the appearance of a strong ARF signal in the samples containing the membranous pellet (Fig. 2, compare lanes 1 and 3). ARF translocation did not occur when translation of poliovirus RNA was inhibited with puromycin (Fig. 2, lane 2). Samples treated with puromycin contained even less ARF in the pellets than samples containing no poliovirus RNA (Fig. 2, compare lanes 2 and 3). We did not pursue this result; however, the background level of membrane-associated ARF may have resulted from some cellular proteins whose synthesis also was inhibited by puromycin. Despite the strong ARF signal in the pellet sample from poliovirus RNA translation, there was no obvious change in the ARF content of the supernatants (Fig. 2, lanes 4 to 6) diluted 10-fold for the PAGE analysis. This indicates that the majority of ARF proteins were not membrane bound in the HeLa cell extract and that only a small portion of the ARF protein present in the extract was activated by poliovirus proteins. In numerous experiments and with different extracts, however, membrane-associated ARF increased at least 5- to 10-fold following synthesis of viral proteins, as judged by analysis of the density of corresponding bands with ImageQuant software (data not shown). The recruitment of ARF proteins to the membranes was not merely a poliovirus translation-dependent, nonspecific aggregation of membrane material, since only some members of the ARF family (ARF3 and -5, but not ARF6) increased their membrane association (see Fig. 6, below) and other cellular membrane-associated proteins, such as GRP1 or PAPα, showed no increase as a result of poliovirus RNA translation.
FIG. 2.

Translocation of ARF to membranes induced by translation of poliovirus proteins in vitro. Poliovirus RNA was translated in HeLa S10 extracts for 3.5 h. Reactions were fractionated by centrifugation, and total pellets (lanes 1 to 3) and one-tenth of the total volume of supernatants (lanes 4 to 6) were analyzed by immunoblotting with anti-ARF antibody. Lanes: 1 and 4, fractions from reactions translating viral RNA; 2 and 5, fractions from reactions in which viral RNA translation was inhibited by 1 mg/ml of puromycin; 3 and 6, fractions from reactions incubated with no RNA.
FIG. 6.

Activation of individual ARFs induced by translation of poliovirus-specific transcripts. RNA transcripts were translated in HeLa S10 extracts for 3.5 h. Reactions were fractionated by centrifugation, and the pellets were analyzed by immunoblotting. The membrane shown in Fig. 5 was stripped and probed sequentially with specific antibodies against ARF3, ARF5, or ARF6.
BFA prevents translocation of ARF to membranes.
Inhibition of poliovirus RNA replication by BFA is well documented in vivo (24, 34, 40) and in vitro (20), although the mechanism of this inhibition is not clear. It has been established, however, that BFA is likely to act on some host factor(s) necessary for replication, but not on the virus replication proteins themselves. We investigated the effect of the drug on ARF association with membranes induced by translation of poliovirus RNA. Translation reaction mixtures were divided into two samples, one of which was treated with BFA, and membranes collected by centrifugation were tested for the presence of ARF and for their ability to support poliovirus RNA synthesis. BFA significantly prevented poliovirus-induced recruitment of ARF to membranes (Fig. 3A), whereas the drug had no effect on translational efficiency or virus polyprotein processing (Fig. 3C), consistent with previously reported data (20, 40). Replication complexes formed during translation of poliovirus RNA in the presence of BFA, lacking associated ARF, were very inefficient in subsequent RNA synthesis (Fig. 3B). Thus, the presence of increased ARF on membranes correlated with efficient RNA replication. To distinguish between BFA inhibition of replication complex formation and possible BFA inhibition of subsequent RNA chain elongation, viral RNA was translated in the absence of the inhibitor and replication complexes were collected by centrifugation and resuspended in replication buffer with BFA (Fig. 3D, lane 5). RNA replication was much less affected in this case than in the sample where BFA was added during translation (Fig. 3D, lane 4), although it was usually slightly lower than in the control sample (Fig. 3D, lane 3), possibly indicating that some ARF activation continues during the RNA replication step of the reaction. Together, these data show that the inhibitory activity of BFA is exerted during translation of viral RNA and membrane association of viral proteins, while ARF is being recruited and replication complexes are being formed. Once the complexes are formed, BFA does not inhibit viral RNA synthesis per se.
FIG. 3.

Inhibition of ARF translocation and viral RNA replication by BFA. Viral RNA was translated in HeLa S10 extracts for 3.5 h. (A to C) Duplicate reactions were fractionated by centrifugation, and the pellets were analyzed by immunoblotting with anti-ARF antibody (A) and assayed for viral RNA replication (B) as described in Materials and Methods. Translation was monitored by SDS-PAGE of [35S]methionine-labeled proteins synthesized during the translation step (C). Lane 1, no BFA was present during either translation or replication steps; lane 2, BFA (80 μg/ml) was present only during the translation step. (D) RNA replication after BFA addition during translation or replication steps. Lane 3, no BFA was present during either translation or replication steps; lane 4, BFA (80 μg/ml) was present only during the translation step; lane 5, BFA (80 μg/ml) was present only during the replication step.
3A and 3CD induce activation of ARF independently.
To determine which viral proteins are responsible for ARF activation, we performed in vitro translation of RNAs coding for individual poliovirus proteins as well as intermediate cleavage precursors. To ensure equal translation potential of these RNAs, we introduced two unique restriction sites in the ribozyme-containing plasmid pXpA (33) that facilitated cloning of any segments of the poliovirus genome in the vector so that the RNAs generated by T7 RNA polymerase would have the same 5′ and 3′ noncoding regions. Introduction of these two additional restriction sites into the poliovirus cDNA had no effect on virus growth in cultured HeLa cells (not shown). First, we tested segments of the polyprotein spanning P2-P3, P2, or P3 regions, as well as individual proteins from the P2 region that have been shown previously to induce membrane alterations when expressed individually (11, 18, 72). After translation, samples were taken for analysis of protein synthesis (Fig. 4B), and membranes with associated proteins were collected by centrifugation and analyzed for ARF by Western blotting (Fig. 4A). Translation of P2-P3 or just the P3 segment of the polyprotein resulted in a positive signal of ARF translocation, similar to that observed upon translation of the full-length RNA (Fig. 4, lanes 1, 2, and 7). Proteins from the P2 region were not active in ARF translocation (Fig. 4, lanes 3 to 6); thus, their ability to induce membrane modification when expressed in cells does not require ARF activation, or is not required in vitro.
FIG. 4.

