Bax forms multispanning monomers that oligomerize to permeabilize membranes during apoptosis (original) (raw)
Abstract
Bax promotes cell death by permeabilizing mitochondrial outer membranes by an unresolved mechanism. However, in cells lacking the gene c-myc, membrane permeabilization by Bax is blocked by changes in the mitochondria that prevent Bax oligomerization. Drug-treated c-myc null cells and cells expressing Myc were used to map the topology of Bax in membranes prior to and after mitochondrial permeabilization. Chemical labeling of single cysteine mutants of Bax using a membrane bilayer impermeant cysteine-specific modifying agent revealed that Bax inserted both the ‘pore domain' (helices α5–α6), and the tail-anchor (helix α9) into membranes prior to oligomerization and membrane permeabilization. Additional topology changes for Bax were not required in Myc-expressing cells to promote oligomerization and cytochrome c release. Our results suggest that unlike most pore-forming proteins, Bax membrane permeabilization results from oligomerization of transmembrane monomers rather than concerted insertion of the pore domains of a preformed oligomer.
Keywords: apoptosis, Bax, membrane topology, membrane permeabilization, Myc
Introduction
The fate of an individual cell is controlled by several factors including the regulation of survival by the Bcl-2 family of apoptosis regulators. Although different Bcl-2 family proteins have been shown to regulate cell death positively or negatively, the precise molecular mechanisms involved remain controversial. An important insight that emerged from the observation that many Bcl-2 proteins require membrane localization to function is that the modulation of membrane properties is crucial to the regulation of apoptosis (Wolter et al, 1997).
The apoptosis-promoting protein Bax remains in the cytoplasm until the induction of cell death, despite containing a predicted tail-anchor sequence expected to bind the protein constitutively to membranes. The structure of the soluble form of Bax revealed that in the nonactivated state, the putative tail-anchor sequence is sequestered in a hydrophobic binding pocket on the surface of the protein (Suzuki et al, 2000). Targeting to mitochondria during cell death requires Bax to be activated via a transient interaction with a BH3 (Bcl-2 homology 3) protein such as tBid that induces a conformational change in Bax that is presumed to expose the tail-anchor sequence. For Bax to selectively target to the outer mitochondrial membrane, the tail-anchor sequence is required (Wolter et al, 1997; Nechushtan et al, 1999). In addition, the central α5 and α6 helices (Nouraini et al, 2000) and regions of the amino-terminus (Goping et al, 1998) contribute to the regulation of Bax membrane binding. Typically, translocation of roughly 20% of the cellular Bax from the cytoplasm to the mitochondrial outer membrane is sufficient to induce apoptosis (Annis et al, 2001). On mitochondrial membranes, Bax permeabilizes membranes either by forming a pore or by modifying an existing pore, resulting in the release of several apoptotic factors including cytochrome c and Smac/Diablo (Antonsson et al, 2000; Kuwana et al, 2002; Arnoult et al, 2003; Belzacq et al, 2003). By itself, Bax can permeabilize a variety of other membranes including liposomes (Kuwana et al, 2002) in which large oligomers of Bax have been visualized (Epand et al, 2002).
Membrane permeabilization has been proposed to occur by Bax oligomerization prior to or concomitant with insertion of a region of the protein (α5–α6) identified as a putative pore-forming domain based on the structural similarity of this region with the pore-forming domain of diphtheria toxin. Although structurally similar, the importance of oligomerization, the size of oligomers and the mechanism by which pores are formed by diphtheria toxin remain uncertain (Sharpe and London, 1999; Steere and Eisenberg, 2000). Colicin, another protein structurally related to Bax, inserts into membranes by a well-defined mechanism but the ion channels formed are much smaller than the pores formed or opened by Bax (Lindeberg et al, 2000) and the mechanism is not consistent with what is known about the biochemical properties of Bax. Although structurally unrelated, Perfringolysin O forms large pores in membranes by a well-defined three-step process: binding of the toxin to the membrane is followed by oligomerization on the surface of the membrane and then a concerted conformational change results in pore formation (Heuck et al, 2000). In this case, oligomerization prior to pore formation is believed to be necessary in order that insertion of the protein into the membrane can rearrange sufficient membrane lipids to form a large pore. In contrast, based on experiments using liposomes and purified proteins, the formation of lipidic pores has been proposed as yet another mechanism for large pore formation in this case by Bax together with a large molar excess of tBid (Kuwana et al, 2002; Terrones et al, 2004). Neither of these models is directly pertinent to membrane permeabilization by Bax in cells in which release of intermembrane space proteins by Bax or Bak can be induced by 100-fold substoichiometric quantities of tBid (Ruffolo and Shore, 2003).
In rat fibroblasts, both the conformational change associated with Bax activation (detected by the monoclonal antibody 6A7) and subsequent release of cytochrome c require expression of the proto-oncogene c-myc: in the absence of c-myc (in HO15.19 cells, referred to here as Myc−/− cells), etoposide treatment caused ∼40% of the cellular Bax to translocate to membranes; however, the 6A7 epitope was not exposed and cytochrome c was not released (Soucie et al, 2001). When Myc was expressed constitutively in isogenic cells (HOmyc3 cells, referred to here as Myc+ cells), etoposide treatment induced Bax translocation (∼30%), activation and release of cytochrome c (Soucie et al, 2001). Inhibition of membrane permeabilization by Bax in Myc−/− cells could be due to either a direct modification of Bax (either by binding to another protein or post-translational modification) or a change in the mitochondrial membrane that arrests Bax prior to permeabilization but subsequent to membrane binding. In either case, comparison of Bax in Myc−/− and Myc+ cell lines can be used to examine the order of events and the mechanism of membrane permeabilization by Bax in drug-treated cells. Our results suggest that in Myc−/− cells, Bax integrates into membranes as a multispanning membrane protein with helices 5–6 and 9 in the bilayer. These Bax monomers are sufficiently proximal that they can be crosslinked in membranes from Myc−/− cells but the protein migrates as a monomer when membranes are solubilized with CHAPS. Thus, in Myc−/− cells, the conformational change that results in the formation of CHAPS-resistant oligomers is blocked and membrane permeability remains intact. This mechanism is most consistent with membrane permeabilization in cells resulting from pores formed by the oligomerization of membrane-embedded Bax monomers rather than concerted insertion of the ‘pore-forming' domains of preformed Bax oligomers.
