Calmodulin-dependent protein kinase IV regulates nuclear export of Cabin1 during T-cell activation (original) (raw)
Abstract
Calcium signaling is critical for activation of T lymphocytes and has been proposed to be transduced through multiple calmodulin target proteins. Whereas the calcineurin–NFAT signaling module is critical for all mammalian T cells, the role of calmodulin-dependent kinase IV (CaMKIV) in mouse naïve CD4+ T-cell activation remains enigmatic. We have applied lentivius-mediated RNA interference of CaMKIV to human T cells and found that knockdown of CaMKIV abrogates T-cell receptor-mediated transcription of the IL-2 gene. We demonstrate that CaMKIV directly phosphorylates Cabin1, a transcriptional corepressor for myocyte enhancer factor 2, creating a docking site for 14-3-3, which causes its nuclear export. CaMKIV-mediated nuclear export of Cabin1 is likely to account for a significant part of the requirement of CaMKIV during human T-cell activation.
Keywords: Cabin1, calcium, CaMKIV, MEF2, T-cell activation
Introduction
The universal second messenger calcium is known to play a pivotal role in T-cell receptor (TCR)-mediated signal transduction leading to either peripheral T-cell activation or thymocyte apoptosis (Imboden et al, 1985; Truneh et al, 1985; Gelfand et al, 1986; Lewis, 2001). Upon an increase in intracellular calcium concentration, the calcium signal is transmitted from the cytosol into the nucleus via multiple calcium signaling modules. A major calcium signaling module in T cells consists of the unique calcium, calmodulin-dependent protein phosphatase calcineurin and its substrate NFAT (Liu, 1993; Crabtree and Clipstone, 1994; Rao et al, 1997). Another calcium signaling module is centered around the transcription factor myocyte enhancer factor (MEF)-2 (McKinsey et al, 2002). Unlike the calcineurin–NFAT signaling module, which is excluded from the nucleus in resting T cells, MEF2 is constitutively bound to its cognate DNA elements in the nucleus and can repress or activate gene transcription in a calcium-dependent fashion.
MEF2 is unique among known transcription factors in its responsiveness to calcium signaling. In the absence of a calcium signal, MEF2 is kept in the ‘off' state through its association with transcriptional repressors, including Cabin1/cain (Lai et al, 1998; Sun et al, 1998; Youn et al, 1999), and the Class II histone deacetylases (HDACs) (Miska et al, 1999; Sparrow et al, 1999). Upon calcium signaling, these repressors are removed from MEF2, allowing the binding of MEF2 to such transcription coactivators as p300 (Youn et al, 2000a) or ERK5 (Yang et al, 1998; Kasler et al, 2000) to switch MEF2 to a transcriptionally ‘on' state. To date, three distinct calcium signaling pathways have been shown to affect the interaction between MEF2 and its transcriptional repressors. One pathway involves the direct competitive binding of calmodulin to Cabin1 and Class II HDACs and the consequent removal of these repressors from MEF2 (Youn et al, 1999, 2000b). A second pathway is mediated by calcineurin (CN)–NFAT; activated NFAT binds to and synergizes with MEF2 to recruit the histone acetyl transferase p300 (Blaeser et al, 2000; Youn et al, 2000a). The third pathway includes calcium/calmodulin-dependent kinase IV (CaMKIV), which has been reported to activate MEF2 by phosphorylating Class II HDACs, leading to their dissociation from MEF2 (McKinsey et al, 2000a, 2000b; Wang et al, 2000; Li et al, 2004).
In contrast to calcineurin, the role of CaMKIV in TCR signaling is less well defined. While studies using transgenic mice overexpressing a catalytically inactive form of CaM kinase IV revealed a requisite role of CaM kinase IV in TCR-mediated IL-2 transcription (Anderson et al, 1997), recent characterization of _Camk4_−/− mice indicated that this kinase is dispensable for TCR signaling in naïve CD4+ T cells (Anderson and Means, 2002). Rather the requirement for CaMKIV in TCR-mediated cytokine induction was restricted to memory CD4+ T cells.
To assess the role of CaMKIV in the activation of human T cells, we generated shRNA against the human enzyme in a lentiviral vector. Surprisingly, we found that knockdown of human CaMKIV led to significant inhibition of TCR-mediated IL-2 transcription. To delineate the mechanism of signal transduction mediated by CaMKIV during TCR signaling, we examined its connection with the MEF2 signaling module. Upon calcium signaling, the C-terminal region of Cabin1 is specifically phosphorylated by CaMKIV, creating a docking site for 14-3-3, which causes the nuclear export of Cabin1. A Cabin1 mutant lacking the CaMKIV phosphorylation site is resistant to CaMKIV-mediated nuclear export. Thus, phosphorylation of Cabin1 by CaMKIV and its subsequent nuclear export play an important role in the full activation of MEF2 and subsequent IL-2 transcription in human T cells.