Determinants specifying ARF translocation map to the P3 region of the poliovirus genome. RNA transcripts were translated in HeLa S10 extracts for 3.5 h. (A) Reactions were fractionated by centrifugation, and the pellets were analyzed by immunoblotting with anti-ARF antibody. (B) Translation of each transcript was monitored by SDS-PAGE of [35S]methionine-labeled proteins. The coding region of the RNA transcripts used to program the translation reaction is indicated above each lane.
These results show that ARF-activating determinants are located in the P3 genomic region. When individual proteins of this region were tested for their ability to induce ARF translocation, unexpectedly we found that two proteins, 3A and 3CD (Fig. 5, lanes 3 and 6), but not 3C or 3D (Fig. 5, lanes 4 and 5), could induce ARF association with membranes. Quantitation of band intensities with ImageQuant software showed a three- to sixfold increase in ARF activation by 3A and four- to sevenfold ARF activation by 3CD in different experiments, compared to levels in controls containing no RNA.
FIG. 5.

Poliovirus proteins 3A and 3CD can induce ARF translocation independently. RNA transcripts were translated in HeLa S10 extracts for 3.5 h. (A) Reactions were fractionated by centrifugation, and the pellets were analyzed by immunoblotting with anti-ARF antibody. (B) Translation was monitored by SDS-PAGE of [35S]methionine-labeled proteins. The coding region of each RNA transcript used to program the translation reaction is indicated above each lane.
Not all ARFs are activated by poliovirus proteins.
The ARF family of small GTPases currently includes six members, which are assigned to three classes based on their primary structures (48, 58). ARF1, -2, and -3 constitute class 1, ARF4 and -5 comprise class 2, and ARF6 is in class 3. Using antibodies specific for individual ARF proteins, we tested their translocation to membranes induced by poliovirus proteins (Fig. 6). Translation of the full-length genomic RNA and RNA coding for 3A and 3CD similarly induced activation of ARF3 and ARF5 (Fig. 6, lanes 3 and 6), which belong to two different classes, while ARF6 was not affected by translation of poliovirus-specific proteins. (Antibodies specific for ARF1, -2, and -4 are not currently available.) This result was not surprising, as ARF3 and ARF5, as well as ARF1, are known to participate in intracellular membrane trafficking (2, 15, 21, 38, 49, 71, 77) while ARF6 is believed to serve mainly in vesicular and cytoskeletal processes and regulation near the plasma membrane (15, 54, 57). The uniform intensity of the ARF6 band in all samples indicated that the amount of material loaded on the gel was the same in all samples and that the difference observed for other ARFs reflects specific translocation of these proteins induced by the poliovirus proteins.
3A-induced ARF translocation is not essential for virus growth in cell culture.
Since two poliovirus proteins induced ARF translocation in vitro, we looked for mutations in those proteins that might affect this property and tested the effect of such mutations on virus growth. A mutation in 3A, called 3A-2, was described by Bernstein and Baltimore (7) and shown by Doedens et al. (25) to prevent the intracellular membrane traffic alterations that are induced by wild-type 3A. The 3A-2 mutant contains an insertion of a serine after position 14 of the 3A sequence, which apparently alters the protein's interaction with membranes; thus, this mutation seemed to be a good candidate to test for ARF translocation. Figure 7 shows that the mutated 3A indeed lost its ability to activate ARF (compare lanes 2 and 3). Interaction of ARF with membrane proteins has been shown to be a central event in remodeling intracellular membranes (3, 16, 28, 50, 51, 65, 68, 70, 75), and the fact that the 3A-2 mutant is unable to induce ARF translocation may at least in part be responsible for its failure to interfere with intracellular membrane traffic. Virus bearing this mutation is, however, viable with growth characteristics close to those of the wild-type virus (7), in spite of the lack of membrane traffic inhibition. We conclude, therefore, that the ability of 3A to induce ARF translocation to membranes is not essential for virus replication in cell culture. Translation of a full-length transcript containing the 3A-2 mutation produced significant ARF translocation, albeit slightly less than wild-type RNA. This was expected, since wild-type 3CD was present to activate ARF. When a mutation that inactivated the 3CD property of ARF activation (see following section) was combined with the 3A-2 mutation, the transcript completely lost its ability to activate ARF (not shown).
FIG. 7.
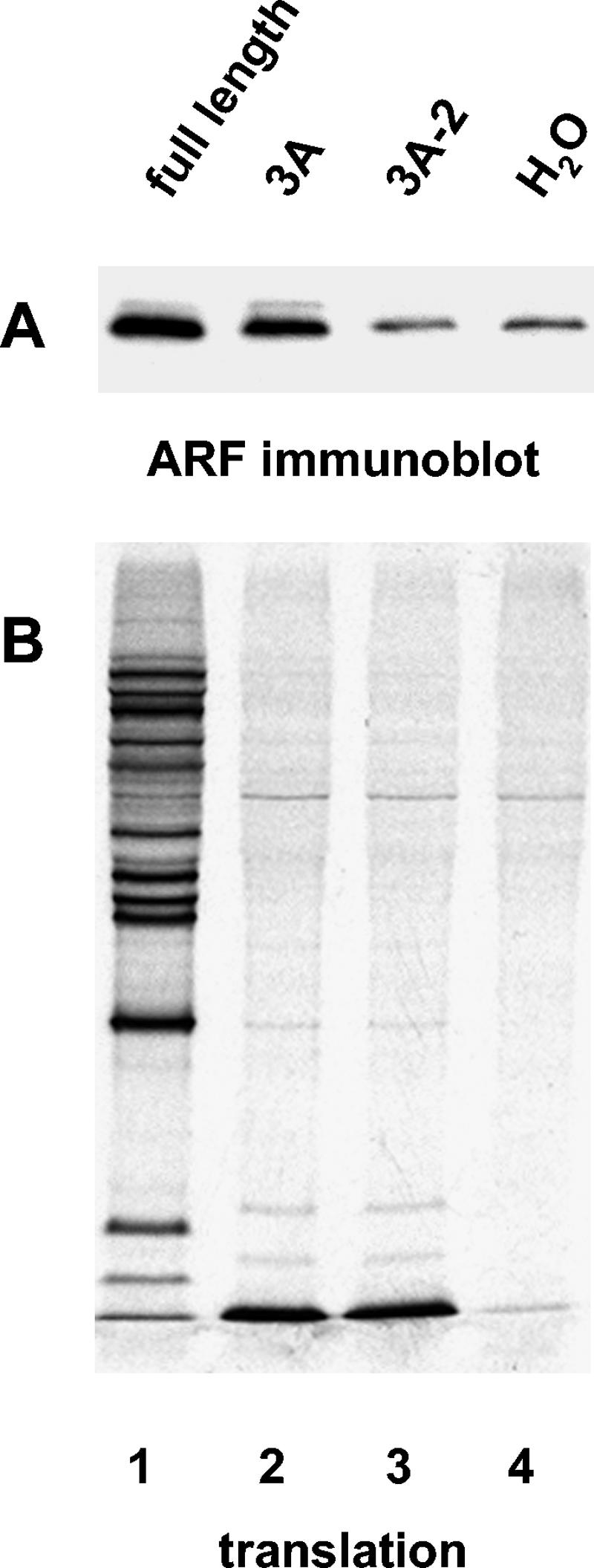
Mutant 3A-2 does not activate ARF. RNA transcripts were translated in HeLa S10 extracts for 3.5 h. (A) Reactions were fractionated by centrifugation, and the pellets were analyzed by immunoblotting with anti-ARF antibodies. (B) Translation was monitored by SDS-PAGE of [35S]methionine-labeled proteins. The coding region of each RNA transcript used to program the translation reaction is indicated above each lane.