Results and discussion
We reported previously that when Myc−/− cells were treated with a variety of apoptotic agonists, Bax migrated to mitochondria but cytochrome c was not released (Soucie et al, 2001). To determine whether the mitochondria from Myc−/− cells are resistant to permeabilization by recombinant (therefore unmodified) Bax, cytochrome c release was assayed by immunoblotting isolated mitochondria treated with recombinant Bax with and without activation by recombinant tBid. As shown in Figure 1, mitochondria from Myc−/− cells are dramatically more resistant to cytochrome c release induced by both Bax and tBid than are mitochondria from Myc+ cells. Both Bax and tBid are required for cytochrome c release, as membrane permeabilization was not observed when mitochondria were incubated with 500 nM Bax or 8 nM tBid individually (Supplementary Figure 1). Cytochrome c is retained in the pellet rather than being released into the supernatant of heavy membrane fractions prepared from Myc−/− cells treated with 100 nM Bax and 1.7 nM tBid (Figure 1A, sample 4), concentrations sufficient to trigger complete cytochrome c release from Myc+ cell mitochondria (Figure 1B, sample 4). Indeed at 100 nM Bax, 0.4 nM tBid is sufficient to induce almost complete release of cytochrome c from Myc+ cell mitochondria (Supplementary Figure 1). In contrast, cytochrome c was only partially released from Myc−/− mitochondria even after incubation with 8 nM tBid and 500 nM Bax, with full release requiring 80 nM tBid at this Bax concentration (Supplementary Figure 1). Moreover, cytosol switching experiments demonstrated that inhibition of Bax pore formation is inherent to the mitochondria rather than being conferred by something in the cytosol of either cell line. Thus, in incubations containing cytosol from Myc+ cells, Bax and tBid, there was no detectable cytochrome c released from Myc−/− cell mitochondria (Figure 1A, sample 3). Conversely, Myc−/− cell lysate did not inhibit tBid- and/or Bax-mediated release of cytochrome c from Myc+ cell mitochondria (Figure 1B, compare samples 3 and 4 with 5 and 6). Mitochondria isolated from Rat-1 fibroblasts that have not been drug treated contain no Bid (Soucie et al, 2001 and data not shown), and the slight increase in endogenous Bax in the heavy membrane fraction of Myc+ cells compared to Myc−/− cells (1.5-fold increase in Myc+ mitochondria; Supplementary Figure 2) does not account for the more than 20-fold increase in sensitivity of the Myc+ cells to recombinant Bax when combined with the same amount of tBid (Supplementary Figure 1). Taken together, these data strongly suggest that the defect in Myc−/− cells is intrinsic to the mitochondria, and that these mitochondria either contain dramatically reduced amounts of a Myc-regulated component necessary for formation of pores by Bax or contain a component that inhibits cytochrome c release by arresting pore formation. To examine this latter alternative, we compared the expression of Bcl-XL and Bcl-2 in the heavy membrane fraction of Myc+ and Myc−/− cells. While we detected no difference in the amount of membrane-bound Bcl-XL, mitochondria from Myc−/− cells contained 1.6 times more Bcl-2 (Supplementary Figure 2). The magnitude of this difference is not large enough to account for the marked difference in sensitivity to recombinant Bax that we detected (Supplementary Figure 1). Thus, additional factors prevent Bax-mediated permeabilization of mitochondrial outer membranes. Nevertheless, in Myc−/− cells, Bax conformational changes are blocked subsequent to membrane binding but prior to membrane permeabilization, affording the opportunity of examining the mechanism of permeabilization in cells rather than cell-free systems. The elucidation of the topography of Bax in the membranes of mitochondria from Myc−/− cells is therefore critical to understanding the regulation of apoptosis.
Figure 1.
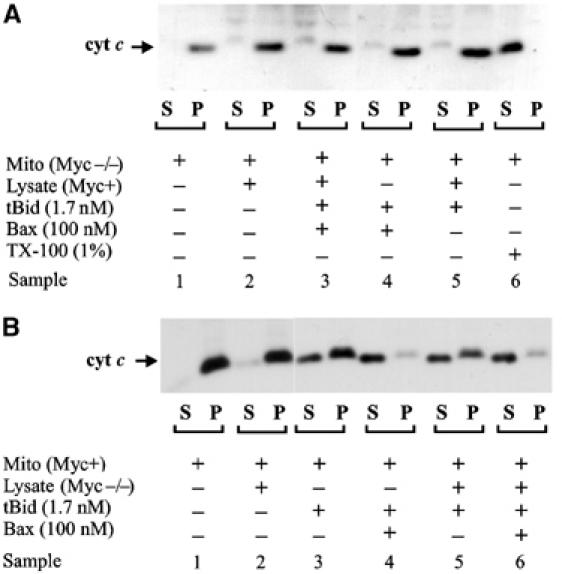
Mitochondria from Myc−/− cells are more resistant than mitochondria from Myc+ cells to induction of cytochrome c release by 100 nM Bax and 1.7 nM tBid. Mitochondria isolated from (A) Myc−/− or (B) Myc+ cells were incubated with tBid, Bax and cytosol as indicated. Cytochrome c release was assayed by pelleting the mitochondria and visualizing the cytochrome c in the supernatant (S) and pellet (P) fractions by SDS–PAGE and immunoblotting. As a control, cytochrome c was released from mitochondria by solubilization using the nonionic detergent Triton X-100 (TX-100).