Results
CaMKIV is required for TCR-mediated IL-2 transcription in primary human T cells
Whereas expression of a kinase-inactive CaMKIV in mouse thymocytes abrogates cytokine production in response to TCR signaling (Anderson et al, 1997), this process remained intact in naïve CD4+ T cells derived from _Camk4_−/− mice (Anderson and Means, 2002). To examine the relevance of CaMKIV in TCR-mediated cytokine production in human T cells, we used lentiviruses to deliver shRNA specific for human CaMKIV (Pan et al, 2004). Three lentiviral constructs were made that target different regions of the human CaMKIV cDNA. Upon testing in Jurkat T cells, one construct, pFUP2-siCaMKIV, exhibited highest efficiency in knocking down CaMKIV expression. Subsequently, lentiviruses were generated from pFUP2-siCaMKIV and used to transduce primary peripheral human CD4+ naïve T cells. At 60 h post-transduction, a considerable downregulation of CaMKIV was observed in primary human T cells (Figure 1A). When the same transduced T cells were stimulated with anti-CD3 plus anti-CD28 antibodies, a large reduction in IL-2 mRNA, as determined by RT–PCR, was seen in comparison to control cells transduced with virus containing an unrelated shRNA (Figure 1B). Similarly, a profound reduction in the level of IL-2 protein secreted into the media was also seen (Figure 1C). These results demonstrate that CaMKIV is necessary for TCR-mediated IL-2 transcription in peripheral human T cells.
Figure 1.
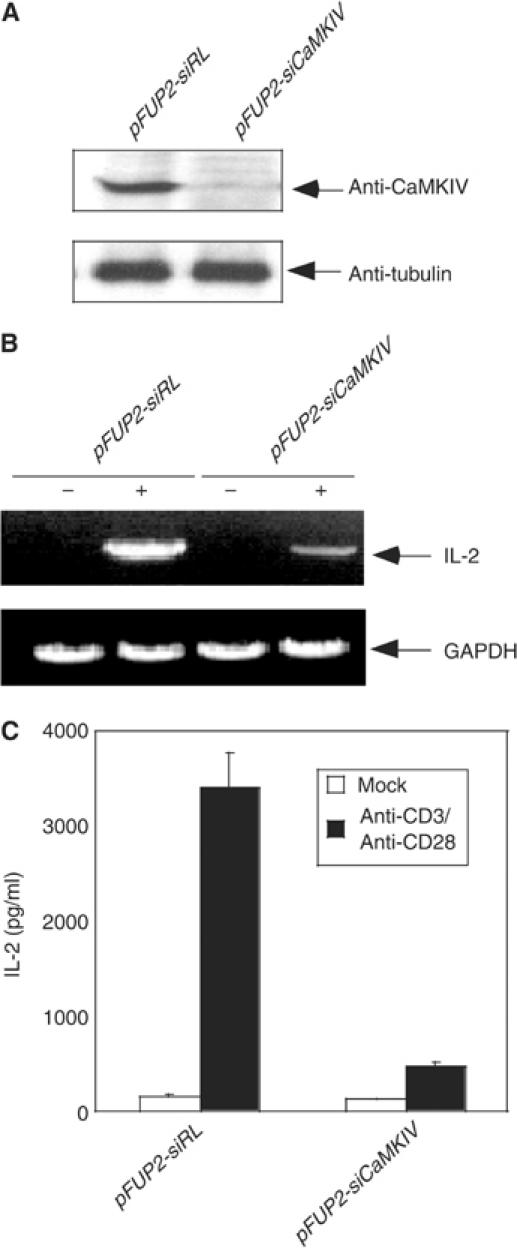
RNA interference of CaMKIV expression leads to inhibition of IL-2 promoter activation. (A) Specific inhibition of CaMKIV expression by CaMKIV siRNA. Lysates prepared from primary human CD4+ naïve T cells transduced with viruses harboring either control pFUP2-RL vector or pFUP2-siCaMKIV were subjected to Western blot analysis using anti-CaMKIV (top panel) or anti-tubulin (bottom panel) antibodies. (B) RNA interference with CaMKIV expression inhibited IL-2 mRNA synthesis in response to stimulation by anti-CD3 and anti-CD28. The two populations of primary human CD4+ naïve T cells were stimulated with a combination of anti-CD3 and anti-CD28 antibodies. Total RNA was prepared from each sample and subjected to RT–PCR analysis. (C) RNA interference with CaMKIV expression inhibited IL-2 secretion in response to stimulation by anti-CD3 plus anti-CD28 antibodies. IL-2 protein secreted into the culture medium was determined by ELISA.
Activated CaMKIV overcomes Cabin1-mediated repression of the transcriptional activity of MEF2
We have recently shown that MEF2, like NFAT, plays an essential role in mediating calcium signaling during T-cell activation (Pan et al, 2004). The major transcriptional repressor for MEF2 in mature T cells appears to be Cabin1, as the deletion of the MEF2-binding domain in Cabin1 led to enhanced activity of MEF2 and consequent upregulation of IL-2 and other cytokines in T cells (Esau et al, 2001; Pan et al, 2004). To determine whether MEF2 is regulated by CaMKIV, we examined whether the inhibition of MEF2-mediated transcription by Cabin1 is affected by overexpression of CaMKIV. We transiently transfected murine T-cell hybridoma DO11.10 with plasmids expressing Cabin1, CaMKIV and a MEF2-driven luciferase reporter gene, along with β-galactosidase reporter plasmid under the control of the CMV immediate-early promoter as an internal control. As previously shown, MEF2 reporter gene activation in response to phorbol-13-myristate-12-acetate (PMA) and ionomycin is dramatically inhibited upon expression of Cabin1-154 (Youn et al, 1999). Coexpression of full-length CaMKIV completely reversed the inhibition of MEF2 reporter gene by Cabin1 (Figure 2). In contrast, expression of full-length CaMKII had no effect on this inhibition. Furthermore, the CaMKIV-mediated reversion of Cabin1 inhibition of MEF2 is blocked by the CaM kinase-selective inhibitor KN62 or the calmodulin-selective inhibitor W-7. The less potent CaM inhibitor W-5 was also a less potent inhibitor of the CaMKIV-mediated reversal of the Cabin1 effect. Finally, FK506, which is a specific inhibitor of calcineurin, had no effect on the CaMKIV-mediated response. These observations revealed a mutually antagonistic crosstalk between Cabin1 and CaMKIV and suggested that CaMKIV mediates calcium signaling, at least in part, through regulation of the interaction between Cabin1 and MEF2.