3CD mutations and ARF translocation.
3CD is an intermediate cleavage product of the poliovirus polyprotein, composed of protease 3C and RNA-dependent RNA polymerase 3D sequences. The uncleaved 3CD protein possesses protease activity with substrate specificity distinct from that of the 3C protease. To test the possibility that 3CD induced ARF translocation by cleaving some cellular factor(s) that triggered the ultimate association of ARF with membranes, we examined a 3CD mutant that had substitutions of two protease active site amino acids, rendering it proteolytically inactive. These mutations had no effect on the ability of the protein to induce ARF association with membranes, nor did a mutation preventing cleavage between 3C and 3D portions of the protein (not shown). During PCR-based cloning of the construct for individual expression of 3CD, we serendipitously isolated a mutant with four changes in amino acid sequence that had lost its ability to activate ARF (not shown). When these mutations were individually reintroduced into the wild-type sequence, one of them, substitution of Phe to Ser in position 441 of 3CD, was found to be solely responsible for the ARF translocation-negative phenotype (Fig. 8A). There were no detectable effects on translational efficiency (Fig. 8B). When this mutation was introduced into the full-length construct, the resulting RNA was very inefficiently replicated in vitro (Fig. 8C) and also showed greatly reduced RNA infectivity after transfection of HeLa cells (Fig. 8E). A few plaques appeared only in wells transfected with the highest dose of RNA, while transfection with the wild-type RNA under the same conditions resulted in complete degeneration of the monolayer. The few plaques formed were about the same size as the wild type; thus, they likely were formed by revertants, which could readily be generated by a single nucleotide substitution to restore the wild-type sequence.
FIG. 8.

Mutation 3CD F441S prevents ARF translocation and inhibits viral RNA replication in vitro and virus growth in vivo. Wild-type (lane 1), mutant (lane 2), or no (lane 3) 3CD RNA transcripts were translated in HeLa S10 extracts for 3.5 h. (A) Reactions were fractionated by centrifugation, and the pellets were analyzed by immunoblotting with anti-ARF antibody. (B) [35S]methionine-labeled translation products. (C) Wild-type (lane 4) or mutant (lane 5) full-length genomic transcripts were translated in HeLa S10 extracts for 3.5 h. Reactions were fractionated by centrifugation, and the pellets were assayed for viral RNA replication. (D) [35S]methionine-labeled translation products. (E) HeLa cell monolayers were transfected with serial dilutions of wild-type or mutant full-length genomic transcripts. Cells were covered after transfection with agarose-solidified medium and stained 48 h later with crystal violet.
Although these results initially suggested that 3CD's ARF activation function was required for viral RNA replication and growth, we also observed slight differences in the processing patterns of polyproteins translated from wild-type and mutant RNAs. The processing defect appears most evident for the P1 region structural proteins, which are preferentially processed by 3CD: a slight decrease in the VP3 band and a corresponding slight increase in the intensity of P1 can be seen in Fig. 8D (compare lanes 4 and 5). Since both proteins 3CD and 3D are known to have multiple functions required for viral RNA replication, any mutation that affects one activity, such as ARF activation, might also affect other activities, such as proteolytic cleavage, 5′-terminal RNA cloverleaf binding, stimulation of cre-dependent VPg uridylylation, or RNA-dependent RNA polymerase activity. To determine whether the F441S mutation had a direct effect on the polymerase function of 3D, we assayed poly(U) polymerase activity, using a poly(A) template and oligo(U) primer. Wild-type and mutant 3D proteins were produced by in vitro translation of mRNAs encoding each protein in HeLa cell extracts, and an aliquot of the translation reaction mixture was used as the source of 3Dpol for the assay. Translation efficiencies were monitored by labeling an aliquot of the reaction with [35S]methionine as described in Materials and Methods and proved to be the same for wild-type and mutated proteins (data not shown). The results showed that the F441S mutation abrogated the polymerase activity of 3D (Table 1). Nevertheless, the appearance of a few plaques after RNA transfection indicates retention of a low level of 3D activity. Unfortunately, the marked defect in RNA polymerase activity of 3D carrying the 3CD F441S mutation, as well as the decreased efficiency of structural protein processing catalyzed by the F441S mutant 3CD, preclude any conclusions about the requirement for ARF activation activity of 3CD for virus growth. Further studies will be required to understand the consequences of 3CD failure to activate ARF on viral RNA replication.
TABLE 1.
Effects of 3CD F441S mutation on poly(A)-dependent poly(U) polymerase activity of 3Dpola
| mRNA translated | [α-32P]UTP incorporation (cpm) |
|---|---|
| Wild-type 3D | 19,407 ± 167 |
| F441S 3D | 837 ± 38 |
| None | 1,011 ± 111 |
DISCUSSION
It is generally accepted that poliovirus replication requires the participation of cellular factors, yet very few host cell proteins have been identified that contribute to specific virus propagation reactions in the virus growth cycle. Most of them have been shown to stimulate internal ribosome entry site (IRES)-dependent translation initiation, and a very few have been postulated to participate in RNA replication itself. Morrow and coworkers reported stimulation of poliovirus replicase activity in vitro by a cellular protein kinase (45), although this finding was neither confirmed nor pursued. Nuclear protein Sam68 was identified in a yeast two-hybrid screen as a protein that interacts with poliovirus polymerase 3D, and the interaction was confirmed by coimmunoprecipitation of Sam68 with 3D from lysates of infected cells (41), although the biological significance of this interaction remains hypothetical. Another nuclear protein, nucleolin, was shown to interact with the 3′ nontranslated region of poliovirus RNA and therefore was speculated to participate in the initiation of synthesis of the complementary negative strand of viral RNA. Cell lysates depleted of nucleolin produced a reduced amount of infectious virus (80). Subsequent investigations showed that nucleolin also interacts with the 5′ nontranslated region and stimulates IRES-dependent translation (35). Another cellular protein, poly(rC) binding protein, was shown to bind specifically to the 5′-terminal cloverleaf structure in plus-strand poliovirus RNA and to stem-loop IV in the poliovirus IRES, and it was demonstrated to be important for both translation and replication of the RNA (12, 52).