Similar to Bcl-2 and other tail-anchor proteins, prior to pore formation, Bax is predicted to adopt a tail-anchored topology in which only helix 9 is embedded in the membrane (Figure 2A). Based on the structural similarity of Bax with colicin and diphtheria toxin, insertion of the α5–α6 region of Bax into the membrane is believed to be necessary for Bax to form a pore and release cytochrome c from mitochondria (Figure 2A, pore forming). A precedent for such a topological change exists for Bcl-2, which initially binds to membranes with a typical tail-anchor topology, but upon induction of apoptosis it undergoes a conformational change in the pore-forming domain (Kim et al, 2004).
Figure 2.
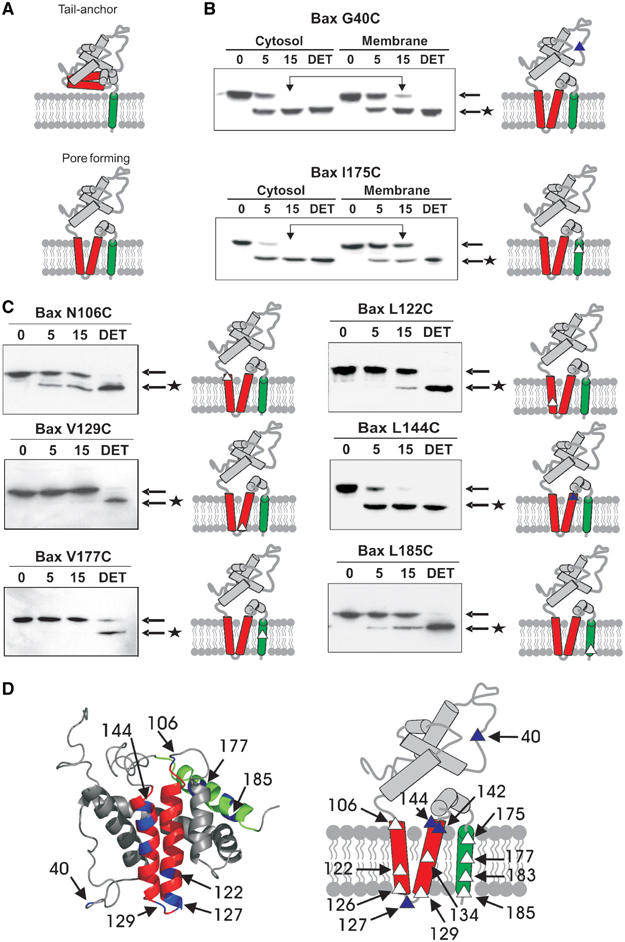
In Myc−/− cells, both the pore-forming and tail-anchor domains of membrane-bound Bax are embedded in the bilayer. (A) Membrane-bound Bax is predicted to adopt either a tail-anchor or pore-forming topology. Helices are diagrammed as solid rods, the tail-anchor (helix 9) is green and the ‘pore-forming' helices (5–6) are red. (B, C) Cytosolic and membrane fractions isolated from etoposide-treated Myc−/− cells expressing the indicated mutants were labeled with IASD (time indicated above the lanes) and labeling was assessed by isoelectric focusing and immunoblotting. Arrows indicate the migration positions of Bax and * indicates IASD-labeled Bax. The location of the single cysteine is indicated as a triangle on the diagram at the right of each panel. Overhead arrows indicate bands representing residues protected from IASD. (B) Labeling of cytoplasmic (Bax G40C) and integrated (Bax I175C) controls. In cytosol and membrane fractions, Bax G40C was labeled by IASD. The cytosolic form of Bax I175C was labeled by IASD, but in membrane fractions this residue was protected from labeling until the bilayer was solubilized with detergent (DET). (C) Labeling reactions for membrane fractions from Myc−/− cells expressing the indicated single cysteine Bax mutants. Interpretive diagrams are presented to the right of each panel. (D) Comparison of the solution structure and the membrane topology of Bax in etoposide-treated Myc−/− and Myc+ cells. Blue residues on the solution structure (left) indicate positions of single cysteine mutants. In the topology diagram (right), closed triangles represent residues modified similarly by IASD in cytosolic and membrane-bound Bax. Open triangles represent residues that were protected from IASD by the membrane.
To determine the membrane topology of Bax in apoptotic cells, cell fractions were prepared from etoposide-treated cells and labeled with the charged cysteine-modifying chemical 4-acetamido-4′-((iodoacetyl) amino) stilbene-2,2′-disulfonic acid (IASD). This reagent will not label cysteine residues located within the bilayer, but efficiently labels residues on the cytoplasmic side of membrane fractions and can label residues located on proteins that are within microsomes or mitochondria by passing through channels in membranes (Kim et al, 2004; Supplementary Figure 3). Thus, IASD labeling adds two negative charges to a membrane protein if the cysteine is located in the cytoplasm, on the luminal side of membranes or within an aqueous channel, but not if the residue is within the bilayer. Therefore, Bax topology can be mapped from cell lysates by assaying the accessibility of residues to IASD by SDS–PAGE on ultra-high-resolution gels (Kim et al, 2004) or more conveniently by isoelectric focusing and immunoblotting, as described in Supplementary data.
The two endogenous cysteine residues in human Bax (amino-acid positions 62 and 126) were replaced with alanine to generate a cysteine-less mutant, from which additional Bax mutants were generated that each contain a single cysteine located at positions distributed throughout the putative pore-forming domain (helices α5–α6). As controls, separate mutants were constructed in which cysteines were positioned in a region predicted to remain exposed to the cytoplasm or within the putative membrane anchor (helix α9). The single cysteine mutants of human Bax were detected after retroviral expression in rat fibroblasts by using a human-specific monoclonal antibody (2D2). The mutants behaved similarly to the endogenous rat Bax in that they were cytoplasmic in growing cells but translocated to membranes following an apoptotic stimulus (Soucie et al, 2001 and data not shown). Moreover, the Bax mutants retained membrane permeabilizing activity in a cell-free assay similar to that shown in Figure 1 in which recombinant mutant Bax protein and the Bax-activating protein tBid were jointly necessary and sufficient to release cytochrome c from isolated mitochondria (data not shown).