Figure 2.
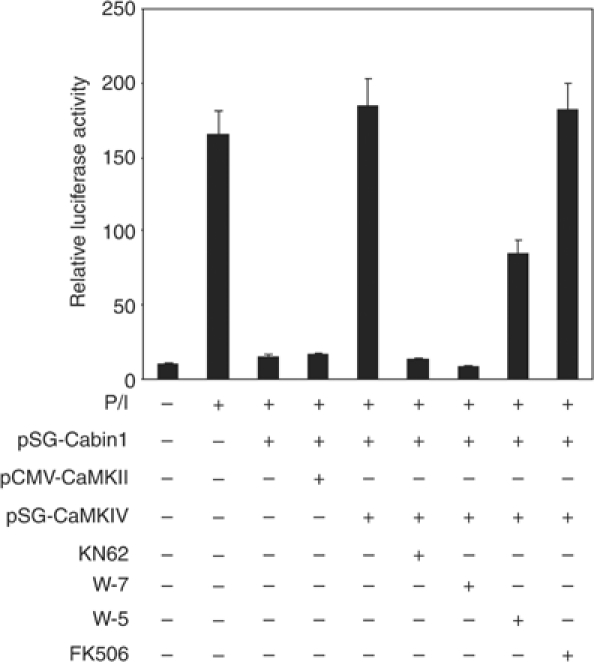
Activated CaMKIV reverses Cabin1-mediated repression of MEF2 transcriptional activity. DO11.10 cells were transfected with MEF2 luciferase reporter plasmid (5 μg) along with plasmids (10 μg each) expressing various proteins as indicated. Transfected cells were allowed to recover for 12 h before they were treated with KN62 (10 μM), W7 (25 μM), W5 (50 μM) or FK506 (1 nM) for 30 min, followed by PMA (40 nM) and ionomycin (1 μM) treatment for another 8 h before cells were lysed for determination of luciferase and β-galactosidase activity.
CaMKIV and calmodulin act in synergy to release Cabin1 from MEF2
The minimal MEF2-binding domain of Cabin1 contains an overlapping CaM-binding domain, and binding of activated CaM to Cabin1 leads to its dissociation from MEF2 (Youn et al, 1999). As CaMKIV could reverse the Cabin1-mediated inhibition of MEF2's transcriptional activity, we speculated that the reversal of the inhibition of MEF2 by Cabin1 might also be mediated through the disruption of the interaction between Cabin1 and MEF2. To test this possibility, we performed a binding assay using [35S]MEF2D generated by in vitro transcription and translation and c-Myc-Cabin1 produced from Jurkat T cells either in the presence or absence of coexpressed constitutively active Flag-CaMKIVΔC. This form of CaMKIV cannot bind Ca2+/calmodulin and therefore any effect of this protein will be independent of its normal activating subunit. As shown in Figure 3A, Cabin1 and MEF2 form a stable complex in the absence of free calcium (lane 1). Upon addition of 2 mM CaCl2, less MEF2D is bound to Cabin1 (Figure 3A, lane 3), presumably due to the competition by Ca2+/calmodulin present in the cell lysates. Coexpression of CaMKIVΔC and Cabin1 decreased, but did not abolish, the binding between Cabin1 and MEF2D (Figure 3A, lane 6 versus 1). However, expression of CaMKIVΔC together with the addition of Ca2+ completely releases Cabin1 from MEF2D (Figure 3A, lane 4), suggesting that Ca2+/calmodulin and active CaMKIV act synergistically to reverse this protein–protein interaction.
Figure 3.