In this study we report poliovirus-specific activation of another class of cellular proteins that is likely to be important for viral RNA replication. We found that cell-free translation of poliovirus genomic RNA or RNAs encoding either of two individual poliovirus proteins from the P3 genomic region—3A and 3CD—results in specific translocation of cellular proteins from the ARF family of small GTPases to membranes. We observed strong ARF signals in Western blot assays of membranous pellets collected after translation of poliovirus RNA with a monoclonal antibody that cross-reacts with several ARFs. Antibodies that recognize individual members of the ARF family revealed that synthesis of poliovirus proteins activates at least two ARFs from different classes—ARF3 and ARF5—while ARF6, which constitutes its own class, showed no response to translation of poliovirus-specific proteins. The behavior of ARFs 1, 2, and 4 could not be determined, due to the absence of specific antibody reagents. The majority of ARF protein in the HeLa cell extract is in the supernatant, and only a small proportion of ARF is activated upon poliovirus translation in vitro. The observation of ARF activation in vitro is consistent with data presented in this study showing that the membrane-associated ARF1-EGFP fusion protein redistributes upon poliovirus infection from the Golgi apparatus to the perinuclear region of infected cells, known to be the site of poliovirus RNA replication (76). ARF1-EGFP fluorescence in intact cells appeared predominantly localized on membranes (Fig. 1), whereas the cell extracts showed a predominantly cytoplasmic distribution of ARF after fractionation (Fig. 2). This dissimilarity may reflect a real difference between ARF distribution in vivo and in vitro, possibly due to poor preservation of membrane structures, leading to ARF dissociation during extract preparation, and/or disorganization of biochemical processes that maintain ARF association with membranes, since ARF normally cycles between the bound and unbound state. Alternatively, multiple ARF species were measured by the antibodies used to test extract fractions, whereas the EGFP fluorescence marks only ARF1. In addition, differences in fluorescence intensity between membrane-bound and cytoplasmic ARF-EGFP in cells cannot be ruled out.
Since ARF function is known to be affected by BFA (55, 62, 63) and the sensitivity of poliovirus infection to BFA also is well documented both in vivo and in vitro (20, 24, 34, 40), we examined the effect of the inhibitor on poliovirus-induced ARF translocation. The first suggestion that suppression of viral RNA replication by BFA may be mediated through its ability to inhibit ARFs was presented by Cuconati et al. (20). We directly showed in this study that BFA, added to a cell-free translation extract, severely inhibited translocation of ARF to membranes. When tested for viral RNA replication, these ARF-deficient membranes failed to support efficient synthesis of viral RNA. On the other hand, if the replication complexes were allowed to form without BFA, they demonstrated high replication activity even when the inhibitor was added later to the replication reaction mixture, in accordance with previous data that BFA inhibits a host factor(s) but not the viral polymerase itself (24). It is tempting to speculate that this lack of membrane-associated ARF mediates the inhibitory effect of BFA on poliovirus replication.
To identify the poliovirus protein(s) responsible for ARF translocation, we tested individual proteins as well as various intermediates of polyprotein processing in an ARF translocation assay in vitro. Surprisingly, we found that synthesis of 3A and 3CD could independently induce association of ARF with membranes. No other poliovirus proteins tested, including 2B, 2C, and 2BC, which have well-known membrane binding and remodeling properties (1, 4, 5, 18, 61, 69, 72), or 3C or 3D showed any effect on ARF distribution. Despite this specificity and the phenomenological similarity, activation of ARF by 3A and 3CD may occur by different mechanisms and play different roles in the virus life cycle. Poliovirus protein 3A is known to inhibit the cell's secretory pathway, and such inhibition is speculated to provide an advantage for the virus to escape the host immune surveillance system (22, 23, 25, 26, 47). Peptides destined for presentation on the plasma membrane or to be secreted into the extracellular medium undergo maturation during their translocation from the ER through the Golgi to the cellular membrane. This intracellular traffic occurs in membranous vesicles and is dependent on ARF activity (13, 27, 39, 46, 58, 66). One can assume that interference with ARF metabolism may at least in part account for inhibition of protein secretion by viral protein 3A. In support of this assumption, the previously described 3A-2 mutant (7) that was shown to be a much less potent inhibitor of intracellular membrane traffic (25) also failed to induce ARF translocation in our assay. Since this 3A mutation exerts no major effects on virus viability except for a diminished replication at 32°C (7), it seems that 3A-dependent ARF activation is dispensable for virus growth, at least in cultured cells.
On the other hand, the ARF-activating property of 3CD appears to correlate with virus replication. 3CD is a multifunctional intermediate cleavage product of the poliovirus polyprotein composed of virus protease 3C and RNA-dependent RNA polymerase 3D sequences that have no reported membrane-binding properties. 3CD manifests protease activity with a substrate specificity distinct from that of 3C and is mainly responsible for processing the structural part of the polyprotein (36, 83). 3CD was also shown to be essential for VPg uridylylation (53) and for binding to the 5′-terminal RNA cloverleaf structure required for RNA replication (31, 79, 82). Since all those activities were documented in biochemically defined systems, they are not dependent on ARF and, therefore, ARF activation is yet another distinct activity of 3CD. The protease function of 3CD is not essential for ARF activation, as substitutions of protease active site amino acids did not diminish its ability to activate ARF. We identified one mutation in the 3D region of 3CD, a substitution of F to S in position 441 of 3CD, which completely abolished activation of ARF and proved to be lethal in vivo and in vitro when introduced into the full-length genome. Since the 3CD structure is not resolved, it is not possible to unambiguously identify the structural feature of the protein that is perturbed by this mutation. However it is generally predicted that the 3CD structure retains relatively unaltered structural domains of the individual 3C and 3D proteins. This assumption is supported by the fact that 3CD manifests the proteolytic activity of 3C, albeit with a slightly modified substrate specificity. If the 3D portion of 3CD forms the structural organization of native 3D, then the F441S mutation, which corresponds to residue 258 in the 3D sequence, is located in the linker region between two α-spirals on the outer side of the fingers domain (74). To our knowledge, no specific functions of the protein have been assigned to this region, but one can speculate that location of this amino acid on the outer surface of the protein makes it possible that this site is involved in interaction with other proteins, such as cellular factors that may attract ARF to membranes. Infectivity of full-length RNA containing the ARF activation-defective mutation was several orders of magnitude lower than that of wild-type RNA. Since this mutation also impaired the elongation activity of 3D and the protease activity of 3CD and may as well have inhibited other known or unknown activities of 3D precursors, it is not possible to assign the ARF-activating defect alone as the cause of the lethal phenotype. Verlinden et al. (79) reported stimulation of RNA synthesis in vitro when full-length viral RNA was cotranslated with RNA coding for 3CD. Recruitment of additional ARF to membranes induced by this additional 3CD could be one of the mechanisms underlying this phenomenon.