To map individual cysteine residues, the extent of IASD labeling of Bax was compared for cytosol and membrane fractions prepared from cells after exposure to 6 μM etoposide for sufficient time to induce translocation of 30–40% of the total cellular Bax in Myc+ and Myc−/− cells. To confirm that protection from IASD was due to the bilayer, detergent (2% CHAPS, 4% IGEPAL) was added to the indicated control reactions to solubilize membranes and expose embedded cysteine residues to IASD. A large increase in labeling of a cysteine after detergent solubilization demonstrated that protection from labeling was due to the lipid bilayer.
A cysteine positioned at Bax residue 40 was selected as an aqueous control as this residue is part of the unstructured loop between helix α1 and α2. As expected, this mutant was modified after 15 min exposure to IASD in both the cytosolic and membrane fractions (Figure 2B, compare bands corresponding to protected species indicated by overhead arrows). Position 175 in the putative tail-anchor of Bax (helix α9) was selected as a membrane-integrated control. In cytosolic fractions prepared from cells expressing this mutant, position 175 was labeled after 15 min exposure to IASD (a 5 min time point is also shown to indicate the extent of labeling at a shorter time (in all reactions labeling did not increase after 15 min; Kim et al, 2004)). However, for membrane-bound Bax protein, the cysteine was largely protected unless detergent (DET) was added to solubilize the membrane (Figure 2B, bands corresponding to protected species indicated by overhead arrows). Thus, this position of Bax is inserted into the lipid bilayer in cells (Figure 2D). The labeling reactions performed on membrane fractions for selected additional single cysteine mutants are shown in Figure 2C, and the results from all the mutants are summarized in Figure 3 and Supplementary Table 1. We were not able to test Bax mutants with a single cysteine at some positions due to low expression levels or difficulty in labeling the residue in cytosolic fractions, as indicated in Supplementary Table 1.
Figure 3.
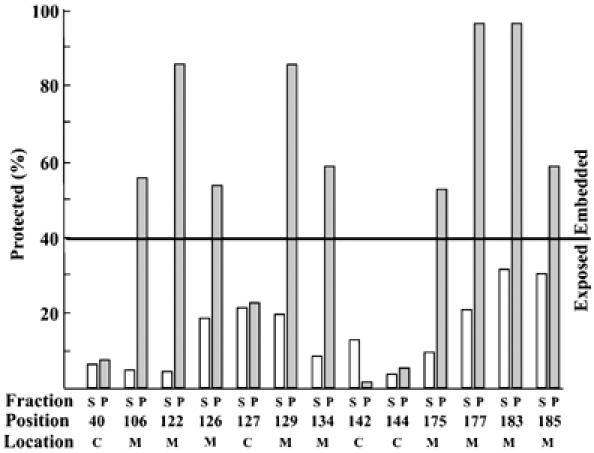
Quantification of labeling efficiency of Bax single cysteine mutants. Open and gray bars indicate supernatant (S) and membrane pellet (P) fractions from etoposide-treated cells, respectively. The position of the cysteine residue is indicated below the lanes. The deduced location of the residue is indicated as exposed to cytoplasm (C) or inserted into the membrane (M). The line at 40% protected represents an arbitrary cutoff that can be used to distinguish exposed cytoplasmic from membrane-embedded cysteines without reference to the supernatant fractions. The approximate error (standard deviation) for each measurement is 10%.
The primary criterion for assigning a residue as cytoplasmic or membrane embedded was the difference in labeling of cytosolic and membrane fractions of drug-treated cells. Where a residue is judged to be membrane inserted, the average protection from labeling of the cytosolic and membrane-bound protein was 17±10% (standard deviation) and 71±19%, respectively. In contrast, for residues judged to be cytoplasmic, protection of cytosolic and membrane-bound protein was 11±8 and 10±9%, respectively. For seven mutants, the expression of Bax was high enough that identification of the bands did not require exposing the blots to film and topology could be deduced after quantifying the extent of labeling using a Typhoon fluorimager to record the chemiluminescence signal from the blots. Quantification of labeling of these mutants revealed that on average membrane-embedded residues were 60±5% protected compared to 12±4% for cytosolic residues, in good agreement with measurements from film. Topology can also be ascribed for membrane fractions without reference to the cytosolic fractions by using 40% protection from labeling as an arbitrary threshold (Figure 3).
The multispanning membrane topology, with helices α5, α6 and α9 integrated into the lipid bilayer, was deduced for Bax by analyzing 13 mutants in etoposide-treated Myc−/− fibroblasts (Figure 2D). The unexpected observation of insertion of the ‘pore domain' of Bax as transmembrane sequences in the absence of cytochrome c release demonstrates that integration of this domain into membranes is not sufficient to permeabilize the outer membrane of mitochondria. The same residues were similarly protected or accessible to IASD in the presence of Myc expression (Myc+ cells) and cytochrome c release, suggesting that Bax adopts the same membrane topology in both cell lines (Supplementary Figure 4). The observation that the residues in helices 5–6 and 9 remain protected from IASD after membrane permeabilization suggests that either these residues do not line the interior of an aqueous pore (as predicted by models favoring lipidic pores; Kuwana et al, 2002) or that the environment of the pore prevents access of IASD or limits the chemical reactivity of the cysteines. The topology shown in Figure 2D represents the minimum number of membrane-embedded segments, as there are other areas of the protein that might also interact with the membrane that we have not yet investigated (Cartron et al, 2003; Garcia-Saez et al, 2004). While it is possible that one or more residues of Bax could be protected from labeling due to a tight interaction between Bax and another protein, binding to another protein would be unlikely to be both sensitive to nonionic detergents and also result in the exact pattern of labeling seen here. In particular, direct binding of the small amount of endogenous Bcl-2 or Bcl-XL found in Myc−/− mitochondria (Supplementary Figure 2) to the retrovirally expressed Bax mutants could not account for the degree of protection from IASD labeling we observed. Furthermore, in cytoplasmic Bax, 13 residues were accessible to the reagent, including several residues predicted from the NMR structure to be buried in the interior of the protein, confirming that IASD can access even the hydrophobic interior of cytoplasmic Bax. A few residues that were not labeled were apparently nonreactive due to local chemistry of the protein, as they were not labeled in either the cytoplasmic or membrane-bound versions of the protein. The residues in Bax that were protected from IASD only when Bax was bound to membranes were all accessible to the reagent when the bilayer was solubilized with nonionic detergents, demonstrating that IASD labels residues in detergent micelles. Based on these results and others published previously (Kim et al, 2004) we conclude that cysteine residues are differentially labeled by IASD between cytoplasmic and membrane versions of a protein when they are inserted into a bilayer. Finally, it is unlikely that binding to another protein masked the protected residues identically in both Myc−/− and Myc+ cells.