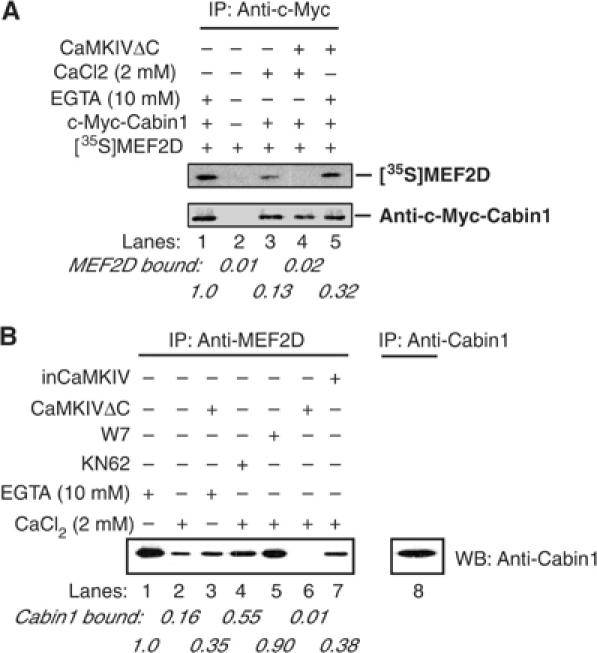
Ca2+/CaM and CaMKIV synergistically release Cabin1 from MEF2D upon calcium signaling. (A) Myc-tagged full-length Cabin1 plasmid alone or with Flag-tagged constitutively active CaMKIVΔC was transfected into DO11.10 cells. The cell lysates were incubated with _in vitro_-transcribed and translated [35S]MEF2D in the presence of either 2 mM CaCl2 or 10 mM EGTA for 2 h. After immunoprecipitation of Cabin1 with anti-Myc antibody, the bound [35S]MEF2D was visualized by autoradiography. The relative intensity of bound MEF2D was quantitated by PhosphoImage analysis. (B) Co-immunoprecipitation of endogenous MEF2D and Cabin1. Lysates were prepared from DO11.10 cells with or without expression of constitutively active CaMKIVΔC or catalytically inactive inCaMKIV and incubated with anti-MEF2D antibody. And 10 μM KN62 or 25 μM W7 was included in the lysis buffer for cells treated with the same concentrations of each inhibitor. The immunoprecipitates were subject to SDS–PAGE followed by Western blot analysis with anti-Cabin1 antibodies.
The synergistic effect of Ca2+/calmodulin and active CaMKIV on the interaction between Cabin1 and MEF2D was confirmed with endogenous proteins. Endogenous MEF2D from DO11.10 T-cell lysates co-immunoprecipitated with Cabin1 in the presence of EGTA (Figure 3B, lane 1), Activated Ca2+/calmodulin considerably, but incompletely, dissociated Cabin1 from MEF2D (Figure 3B, lane 2), an effect similar to the expression of active CaMKIV (Figure 3B, lane 3). However, Ca2+/calmodulin together with active CaMKIV abrogated the interaction between Cabin1 and MEF2D completely (Figure 3B, lane 6), and this effect could be partially restored by the presence of the CaM kinase inhibitor KN62 or the calmodulin antagonist W7 (Figure 3B, lanes 4 and 5). Similar results were seen when CaMKIVΔC and endogenous CaMKIV were knocked down by siRNA (Supplementary Figure 1). Thus, activated CaMKIV and Ca2+/calmodulin can act synergistically to dissociate Cabin1 from MEF2D.
Cabin1 is a substrate for CaMKIV
To test whether Cabin1 could serve as a substrate for CaMKIV, we coexpressed Myc-tagged Cabin1 with full-length CaMKIV in DO11.10 cells, and performed in vivo labeling of Cabin1 with 32P. Cabin1 was immunoprecipitated using anti-c-Myc antibodies and resolved by SDS–PAGE, followed by autoradiography to determine the extent of its phosphorylation. Treatment of DO11.10 cells with PMA plus ionomycin or ionomycin alone led to an increase in the phosphorylation of Cabin1 (Figure 4A, lanes 1–3). PMA alone did not lead to hyperphosphorylation of Cabin1, indicating that the enhanced phosphorylation of Cabin1 is calcium dependent (Figure 4A, lane 4). This enhanced phosphorylation of Cabin1 is inhibited upon downregulation of endogenous CaMKIV by siRNA (Figure 4A, lane 5). Expression of exogenous CaMKIV restored the Ca2+-dependent phosphorylation of Cabin1 (Figure 4A, lane 6), which is sensitive to inhibition by the CaM kinase inhibitor KN62, but not by the calcineurin inhibitor FK506 (Figure 4A, lanes 7 and 8). These results clearly show that CaMKIV can participate in the calcium-dependent phosphorylation of Cabin1 in DO11.10 cells.
Figure 4.
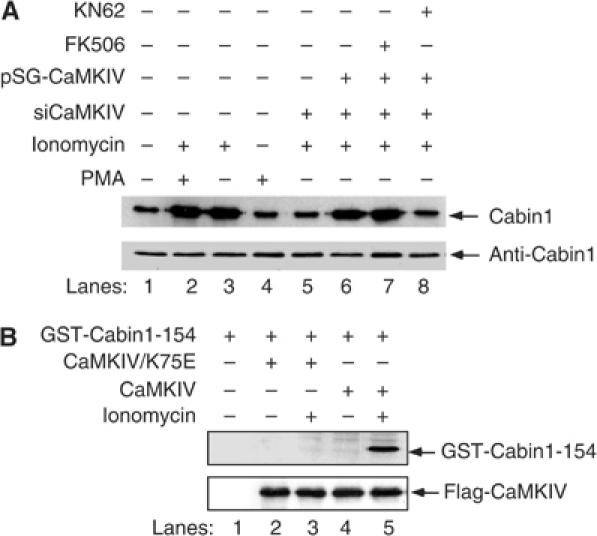
Cabin1 is phosphorylated by CaMKIV in vivo and in vitro. (A) Ionomycin treatment increases Cabin1 phosphorylation. Three cell populations, including (1) DO11.10 cells, (2) DO11.10 CaMKIV knockdown (siCaMKIV) and (3) DO11.10 CaMKIV knockdown cells transfected with 10 μg exogenous wild-type CaMKIV, were cultured in media containing 32P for 3 h, followed by either mock or FK506 (1 nM) or KN62 (10 μM) treatment for 30 min prior to treatment with 40 nM PMA and/or 1 μM ionomycin for another 3 h. The cells were lysed, and Cabin1 was immunoprecipitated from each cell extract and analyzed by SDS–PAGE and autoradiography. The total amount of Cabin1 present in each immunoprecipitate was determined by immunoblotting with the Cabin1-specific polyclonal antibodies. (B) In vitro kinase assay. DO11.10 cells were transfected with empty vector alone or with constructs (10 μg each) encoding either human Flag-tagged wild-type CaMKIV or catalytically inactive kinase mutant Flag-CaMKIV/K75E. After a 24 h incubation, cells were treated with 1 μM ionomycin for another 5 min, following which recombinant kinase proteins were immunoprecipitated and incubated with GST-Cabin1-154 (3 μg) in the presence of Ca2+, calmodulin and [γ-32P]ATP.