In spite of the requirement for membranes for replication, which appears to be common to all known positive-strand RNA viruses, the specific mechanisms underlying conversion of cellular membranes into different viral replication sites seem to vary greatly. Picornaviruses, for example, show various sensitivities to BFA from complete inhibition of replication, as with poliovirus (24, 34, 40) and echovirus 11 (32), through partial sensitivity, as with parechovirus 1 (32), to complete resistance to the drug, as shown by encephalomyocarditis virus (34, 40). This indicates that even within one family, viruses may exploit different pathways of remodeling cellular membranes. Although data presented here suggest that BFA-sensitive ARF activation is essential for poliovirus replication, Crotty et al. recently reported isolation of poliovirus mutants resistant to BFA (19). Mutations conferring partial resistance to the drug were found in proteins 2C and 3A; when combined, these mutations made virus completely resistant to the inhibitor. One can assume that such mutations enable virus to escape ARF dependence by switching to an alternative mode of modification of host membranes.
What function can ARF perform in the formation of functional poliovirus replication complexes? The available evidence suggests that ARFs are key components promoting formation of coated membranous transport vesicles from different intracellular compartments through their interactions with coat and adaptor proteins (3, 16, 28, 50, 51, 65, 68, 70, 75) as well as through stimulation of activity of phospholipase D (14, 17, 37, 58, 59, 67, 81). Transport vesicles may show some morphological similarity to those induced during poliovirus infection; therefore, one of the roles of ARF recruitment by poliovirus proteins may be to stimulate formation of membranous structures necessary for virus replication. In support of this hypothesis, we found that synthesis of poliovirus proteins activates ARF3 and -5, which participate in formation of transport vesicles from intracellular organelles (2, 15, 21, 38, 49, 71, 77) but not ARF6, which is believed to be primarily involved in plasma membrane metabolism (15, 54, 57). The origin and formation of poliovirus-induced replication vesicles are poorly understood, but available data indicate that there are probably different pathways of converting cellular membranes into poliovirus replication vesicles. Some data suggest that ER membranes may participate in formation of poliovirus-induced vesicles. Rust et al. (60), based on colocalization studies, postulated that cellular components of the COPII membrane traffic pathway known to be responsible for transfer of material from the ER to Golgi contribute to formation of poliovirus replication complexes, at least at the early stages of infection. It was also shown that expression of poliovirus proteins from the P2 region or in conjunction with 3A may induce ER membrane alterations sometimes very similar morphologically to native poliovirus vesicles (11, 18, 72). Other investigations concluded that an autophagy-like process was involved in generation of poliovirus replication vesicles (64, 69) because of their morphology and biochemical markers. Neither ER modification nor autophagy is sensitive to BFA. Therefore, if the only involvement of ARF in poliovirus replication were to generate a structural scaffold, it would be difficult to explain the severe inhibition of replication by BFA because of the likely existence of alternative pathways for remodeling membranes. The other possibility, not necessarily excluding the requirement of ARF for generation of vesicles, is that ARF may participate in the replication process itself, by attracting other cellular proteins to the replication sites. ARFs are known to interact directly with many regulatory and structural proteins and to affect through such interactions the behavior of other proteins (48, 58). It has been recently shown, for example, that ARF activation is necessary for retention of a number of proteins on Golgi membranes, including those not directly involved in membrane traffic (2). To discriminate between these possibilities, further investigations are necessary.
Acknowledgments
We thank all members of the laboratory for help in preparing reagents. We are grateful to O. Richards, K. Green, M. Vaughn, J. Moss, G. Pacheco, J. Lippincott-Schwartz, and N. Altan-Bonnet for stimulating and helpful discussions and technical advice and critical reading of the manuscript.
REFERENCES
- 1.Aldabe, R., and L. Carrasco. 1995. Induction of membrane proliferation by poliovirus proteins 2C and 2BC. Biochem. Biophys. Res. Commun. 206**:**64-76. [DOI] [PubMed] [Google Scholar]
- 2.Altan-Bonnet, N., R. D. Phair, R. S. Polishchuk, R. Weigert, and J. Lippincott-Schwartz. 2003. A role for Arf1 in mitotic Golgi disassembly, chromosome segregation, and cytokinesis. Proc. Natl. Acad. Sci. USA 100**:**13314-13319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balch, W. E., R. A. Kahn, and R. Schwaninger. 1992. ADP-ribosylation factor is required for vesicular trafficking between the endoplasmic reticulum and the cis-Golgi compartment. J. Biol. Chem. 267**:**13053-13061. [PubMed] [Google Scholar]
- 4.Barco, A., and L. Carrasco. 1995. A human virus protein, poliovirus protein 2BC, induces membrane proliferation and blocks the exocytic pathway in the yeast Saccharomyces cerevisiae. EMBO J. 14**:**3349-3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barco, A., and L. Carrasco. 1998. Identification of regions of poliovirus 2BC protein that are involved in cytotoxicity. J. Virol. 72**:**3560-3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barton, D. J., E. P. Black, and J. B. Flanegan. 1995. Complete replication of poliovirus in vitro: preinitiation RNA replication complexes require soluble cellular factors for the synthesis of VPg-linked RNA. J. Virol. 69**:**5516-5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berstein, H. D., and D. Baltimore. 1988. Poliovirus mutant that contains a cold-sensitive defect in viral RNA synthesis. J. Virol. 62**:**2922-2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bienz, K., D. Egger, and T. Pfister. 1994. Characteristics of the poliovirus replication complex. Arch. Virol. Suppl. 9**:**147-157. [DOI] [PubMed] [Google Scholar]