Our results suggest that cytochrome c release in cells is not regulated by changes in the membrane topology of Bax but that some other conformational change is required that is blocked in Myc−/− cells. To determine if Myc expression is required for Bax oligomerization, mitochondria were isolated from etoposide-treated Myc−/− and Myc+ cells, solubilized in CHAPS and analyzed by gel filtration chromatography. Consistent with the reported cytochrome _c_-releasing activity of Bax in cells expressing Myc, Bax formed higher molecular weight complexes following etoposide treatment (Figure 4). However, in the absence of Myc, Bax remained monomeric on the mitochondrial membrane after etoposide treatment (Figure 4A). To independently assess the oligomeric state of Bax in these cells and to detect Bax complexes that may not be stable in the detergent required for membrane solubilization prior to gel filtration, we performed crosslinking studies on cytosolic and membrane fractions of Myc−/− and Myc+ cells before and after exposure to etoposide. As expected, no crosslinking was observed for cytosolic Bax, consistent with the protein being monomeric (data not shown). After exposure to etoposide, Bax proteins in membrane fractions were sufficiently proximal to crosslink as dimers in both cell types. The very small amount of Bax on membranes before exposure to drug was also crosslinked by 1,6-bismaleimidohexane (BMH) (Figure 4B), indicating that these proteins are also in close proximity (and may be due to the small percentage of cells dying in culture). Thus, in Myc−/− cells treated with etoposide, membrane-bound Bax can be dimerized by the BMH crosslinker, but these complexes are not stable in the detergent conditions used for gel filtration chromatography. We found no evidence of hetero-oligomers between Bax and Bcl-XL in the membrane fraction, which would result in bands between those indicated to the right of the panels (Figure 4B) although Bax and Bcl-XL recombinant proteins can be readily crosslinked by BMH in vitro (data not shown). Taken together, our gel filtration chromatography and crosslinking experiments indicate that the defect in Bax activation in mitochondria from Myc−/− cells occurs at the stage of the formation of detergent-resistant oligomers. These complexes may result from further oligomerization of smaller complexes or from a conformational change that makes the oligomer resistant to detergent. Together with our IASD labeling experiments, this demonstrates that helices 5–6 and 9 of monomeric or detergent-sensitive oligomers and detergent-resistant oligomers of Bax adopt the same topology in the membrane.
Figure 4.
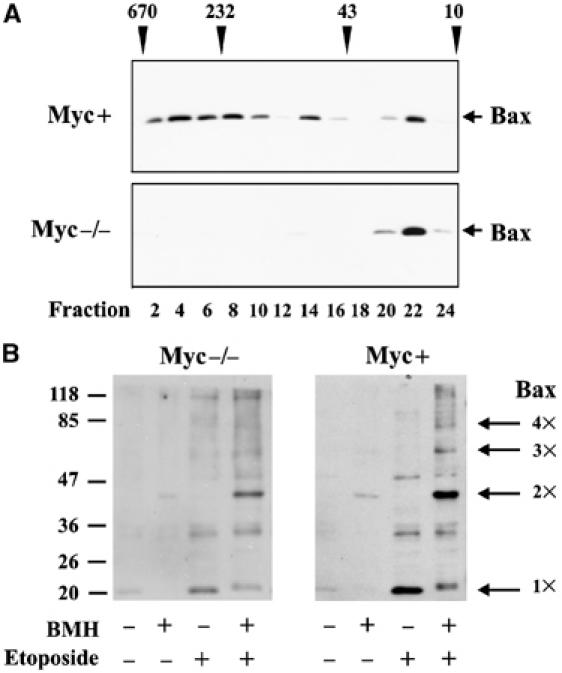
Myc expression is required for Bax oligomerization. Oligomerization of Bax was measured by (A) gel filtration chromatography and (B) crosslinking with BMH of membrane fractions. The membrane fraction was solubilized in CHAPS for chromatography. Fraction number and the migration positions of molecular weight standards are indicated below and above the immunoblots in (A). Crosslinking was performed in cell lysates prepared by nitrogen cavitation and then membrane fractions were collected by centrifugation. Migration positions for molecular weight standards are shown to the left of the panels. Deduced migration positions for Bax monomer and oligomers are indicated to the right of the panels in (B).