To assess whether CaMKIV can directly phosphorylate Cabin1 and to map the potential phosphorylation sites in Cabin1, different fragments of Cabin1 were generated by in vitro transcription/translation, followed by incubation with [γ-32P]ATP in the presence of activated CaMKIV or CaMKII. It was found that the C-terminal region of Cabin1 (amino acids 2036–2220) contains the sites of phosphorylation by CaMKIV (data not shown). To verify this observation, in vitro kinase assays were carried out using a recombinant GST-Cabin1-154 fusion protein, which contains amino acids 2036–2220 of Cabin1, as a substrate. The wild type and catalytically inactive CaMKIV mutant tagged with a Flag epitope were expressed in DO11.10 T cells. Upon stimulation with ionomycin (to activate CaMKIV), Flag-CaMKIV was immunoprecipitated using an anti-Flag antibody and incubated with recombinant GST-Cabin1-154 in vitro in the presence of [γ-32P]ATP. The reaction mixtures were resolved by SDS–PAGE and the phosphorylation state of GST-Cabin1-154 was determined by autoradiography. Wild-type CaMKIV phosphorylated GST-Cabin1-154 (Figure 4B, lane 5) and this phosphorylation required treatment of cells with ionomycin (Figure 4B, lane 4). The catalytically inactive mutant of CaMKIV (CaMKIV/K75E) did not phosphorylate GST-Cabin1-154 (Figure 4B, lanes 3), ruling out the possibility that another kinase bound to CaMKIV could be responsible for the Ca2+-dependent phosphorylation of GST-Cabin1-154. Thus, Cabin1-154 can serve as a direct substrate for CaMKIV in vitro.
Identification of CaMKIV phosphorylation sites in Cabin1
CaMKIV phosphorylation sites are known to share the consensus sequence ‘RXXS' (White et al, 1998; Hook and Means, 2001). One such sequence in Cabin1-154 is R2123AKS2126. To determine whether S2126 is the site of phosphorylation by CaMKIV, we mutated this residue to A in GST-Cabin1-154 and subjected this mutant to the in vitro kinase assay. In contrast to the wild-type Cabin1-154 (Figure 5, lane 2), the S2126A mutant can no longer be phosphorylated by CaMKIV (Figure 5, lane 4), suggesting that S2126 is a CaMKIV-dependent phosphorylation site in vitro. In addition to CaMKIV, we also tested CaMKII for its ability to phosphorylate GST-Cabin1-154 in vitro. No phosphorylation of GST-Cabin1-154 was seen in the presence of CaMKII, indicating that Cabin1-154 is at least a selective substrate for CaMKIV.
Figure 5.
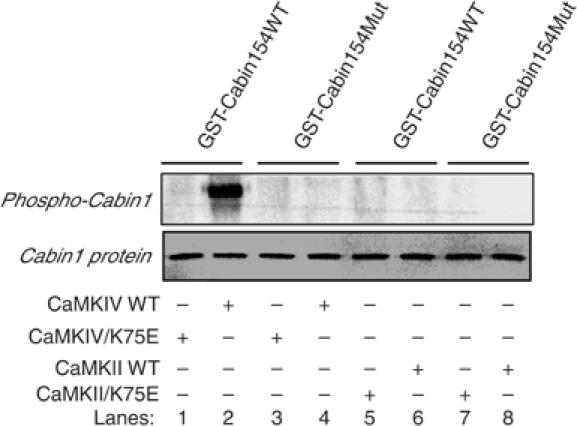
Identification and verification of CaMKIV phosphorylation site in Cabin1-154. Constitutively active CaMKIV or CaMKII or the corresponding catalytically inactive mutants CaMKIV/K75E or CaMKII/K75E were prepared by using the same protocol described in Figure 4B. Recombinant GST-Cabin154 and GST-Cabin154/S2126A were purified from Escherichia coli lysates by using glutathione-Sepharose beads. The in vitro kinase assay was carried out using the same procedure as described in Figure 4 legend. The GST-Cabin154 or GST-Cabin154/S2126A proteins in each lane were visualized by Coomassie blue staining.