- 9.Bienz, K., D. Egger, T. Pfister, and M. Troxler. 1992. Structural and functional characterization of the poliovirus replication complex. J. Virol. 66**:**2740-2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bienz, K., D. Egger, Y. Rasser, and W. Bossart. 1983. Intracellular distribution of poliovirus proteins and the induction of virus-specific cytoplasmic structures. Virology 131**:**39-48. [DOI] [PubMed] [Google Scholar]
- 11.Bienz, K., D. Egger, M. Troxler, and L. Pasamontes. 1990. Structural organization of poliovirus RNA replication is mediated by viral proteins of the P2 genomic region. J. Virol. 64**:**1156-1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blyn, L. B., J. S. Towner, B. L. Semler, and E. Ehrenfeld. 1997. Requirement of poly(rC) binding protein 2 for translation of poliovirus RNA. J. Virol. 71**:**6243-6246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boman, A. L., and R. A. Kahn. 1995. Arf proteins: the membrane traffic police? Trends Biochem. Sci. 20**:**147-150. [DOI] [PubMed] [Google Scholar]
- 14.Brown, H. A., S. Gutowski, C. R. Moomaw, C. Slaughter, and P. C. Sternweis. 1993. ADP-ribosylation factor, a small GTP-dependent regulatory protein, stimulates phospholipase D activity. Cell 75**:**1137-1144. [DOI] [PubMed] [Google Scholar]
- 15.Cavenagh, M. M., J. A. Whitney, K. Carroll, C. Zhang, A. L. Boman, A. G. Rosenwald, I. Mellman, and R. A. Kahn. 1996. Intracellular distribution of Arf proteins in mammalian cells. Arf6 is uniquely localized to the plasma membrane. J. Biol. Chem. 271**:**21767-21774. [DOI] [PubMed] [Google Scholar]
- 16.Chen, Y. G., and D. Shields. 1996. ADP-ribosylation factor-1 stimulates formation of nascent secretory vesicles from the trans-Golgi network of endocrine cells. J. Biol. Chem. 271**:**5297-5300. [DOI] [PubMed] [Google Scholar]
- 17.Chen, Y. G., A. Siddhanta, C. D. Austin, S. M. Hammond, T. C. Sung, M. A. Frohman, A. J. Morris, and D. Shields. 1997. Phospholipase D stimulates release of nascent secretory vesicles from the trans-Golgi network. J. Cell Biol. 138**:**495-504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cho, M. W., N. Teterina, D. Egger, K. Bienz, and E. Ehrenfeld. 1994. Membrane rearrangement and vesicle induction by recombinant poliovirus 2C and 2BC in human cells. Virology 202**:**129-145. [DOI] [PubMed] [Google Scholar]
- 19.Crotty, S., M. C. Saleh, L. Gitlin, O. Beske, and R. Andino. 2004. The poliovirus replication machinery can escape inhibition by an antiviral drug that targets a host cell protein. J. Virol. 78**:**3378-3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cuconati, A., A. Molla, and E. Wimmer. 1998. Brefeldin A inhibits cell-free, de novo synthesis of poliovirus. J. Virol. 72**:**6456-6464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dascher, C., and W. E. Balch. 1994. Dominant inhibitory mutants of ARF1 block endoplasmic reticulum to Golgi transport and trigger disassembly of the Golgi apparatus. J. Biol. Chem. 269**:**1437-1448. [PubMed] [Google Scholar]
- 22.Deitz, S. B., D. A. Dodd, S. Cooper, P. Parham, and K. Kirkegaard. 2000. MHC I-dependent antigen presentation is inhibited by poliovirus protein 3A. Proc. Natl. Acad. Sci. USA 97**:**13790-13795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dodd, D. A., T. H. Giddings, Jr., and K. Kirkegaard. 2001. Poliovirus 3A protein limits interleukin-6 (IL-6), IL-8, and beta interferon secretion during viral infection. J. Virol. 75**:**8158-8165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doedens, J., L. A. Maynell, M. W. Klymkowsky, and K. Kirkegaard. 1994. Secretory pathway function, but not cytoskeletal integrity, is required in poliovirus infection. Arch. Virol. Suppl. 9**:**159-172. [DOI] [PubMed] [Google Scholar]
- 25.Doedens, J. R., T. H. Giddings, Jr., and K. Kirkegaard. 1997. Inhibition of endoplasmic reticulum-to-Golgi traffic by poliovirus protein 3A: genetic and ultrastructural analysis. J. Virol. 71**:**9054-9064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doedens, J. R., and K. Kirkegaard. 1995. Inhibition of cellular protein secretion by poliovirus proteins 2B and 3A. EMBO J. 14**:**894-907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Donaldson, J. G., H. Radhakrishna, and P. J. Peters. 1995. The ARF GTPases: defining roles in membrane traffic and organelle structure. Cold Spring Harbor Symp. Quant. Biol. 60**:**229-234. [DOI] [PubMed] [Google Scholar]
- 28.Elazar, Z., L. Orci, J. Ostermann, M. Amherdt, G. Tanigawa, and J. E. Rothman. 1994. ADP-ribosylation factor and coatomer couple fusion to vesicle budding. J. Cell Biol. 124**:**415-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fensome, A., E. Cunningham, O. Troung, and S. Cockcroft. 1994. ARF1(2-17) does not specifically interact with ARF1-dependent pathways. Inhibition by peptide of phospholipases C beta, D and exocytosis in HL60 cells. FEBS Lett. 349**:**34-38. [DOI] [PubMed] [Google Scholar]
- 30.Fogg, M. H., N. L. Teterina, and E. Ehrenfeld. 2003. Membrane requirements for uridylylation of the poliovirus VPg protein and viral RNA synthesis in vitro. J. Virol. 77**:**11408-11416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gamarnik, A. V., and R. Andino. 1998. Switch from translation to RNA replication in a positive-stranded RNA virus. Genes Dev. 12**:**2293-2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gazina, E. V., J. M. Mackenzie, R. J. Gorrell, and D. A. Anderson. 2002. Differential requirements for COPI coats in formation of replication complexes among three genera of Picornaviridae. J. Virol. 76**:**11113-11122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Herold, J., and R. Andino. 2000. Poliovirus requires a precise 5′ end for efficient positive-strand RNA synthesis. J. Virol. 74**:**6394-6400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Irurzun, A., L. Perez, and L. Carrasco. 1992. Involvement of membrane traffic in the replication of poliovirus genomes: effects of brefeldin A. Virology 191**:**166-175. [DOI] [PubMed] [Google Scholar]