These observations suggest that unlike other pore-forming proteins for which integration into the membrane is sufficient to generate a pore, the critical event for Bax membrane permeabilization function is the transition from a membrane-integrated ‘monomer' to a detergent-resistant oligomeric form (Figure 4). Previously, we and others have demonstrated that a conformational change in Bax detected by the conformation-sensitive 6A7 antibody correlates with activation of Bax in cells (Duelli and Lazebnik, 2000; Soucie et al, 2001). The location of the epitope recognized by this antibody is near the amino-terminus of the protein, suggesting that conformational changes in this region of the protein regulate oligomerization. Recently, we have shown that Bax proteins that are not integrated into membranes can undergo the conformational change detected by this antibody in a reversible manner without oligomerizing (Yethon et al, 2003). Thus, it appears that the conformational change detected by 6A7 triggers Bax oligomerization only if the pore-forming domain has been exposed by prior integration of the protein into the membrane. Together, these results strongly suggest that during apoptosis, Bax permeabilizes membranes via discrete steps beginning with the integration of Bax monomers into outer mitochondrial membranes and followed by a conformational change at the amino-terminus resulting in oligomerization (Figure 5). We speculate that this results in pore formation in the plane of the bilayer that is actually driven by this oligomerization step (Figure 5). This mechanism for reorganizing the lipids to create a pore is different from the steps involved in pore formation by cytolytic toxins (Figure 5).
Figure 5.
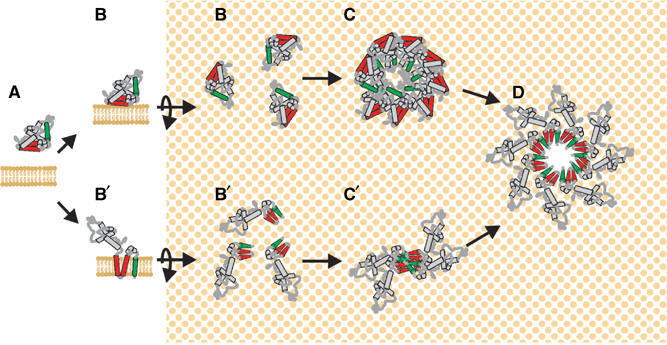
Comparison of pore-forming models for Bax. (A) In untreated cells, Bax is a cytoplasmic monomer. In the model based on cytolytic toxins, (B) Bax binds to membranes peripherally or by insertion of helix 9. (C) Membrane-bound monomers oligomerize to generate a structure capable of forming a pore in the membrane. (D) A concerted conformational change inserts helices 5–6 and 9 (unless helix 9 is already embedded) and reorganizes lipids to form a large hole in the membrane. In the oligomerization within the membrane model, based on the data presented here, (B′) Bax inserts helices 5–6 and 9 into the bilayer as a monomer. (C′) Monomers undergo a conformational change at the amino-terminus initiating oligomerization. In Myc−/− cells, the process is blocked at this step. (D) Ongoing oligomerization reorganizes the bilayer to form a large hole in the membrane. For simplicity, the pore formed here is depicted as arising only from helices 5–6 and 9; however, other regions of the protein and lipid structures may also contribute to membrane permeabilization. The number of Bax molecules per pore is arbitrary and was chosen for illustrative purposes only.
In the model in Figure 5, the pore is shown to be composed exclusively of Bax protein to permit comparison of the steps identified here with those of cytolytic toxins. However, because the mechanism of membrane permeabilization is unlike that of better-characterized pores, it is possible that lipids or other membrane proteins that form pre-existing channels are a major component of pore structure as hypothesized previously (Marzo et al, 1998; Epand et al, 2002; Kuwana et al, 2002; Terrones et al, 2004). The identification of these discrete steps in pore formation by Bax will hopefully provide additional avenues for pharmacologic intervention to either accelerate apoptosis in tumors or inhibit apoptosis in diseases such as stroke.
Materials and methods
Single cysteine Bax mutants
Mutations were made to the cDNA sequence for cysteine-less human Bax (pMac-1529) using Quick-Change™ mutagenesis (Stratagene). The standard nomenclature for additional mutations made in this construct is the original residue in single-letter code followed by the position in the amino-acid sequence and then the single-letter code for the new residue. The following mutants were used in this study: Bax G40C, N106C, L122C, 126C, T127C, V129C, R134C, D142C, L144C, I175C, V177C, A183C and L185C. Bax 126C is a mutant in which one of the two endogenous cysteines was converted to alanine leaving only the cysteine at position 126. After mutagenesis, all constructs were sequenced and the Bax coding sequence was excised using _Bgl_II and _Eco_RI restriction enzymes. The resulting fragment was gel purified and subcloned into the _Bam_HI and _Eco_RI sites of pBABE-mnIRES-GFP (pMac-1248; GFP: green fluorescence protein). These constructs were sequenced using primers MAC-781 and MAC-782. The resulting constructs were then packaged and the virus used to infect the Myc−/− and Myc+ cells. Cells expressing GFP were selected using a fluorescence activated cell sorter and cell lines established from the pooled populations.
Preparation of subcellular fractions from cell extracts by nitrogen cavitation
For Myc+ and Myc−/− cells, 10 and 20 100 mm Petri dishes of cultured cells were harvested, respectively. The cells were pelleted by centrifugation in a clinical centrifuge for 3 min at 4°C. The cell pellet was washed twice with cell buffer (250 mM sucrose, 20 mM HEPES pH 7.5, 2 mM MgCl2, 1 mM Na EDTA, 1 mM PMSF, 1 mM DTT and protease inhibitors (0.2 μg/ml of chymostatin, antipain, leupeptin and pepstatin and 0.4 μg/ml of aprotinin). The final pellet was resuspended in an equal volume of cell buffer. This suspension was held at 150 psi for 15 min on ice in a 45 ml Nitrogen Bomb (Parr Instruments) (Annis et al, 2001), and cells were disrupted by releasing the pressure. The nuclei and cell debris in the expelled lysate were removed by centrifugation at 500 g for 2 min in a microcentrifuge (Eppendorf). The resulting supernatant was split into a post-heavy membrane supernatant (S1) and pellet (P1) fraction enriched in mitochondria by centrifugation at 1000 g for 25 min in a Beckman TLA100 rotor at 4°C (Annis et al, 2001). Pellet fractions were resuspended in cell buffer snap-frozen in liquid nitrogen and stored at −80°C. Protein in samples was quantified using a standard Bradford assay (Bio-Rad).