Phosphorylation of Cabin1 by CaMKIV leads to its association with 14-3-3
The CaMKIV phosphorylation site of Cabin1 is also similar, albeit not identical, to a consensus binding motif (RXXSXP) for 14-3-3 if expanded to include amino acids 2127 and 2128 (R2123AKSRP2128) (Rittinger et al, 1999). To determine whether Cabin1 is capable of binding to 14-3-3 upon phosphorylation by CaMKIV, Myc-tagged Cabin1-154 was expressed in DO11.10 cells either alone or in combination with Flag-tagged CaMKIV. Upon stimulation with ionomycin, Cabin1 was immunoprecipitated with an anti-Myc antibody and the presence of bound 14-3-3 protein was evaluated by Western blot analysis using antibodies against 14-3-3τ, the most abundant isoform of 14-3-3 present in T cells (Meller et al, 1996). As shown in Figure 6 (lane 3 versus 2), 14-3-3τ was found to be associated with Cabin1 only in stimulated T cells. Treatment of T cells with KN62 abolished the binding of 14-3-3τ to Cabin1 (Figure 6, lane 4). In contrast, overexpression of CaMKIV enhanced the binding of 14-3-3τ to Cabin1 (Figure 6, lane 6), and this interaction is sensitive to inhibition by KN62 (Figure 6, lane 7). Thus, 14-3-3 binds to Cabin1 in a CaMKIV-dependent manner.
Figure 6.
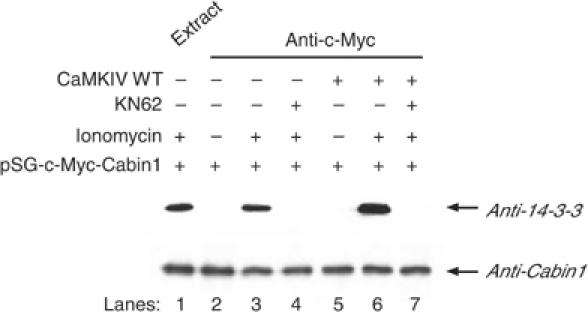
Phosphorylated Cabin1 interacts with 14-3-3. DO11.10 cells were transfected with Myc-tagged Cabin1 plasmid (10 μg) alone or together with CaMKIV expression plasmid (10 μg). At 30 h post-transfection, cells were treated with either mock or 10 μM KN62 for 30 min prior to treatment with 1 μM ionomycin for an additional 8 h. The cells were harvested, lysed and immunoprecipitated with anti-Myc antibody. Cabin1-containing immunoprecipitates were resolved on a 5% SDS–polyacrylamide gel and immunoblotted with anti-Cabin1 antibodies (lower panel) or with 14-3-3τ antibodies (upper panel).
Activated CaM and 14-3-3_τ_ can simultaneously bind to Cabin1 in T cells upon stimulation with ionomycin
The CaM-binding site of Cabin1 is also located in the C-terminus, spanning amino acids 2157 and 2220 (Youn et al, 1999). As the 14-3-3-binding site at 2126 is quite close to the CaM- and MEF2-bindig sites, we questioned whether both calmodulin and 14-3-3 could be bound to Cabin1 simultaneously. To examine this possibility, CaM-Sepharose beads were incubated with DO11.10 cell lysates containing Myc-tagged Cabin1 alone or together with activated CaMKIV in the presence of either CaCl2 (2 mM) or EGTA (10 mM) for 2 h to allow for the association of Cabin1 along with its partner proteins. The proteins bound by CaM-Sepharose were subjected to SDS–PAGE followed by Western blotting with anti-14-3-3τ antibodies. As shown in Figure 7, Cabin1 and 14-3-3τ are both retained on CaM-Sepharose in the presence of Ca2+ (lane 7). Activated CaMKIV significantly enhanced 14-3-3 binding to Cabin1 (Figure 7, lane 6), whereas KN62 abolished the interaction between Cabin1 and 14-3-3τ (Figure 7, lane 8). Furthermore, depletion of endogenous Cabin1 using anti-Cabin1 antibodies also depleted endogenous 14-3-3 (Figure 7, lane 9). These observations are consistent with the possibility that Cabin1 can form a ternary complex with 14-3-3 and Ca2+/calmodulin.
Figure 7.
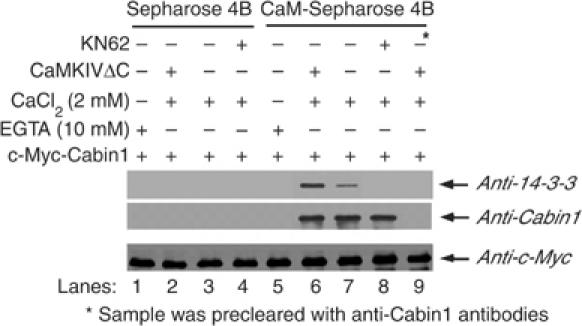
CaM and 14-3-3 bind to Cabin1 simultaneously upon calcium signaling. DO11.10 cells were transiently transfected with 10 μg of pSG-Myc-Cabin1 plasmid alone or along with 10 μg pSG-CaMKIVΔC. After recovery for 24 h, cells were treated with either mock or 10 μM KN62 for 30 min before they were harvested and lysates were prepared. One-tenth of the total cell lysates in each group was set aside as loading control (bottom panel, detected by anti-Myc antibody). The remaining lysates were incubated with either CaM-Sepharose or Sepharose 4B control beads in the presence of either 10 mM EGTA or 2 mM CaCl2 for 2 h. Precipitates were subjected to SDS–PAGE, followed by Western blot analysis using anti-Cabin1 and anti-14-3-3 antibodies.