- 35.Izumi, R. E., B. Valdez, R. Banerjee, M. Srivastava, and A. Dasgupta. 2001. Nucleolin stimulates viral internal ribosome entry site-mediated translation. Virus Res. 76**:**17-29. [DOI] [PubMed] [Google Scholar]
- 36.Jore, J., B. De Geus, R. J. Jackson, P. H. Pouwels, and B. E. Enger-Valk. 1988. Poliovirus protein 3CD is the active protease for processing of the precursor protein P1 in vitro. J. Gen. Virol. 69**:**1627-1636. [DOI] [PubMed] [Google Scholar]
- 37.Ktistakis, N. T., H. A. Brown, M. G. Waters, P. C. Sternweis, and M. G. Roth. 1996. Evidence that phospholipase D mediates ADP ribosylation factor-dependent formation of Golgi coated vesicles. J. Cell Biol. 134**:**295-306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liang, J. O., and S. Kornfeld. 1997. Comparative activity of ADP-ribosylation factor family members in the early steps of coated vesicle formation on rat liver Golgi membranes. J. Biol. Chem. 272**:**4141-4148. [DOI] [PubMed] [Google Scholar]
- 39.Lippincott-Schwartz, J., N. B. Cole, and J. G. Donaldson. 1998. Building a secretory apparatus: role of ARF1/COPI in Golgi biogenesis and maintenance. Histochem. Cell Biol. 109**:**449-462. [DOI] [PubMed] [Google Scholar]
- 40.Maynell, L. A., K. Kirkegaard, and M. W. Klymkowsky. 1992. Inhibition of poliovirus RNA synthesis by brefeldin A. J. Virol. 66**:**1985-1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McBride, A. E., A. Schlegel, and K. Kirkegaard. 1996. Hum. protein Sam68 relocalization and interaction with poliovirus RNA polymerase in infected cells. Proc. Natl. Acad. Sci. USA 93**:**2296-2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Molla, A., A. V. Paul, and E. Wimmer. 1991. Cell-free, de novo synthesis of poliovirus. Science 254**:**1647-1651. [DOI] [PubMed] [Google Scholar]
- 43.Morinaga, N., Y. Kaihou, N. Vitale, J. Moss, and M. Noda. 2001. Involvement of ADP-ribosylation factor 1 in cholera toxin-induced morphological changes of Chinese hamster ovary cells. J. Biol. Chem. 276**:**22838-22843. [DOI] [PubMed] [Google Scholar]
- 44.Morinaga, N., S. C. Tsai, J. Moss, and M. Vaughan. 1996. Isolation of a brefeldin A-inhibited guanine nucleotide-exchange protein for ADP ribosylation factor (ARF) 1 and ARF3 that contains a Sec7-like domain. Proc. Natl. Acad. Sci. USA 93**:**12856-12860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morrow, C. D., G. F. Gibbons, and A. Dasgupta. 1985. The host protein required for in vitro replication of poliovirus is a protein kinase that phosphorylates eukaryotic initiation factor-2. Cell 40**:**913-921. [DOI] [PubMed] [Google Scholar]
- 46.Moss, J., and M. Vaughan. 1998. Molecules in the ARF orbit. J. Biol. Chem. 273**:**21431-21434. [DOI] [PubMed] [Google Scholar]
- 47.Neznanov, N., A. Kondratova, K. M. Chumakov, B. Angres, B. Zhumabayeva, V. I. Agol, and A. V. Gudkov. 2001. Poliovirus protein 3A inhibits tumor necrosis factor (TNF)-induced apoptosis by eliminating the TNF receptor from the cell surface. J. Virol. 75**:**10409-10420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nie, Z., D. S. Hirsch, and P. A. Randazzo. 2003. Arf and its many interactors. Curr. Opin. Cell Biol. 15**:**396-404. [DOI] [PubMed] [Google Scholar]
- 49.Ooi, C. E., E. C. Dell'Angelica, and J. S. Bonifacino. 1998. ADP-ribosylation factor 1 (ARF1) regulates recruitment of the AP-3 adaptor complex to membranes. J. Cell Biol. 142**:**391-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Orcl, L., D. J. Palmer, M. Amherdt, and J. E. Rothman. 1993. Coated vesicle assembly in the Golgi requires only coatomer and ARF proteins from the cytosol. Nature 364**:**732-734. [DOI] [PubMed] [Google Scholar]
- 51.Ostermann, J., L. Orci, K. Tani, M. Amherdt, M. Ravazzola, Z. Elazar, and J. E. Rothman. 1993. Stepwise assembly of functionally active transport vesicles. Cell 75**:**1015-1025. [DOI] [PubMed] [Google Scholar]
- 52.Parsley, T. B., J. S. Towner, L. B. Blyn, E. Ehrenfeld, and B. L. Semler. 1997. Poly(rC) binding protein 2 forms a ternary complex with the 5′-terminal sequences of poliovirus RNA and the viral 3CD proteinase. RNA 3**:**1124-1134. [PMC free article] [PubMed] [Google Scholar]
- 53.Paul, A. V., E. Rieder, D. W. Kim, J. H. van Boom, and E. Wimmer. 2000. Identification of an RNA hairpin in poliovirus RNA that serves as the primary template in the in vitro uridylylation of VPg. J. Virol. 74**:**10359-10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Peters, P. J., V. W. Hsu, C. E. Ooi, D. Finazzi, S. B. Teal, V. Oorschot, J. G. Donaldson, and R. D. Klausner. 1995. Overexpression of wild-type and mutant ARF1 and ARF6: distinct perturbations of nonoverlapping membrane compartments. J. Cell Biol. 128**:**1003-1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Peyroche, A., B. Antonny, S. Robineau, J. Acker, J. Cherfils, and C. L. Jackson. 1999. Brefeldin A acts to stabilize an abortive ARF-GDP-Sec7 domain protein complex: involvement of specific residues of the Sec7 domain. Mol. Cell 3**:**275-285. [DOI] [PubMed] [Google Scholar]
- 56.Purhonen, P., K. Pursiainen, and H. Reunanen. 1997. Effects of brefeldin A on autophagy in cultured rat fibroblasts. Eur. J. Cell Biol. 74**:**63-67. [PubMed] [Google Scholar]
- 57.Radhakrishna, H., and J. G. Donaldson. 1997. ADP-ribosylation factor 6 regulates a novel plasma membrane recycling pathway. J. Cell Biol. 139**:**49-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Randazzo, P. A., Z. Nie, K. Miura, and V. W. Hsu. 2000. Molecular aspects of the cellular activities of ADP-ribosylation factors. Sci STKE 2000**:**RE1. [DOI] [PubMed] [Google Scholar]
- 59.Roth, M. G., K. Bi, N. T. Ktistakis, and S. Yu. 1999. Phospholipase D as an effector for ADP-ribosylation factor in the regulation of vesicular traffic. Chem. Phys. Lipids 98**:**141-152. [DOI] [PubMed] [Google Scholar]