Purification of tBid and Bax
Bid was expressed and purified as described previously (Desagher et al, 1999). The purified protein was cleaved to tBid using His-tagged caspase 8 (Biomol) and purified as published (Zha et al, 2000). This procedure yields highly purified tBid without contaminating pro-peptide or additional amino acids from a tag sequence. Full-length untagged Bax was expressed and purified as described (Yethon et al, 2003).
Cytochrome c release assay
Heavy membranes enriched in mitochondria were isolated essentially as described by Eskes et al (2000). Cells were harvested, washed twice in mitochondrial buffer (MB) (210 mM mannitol, 70 mM sucrose, 1 mM EDTA, 10 mM HEPES, pH 7.5), resuspended in MB supplemented with Complete protease inhibitor cocktail (Roche) and lysed by nitrogen cavitation at 150 psi for 15 min on ice in a 45 ml Nitrogen Bomb (Parr Instrument) (Annis et al, 2001). The debris was separated from the lysate by centrifugation at 2000 g for 4 min. Mitochondria and other heavy membranes were pelleted by centrifugation at 13 000 g for 10 min at 4°C. The pellet was resuspended in MB–EGTA (MB with 0.5 mM EGTA instead of EDTA and 150 mM KCl instead of sucrose). Isolated heavy membranes were diluted to a concentration of 1 mg/ml in buffer with a final composition of 210 mM mannitol, 150 mM KCl, 0.5 mM EGTA, 10 mM HEPES, pH 7.5, 2 mM MgCl2, 2.5 mM Na2PO4, 2.5 mM succinate and 2.5 μM rotenone and incubated with purified proteins for 1 h at 30°C. The reactions were then centrifuged at 13 000 g for 10 min and the resulting supernatants and pellets were analyzed by SDS–PAGE and immunoblotting with sheep anti-cytochrome c antibodies.
Gel filtration chromatography
A 200 μg portion of membrane protein (P1) was solubilized in cell buffer containing 300 mM NaCl and 2% CHAPS for 30 min on ice. Samples were applied to a Superdex 200 HR 10/30 column (Amersham Biosciences) equilibrated in 20 mM Na HEPES pH 7.5, 300 mM NaCl, 0.2 mM DTT and 2% (w/v) CHAPS. Fractions (400 μl) were collected starting at the column void volume; proteins were precipitated with trichloroacetic acid and analyzed by SDS–PAGE and immunoblotting.
IASD labeling reactions
Myc−/− or Myc+ cells expressing the Bax mutants were treated with etoposide for times at which 30–40% of the total cellular Bax is bound to mitochondria (48 and 18 h, respectively), lysed by nitrogen cavitation and subcellular fractions were prepared as above in the cell buffer without DTT to separate soluble and membrane-bound Bax. Aliquots (100 μg) of protein from both the supernatant and membrane fraction were labeled with 150 nmol of IASD. Samples were removed 5 and 15 min after the addition of IASD. Control samples were either left unlabeled or detergent solubilized (2% CHAPS/4% IGEPAL-630) prior to IASD labeling. Labeling was quenched with 5 μmol of DTT and samples were resolved by isoelectric focusing using a pH 4–6 gel (1.5% 4–6 ampholytes (Bio-Rad), 0.5% 3–10 ampholytes (Bio-Rad), 5% acrylamide (40%T/3%C BioShop), 8 M urea (BioShop), 4% IGEPAL-630 (Sigma), 2% CHAPS (BioShop)) and then transferred to PVDF in 1% acetic acid/4% SDS using a semidry transfer apparatus (50 mA per gel for 1 h with cooling). The specificity of the reaction for cysteine under these conditions was demonstrated by the lack of labeling of a Bax mutant with no cysteine (data not shown). Proteins were identified on membranes using mouse anti-Bax antibody (2D2, Exalpha Biologicals) and developed after incubation with peroxidase-conjugated donkey anti-mouse antibodies (Jackson Laboratories) using enhanced chemiluminescence (Perkin-Elmer). For quantification, chemiluminescence reactions were performed directly on a Typhoon fluorimager (Molecular Devices).
Crosslinking of Bax in cell lysates
Cells treated with buffer or 6 μM of etoposide for either 18 h (Myc+) or 48 h (Myc−/−) were washed twice with cell buffer without protease inhibitors (250 mM sucrose, 20 mM HEPES, pH 7.5, 2 mM MgCl2, 1 mM EDTA), and lysed by nitrogen cavitation and cell debris and nuclei removed by centrifugation as described above. The supernatant was incubated with 1 mM BMH in 10% DMSO or DMSO alone for 30 min at 25°C. After centrifugation at 5000 g for 25 min at 4°C, the reaction was split into supernatant and pellet fractions. The pelleted material (10 μg total protein) was separated by SDS–PAGE and immunoblotted with anti-Bax 1D1 antibody.
Supplementary Material
Supplementary Information
Acknowledgments
This work was supported by grants from the Canadian Institutes of Health Research FRN12517 to DWA and BL as well as from the National Cancer Institute of Canada with funds from the Terry Fox Foundation to LZP. ES held a studentship from CIHR. DWA holds the Canada Research Chair in Membrane Biogenesis.