CaMKIV promotes nuclear export of Cabin1 and acts in synergy with calmodulin to dissociate Cabin1 from MEF2
Since Cabin1 is phosphorylated by CaMKIV, which leads to its association with 14-3-3, we examined whether Cabin1 is exported from the nucleus to the cytosol upon Ca2+-mediated signaling. We thus transfected plasmids encoding GFP-Cabin1 or GFP-Cabin1-S2126A alone or in combination with the constitutively active CaMKIVΔC into immobilized Jurkat T cells in the absence or presence of ionomycin treatment. In unstimulated Jurkat T cells, GFP-Cabin1 was predominantly localized in the nucleus (Figure 8A, B and M). Interestingly, the number of cells with cytoplasmic Cabin1 was increased in the presence of constitutively active CaMKIV or upon treatment with ionomycin (Figure 8C–F and M). In contrast, neither constitutively active CaMKIV (Figure 8I, J and M) nor ionomycin treatment (Figure 8K–M) caused nuclear export of GFP-Cabin1-S2126A (Figure 8G, H and M), consistent with the idea that the Ca2+- and CaMKIV-dependent nuclear export of Cabin1 requires its phosphorylation by CaMKIV and likely binding of 14-3-3. In addition to the transiently expressed GFP-Cabin1 fusion protein, we also determined the subcellular localization of endogenous Cabin1 by cellular fractionation followed by Western blot. We observed both calcium- and CaMKIV-dependent nuclear export of endogenous Cabin1 (Supplementary Figure 2), consistent with the observations made with the GFP fusion protein. We also investigated the time course of calcium- and CaMKIV-dependent nuclear export of GFP-Cabin1. As shown in Figure 8N, nuclear export of GFP-Cabin1 starts as early as 2 min after ionomycin treatment and is completed by 30 min, consistent with the relatively rapid activation of CaMKIV by calcium signals.
Figure 8.
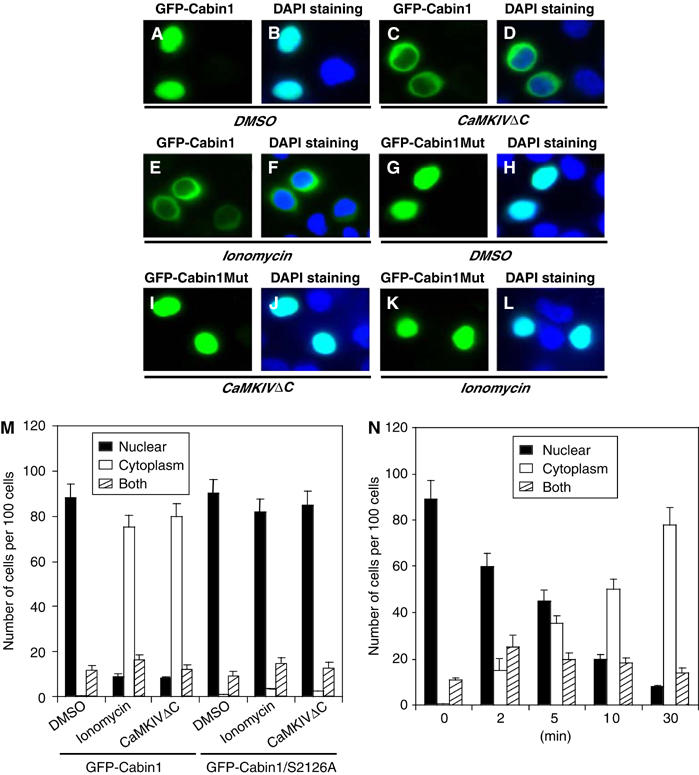
Nuclear export of Cabin1 in response to calcium signaling. Jurkat T cells were transiently transfected with plasmids (10 μg each) encoding GFP-Cabin1 or GFP-Cabin1/S2126A or together with CaMKIVΔC. At 24 h after transfection, cells were treated with DMSO or ionomycin (1 μM) for an additional 6 h, followed by visualization of green GFP (A, C, E, G, I, K) or merged green GFP and blue DAPI staining (B, D, F, H, J, L). (A, B) GFP-Cabin1 is predominantly localized in the nucleus of untreated Jurkat T cells. (C, D) Constitutively active CaMKIV promotes Cabin1 nuclear export. (E, F) Ionomycin treatment causes translocation of GFP-Cabin1 from the nucleus to the cytosol. (G–L) GFP-Cabin1/S2126A following similar treatments as described for wild-type GFP-Cabin1 in panels A–F. GFP-Cabin1/S2126A remains in the nucleus with either CaMKIVΔC coexpression (I, J) or ionomycin treatment (K, L). (M) Quantification of subcellular localization of GFP-Cabin1 and GFP-Cabin1/S2126A mutant. A total of 100 cells were counted for the subcellular distribution of the GFP fusion proteins. (N) Time course of nuclear export of GFP-Cabin1 in response to stimulation by ionomycin in Jurkat T cells.