- 60.Rust, R. C., L. Landmann, R. Gosert, B. L. Tang, W. Hong, H. P. Hauri, D. Egger, and K. Bienz. 2001. Cellular COPII proteins are involved in production of the vesicles that form the poliovirus replication complex. J. Virol. 75**:**9808-9818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sandoval, I. V., and L. Carrasco. 1997. Poliovirus infection and expression of the poliovirus protein 2B provoke the disassembly of the Golgi complex, the organelle target for the antipoliovirus drug Ro-090179. J. Virol. 71**:**4679-4693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sata, M., J. G. Donaldson, J. Moss, and M. Vaughan. 1998. Brefeldin A-inhibited guanine nucleotide-exchange activity of Sec7 domain from yeast Sec7 with yeast and mammalian ADP ribosylation factors. Proc. Natl. Acad. Sci. USA 95**:**4204-4208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sata, M., J. Moss, and M. Vaughan. 1999. Structural basis for the inhibitory effect of brefeldin A on guanine nucleotide-exchange proteins for ADP-ribosylation factors. Proc. Natl. Acad. Sci. USA 96**:**2752-2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schlegel, A., T. H. Giddings, Jr., M. S. Ladinsky, and K. Kirkegaard. 1996. Cellular origin and ultrastructure of membranes induced during poliovirus infection. J. Virol. 70**:**6576-6588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Serafini, T., L. Orci, M. Amherdt, M. Brunner, R. A. Kahn, and J. E. Rothman. 1991. ADP-ribosylation factor is a subunit of the coat of Golgi-derived COP-coated vesicles: a novel role for a GTP-binding protein. Cell 67**:**239-253. [DOI] [PubMed] [Google Scholar]
- 66.Spang, A. 2002. ARF1 regulatory factors and COPI vesicle formation. Curr. Opin. Cell Biol. 14**:**423-427. [DOI] [PubMed] [Google Scholar]
- 67.Stamnes, M., G. Schiavo, G. Stenbeck, T. H. Sollner, and J. E. Rothman. 1998. ADP-ribosylation factor and phosphatidic acid levels in Golgi membranes during budding of coatomer-coated vesicles. Proc. Natl. Acad. Sci. USA 95**:**13676-13680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stamnes, M. A., and J. E. Rothman. 1993. The binding of AP-1 clathrin adaptor particles to Golgi membranes requires ADP-ribosylation factor, a small GTP-binding protein. Cell 73**:**999-1005. [DOI] [PubMed] [Google Scholar]
- 69.Suhy, D. A., T. H. Giddings, Jr., and K. Kirkegaard. 2000. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: an autophagy-like origin for virus-induced vesicles. J. Virol. 74**:**8953-8965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Taylor, T. C., M. Kanstein, P. Weidman, and P. Melancon. 1994. Cytosolic ARFs are required for vesicle formation but not for cell-free intra-Golgi transport: evidence for coated vesicle-independent transport. Mol. Biol. Cell 5**:**237-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Teal, S. B., V. W. Hsu, P. J. Peters, R. D. Klausner, and J. G. Donaldson. 1994. An activating mutation in ARF1 stabilizes coatomer binding to Golgi membranes. J. Biol. Chem. 269**:**3135-3138. [PubMed] [Google Scholar]
- 72.Teterina, N. L., A. E. Gorbalenya, D. Egger, K. Bienz, and E. Ehrenfeld. 1997. Poliovirus 2C protein determinants of membrane binding and rearrangements in mammalian cells. J. Virol. 71**:**8962-8972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Teterina, N. L., W. D. Zhou, M. W. Cho, and E. Ehrenfeld. 1995. Inefficient complementation activity of poliovirus 2C and 3D proteins for rescue of lethal mutations. J. Virol. 69**:**4245-4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Thompson, A. A., and O. B. Peersen. 2004. Structural basis for proteolysis-dependent activation of the poliovirus RNA-dependent RNA polymerase. EMBO J. 23**:**3462-3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Traub, L. M., J. A. Ostrom, and S. Kornfeld. 1993. Biochemical dissection of AP-1 recruitment onto Golgi membranes. J. Cell Biol. 123**:**561-573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Troxler, M., D. Egger, T. Pfister, and K. Bienz. 1992. Intracellular localization of poliovirus RNA by in situ hybridization at the ultrastructural level using single-stranded riboprobes. Virology 191**:**687-697. [DOI] [PubMed] [Google Scholar]
- 77.Tsai, S. C., R. Adamik, R. S. Haun, J. Moss, and M. Vaughan. 1993. Effects of brefeldin A and accessory proteins on association of ADP-ribosylation factors 1, 3, and 5 with Golgi. J. Biol. Chem. 268**:**10820-10825. [PubMed] [Google Scholar]
- 78.Vasudevan, C., W. Han, Y. Tan, Y. Nie, D. Li, K. Shome, S. C. Watkins, E. S. Levitan, and G. Romero. 1998. The distribution and translocation of the G protein ADP-ribosylation factor 1 in live cells is determined by its GTPase activity. J. Cell Sci. 111**:**1277-1285. [DOI] [PubMed] [Google Scholar]
- 79.Verlinden, Y., A. Cuconati, E. Wimmer, and B. Rombau. 2002. The viral protein 3CD induces an equilibrium between the viral protein and RNA synthesis in a cell-free system for poliovirus replication. Arch. Virol. 147**:**731-744. [DOI] [PubMed] [Google Scholar]
- 80.Waggoner, S., and P. Sarnow. 1998. Viral ribonucleoprotein complex formation and nucleolar-cytoplasmic relocalization of nucleolin in poliovirus-infected cells. J. Virol. 72**:**6699-6709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.West, M. A., N. A. Bright, and M. S. Robinson. 1997. The role of ADP-ribosylation factor and phospholipase D in adaptor recruitment. J. Cell Biol. 138**:**1239-1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xiang, W., K. S. Harris, L. Alexander, and E. Wimmer. 1995. Interaction between the 5′-terminal cloverleaf and 3AB/3CDpro of poliovirus is essential for RNA replication. J. Virol. 69**:**3658-3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ypma-Wong, M. F., P. G. Dewalt, V. H. Johnson, J. G. Lamb, and B. L. Semler. 1988. Protein 3CD is the major poliovirus proteinase responsible for cleavage of the P1 capsid precursor. Virology 166**:**265-270. [DOI] [PubMed] [Google Scholar]