References
- Annis MG, Zamzami N, Zhu W, Penn LZ, Kroemer G, Leber B, Andrews DW (2001) Endoplasmic reticulum localized Bcl-2 prevents apoptosis when redistribution of cytochrome c is a late event. Oncogene 20: 1939–1952 [DOI] [PubMed] [Google Scholar]
- Antonsson B, Montessuit S, Lauper S, Eskes R, Martinou JC (2000) Bax oligomerization is required for channel-forming activity in liposomes and to trigger cytochrome c release from mitochondria. Biochem J 345: 271–278 [PMC free article] [PubMed] [Google Scholar]
- Arnoult D, Gaume B, Karbowski M, Sharpe JC, Cecconi F, Youle RJ (2003) Mitochondrial release of AIF and EndoG requires caspase activation downstream of Bax/Bak-mediated permeabilization. EMBO J 22: 4385–4399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belzacq AS, Vieira HL, Verrier F, Vandecasteele G, Cohen I, Prevost MC, Larquet E, Pariselli F, Petit PX, Kahn A, Rizzuto R, Brenner C, Kroemer G (2003) Bcl-2 and Bax modulate adenine nucleotide translocase activity. Cancer Res 63: 541–546 [PubMed] [Google Scholar]
- Cartron PF, Priault M, Oliver L, Meflah K, Manon S, Vallette FM (2003) The N-terminal end of Bax contains a mitochondrial-targeting signal. J Biol Chem 278: 11633–11641 [DOI] [PubMed] [Google Scholar]
- Desagher S, Osen-Sand A, Nichols A, Eskes R, Montessuit S, Lauper S, Maundrell K, Antonsson B, Martinou JC (1999) Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J Cell Biol 144: 891–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duelli DM, Lazebnik YA (2000) Primary cells suppress oncogene-dependent apoptosis. Nat Cell Biol 2: 859–862 [DOI] [PubMed] [Google Scholar]
- Epand RF, Martinou JC, Montessuit S, Epand RM, Yip CM (2002) Direct evidence for membrane pore formation by the apoptotic protein Bax. Biochem Biophys Res Commun 298: 744–749 [DOI] [PubMed] [Google Scholar]
- Eskes R, Desagher S, Antonsson B, Martinou JC (2000) Bid induces the oligomerization and insertion of Bax into the outer mitochondrial membrane. Mol Cell Biol 20: 929–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Saez AJ, Mingarro I, Perez-Paya E, Salgado J (2004) Membrane-insertion fragments of Bcl-xL, Bax, and Bid. Biochemistry 43: 10930–10943 [DOI] [PubMed] [Google Scholar]
- Goping IS, Gross A, Lavoie JN, Nguyen M, Jemmerson R, Roth K, Korsmeyer SJ, Shore GC (1998) Regulated targeting of BAX to mitochondria. J Cell Biol 143: 207–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuck AP, Hotze EM, Tweten RK, Johnson AE (2000) Mechanism of membrane insertion of a multimeric beta-barrel protein: perfringolysin O creates a pore using ordered and coupled conformational changes. Mol Cell 6: 1233–1242 [DOI] [PubMed] [Google Scholar]
- Kim PK, Annis MG, Dlugosz PJ, Leber B, Andrews DW (2004) During apoptosis Bcl-2 changes membrane topology at both the endoplasmic reticulum and mitochondria. Mol Cell 14: 523–529 [DOI] [PubMed] [Google Scholar]
- Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, Green DR, Newmeyer DD (2002) Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell 111: 331–342 [DOI] [PubMed] [Google Scholar]
- Lindeberg M, Zakharov SD, Cramer WA (2000) Unfolding pathway of the colicin E1 channel protein on a membrane surface. J Mol Biol 295: 679–692 [DOI] [PubMed] [Google Scholar]
- Marzo I, Brenner C, Zamzami N, Jurgensmeier JM, Susin SA, Vieira HL, Prevost MC, Xie Z, Matsuyama S, Reed JC, Kroemer G (1998) Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science 281: 2027–2031 [DOI] [PubMed] [Google Scholar]
- Nechushtan A, Smith CL, Hsu YT, Youle RJ (1999) Conformation of the Bax C-terminus regulates subcellular location and cell death. EMBO J 18: 2330–2341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nouraini S, Six E, Matsuyama S, Krajewski S, Reed JC (2000) The putative pore-forming domain of Bax regulates mitochondrial localization and interaction with Bcl-X(L). Mol Cell Biol 20: 1604–1615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffolo SC, Shore GC (2003) BCL-2 selectively interacts with the BID-induced open conformer of BAK, inhibiting BAK auto-oligomerization. J Biol Chem 278: 25039–25045 [DOI] [PubMed] [Google Scholar]
- Sharpe JC, London E (1999) Diphtheria toxin forms pores of different sizes depending on its concentration in membranes: probable relationship to oligomerization. J Membr Biol 171: 209–221 [DOI] [PubMed] [Google Scholar]
- Soucie EL, Annis MG, Sedivy J, Filmus J, Leber B, Andrews DW, Penn LZ (2001) Myc potentiates apoptosis by stimulating Bax activity at the mitochondria. Mol Cell Biol 21: 4725–4736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steere B, Eisenberg D (2000) Characterization of high-order diphtheria toxin oligomers. Biochemistry 39: 15901–15909 [DOI] [PubMed] [Google Scholar]
- Suzuki M, Youle RJ, Tjandra N (2000) Structure of Bax: coregulation of dimer formation and intracellular localization. Cell 103: 645–654 [DOI] [PubMed] [Google Scholar]
- Terrones O, Antonsson B, Yamaguchi H, Wang H-G, Liu J, Lee RM, Herrmann A, Basanez G (2004) Lipidic pore formation by the concerted action of proapoptotic BAX and tBID. J Biol Chem 279: 30081–30091 [DOI] [PubMed] [Google Scholar]
- Wolter KG, Hsu YT, Smith CL, Nechushtan A, Xi XG, Youle RJ (1997) Movement of Bax from the cytosol to mitochondria during apoptosis. J Cell Biol 139: 1281–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yethon JA, Epand RF, Leber B, Epand RM, Andrews DW (2003) Interaction with a membrane surface triggers a reversible conformational change in Bax normally associated with induction of apoptosis. J Biol Chem 278: 48935–48941 [DOI] [PubMed] [Google Scholar]
- Zha J, Weiler S, Oh KJ, Wei MC, Korsmeyer SJ (2000) Posttranslational N-myristoylation of BID as a molecular switch for targeting mitochondria and apoptosis. Science 290: 1761–1765 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information