The results shown above clearly indicate that Cabin1 is regulated by CaMKIV and undergoes nuclear export upon calcium signaling. To a first approximation, this appears to be functionally redundant to the Ca2+/calmodulin-mediated dissociation of Cabin1 from MEF2D. To determine the contribution of Ca2+/calmodulin and CaMKIV to the dissociation of Cabin1 from MEF2D, we employed the mammalian two-hybrid assay in which a Gal4-luciferase reporter gene is activated upon reconstitution of a split Gal4 transcription factor upon binding of a Gal4 DNA-binding domain-MEF2D fusion protein to a Cabin1-VP16 fusion protein. As expected, the interaction between MEF2D and Cabin1 is significantly decreased upon stimulation of T cells with ionomycin (Figure 9A). Treatment of T cells with the CaM kinase inhibitor KN62 reversed the decrease by approximately two-fold, suggesting that CaMKIV kinase activity contributed to the weakening of the Cabin1–MEF2D interaction in response to ionomycin treatment. Overexpression of the constitutively active CaMKIV, like ionomycin treatment, also led to a decrease in Cabin1–MEF2D interaction. A combination of ionomycin treatment and CaMKIV overexpression almost completely abolished the interaction between Cabin1 and MEF2, suggesting that CaMKIV and calmodulin can work synergistically to cause dissociation of Cabin1 from MEF2D. When wild-type Cabin1-154 is replaced by Cabin1-154/S2162A mutant, the interaction between the Cabin1 mutant and MEF2D is no longer sensitive to either the CaM kinase inhibitor KN62 or the constitutively active CaMKIV (Figure 9A).
Figure 9.
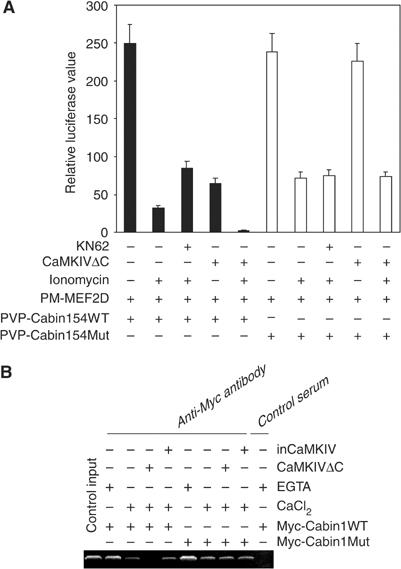
Calmodulin and CaMKIV synergistically dissociate Cabin1 from MEF2 in vivo. (A) Effects of calcium and CaMKIV on the interaction between wild-type or S2126A mutant Cabin1 and MEF2 in a mammalian two-hybrid assay. DO11.10 cells were transfected with 5 μg Gal4-luciferase reporter plasmid along with plasmids (5 μg each) expressing various proteins as indicated. After an overnight recovery, cells were treated with 10 μM KN62 for 30 min prior to treatment with 1 μM ionomycin for an additional 8 h. Cells were harvested and lysed for measurement of luciferase and β-galactosidase activities. (B) Effects of calcium and CaMKIV on association of Cabin1 with MEF2 bound to the endogenous IL-2 promoter. Jurkat T cells were transfected with plasmids (10 μg each) expressing either wild-type or mutant Cabin1 with an N-terminal Myc tag together with plasmids (10 μg each) encoding either constitutively active CaMKIV or an inactive CaMKIV mutant. The transfected cells were incubated at 37°C for 24 h before they were stimulated with ionomycin (1 μM), where indicated, for another 6 h. The cells were then subject to the CHIP assay as described previously with the same PCR primers for DNA fragment containing the MEF2-binding site on the IL-2 promoter (Pan et al, 2004).
To further examine the role of CaMKIV in the regulation of the interaction between Cabin1 and MEF2 in its natural cellular context, we performed the CHIP assay under a variety of conditions with perturbed CaMKIV activity. Thus, ectopically expressed Myc-Cabin1 is associated with the endogenous IL-2 promoter via its interaction with MEF2 that is known to be constitutively bound to the IL-2 promoter (Figure 9B, lane 2) (Pan et al, 2004). Upon treatment with ionomycin, Cabin1 dissociates from MEF2 and the IL-2 promoter (Figure 9B, lane 3). Importantly, expression of the constitutively active mutant CaMKIVΔC, but not the corresponding catalytically inactive mutant, inCaMKIV, caused a further decrease in the amount of Cabin1 bound to endogenous MEF2 (Figure 9B, lanes 4 and 5). Unlike the wild-type Myc-Cabin1, the Cabin1/S2126A mutant did not undergo further dissociation from the IL-2 promoter in response to CaMKIVΔC expression (Figure 9B, lane 8 versus 4), indicating that the dissociation of Cabin1 from the IL-2 promoter in response to CaMKIV is mediated through the phosphorylation of S2126. In addition to the CHIP assay, we also determined the effects of Cabin1-154 and Cabin1-154/S2126A mutant on the activation of a MEF2-luciferase reporter gene by PMA and ionomycin (Supplementary Figure 3). Cabin1-154 blocked the activation of MEF2 reporter gene, which can be reversed by the coexpression of the constitutively active CaMKIVΔC, but not the catalytically inactive mutant inCaMKIV. In contrast, the inhibition of MEF2 reporter gene activation by Cabin1-154/S2126A mutant is insensitive to coexpression of CaMKIVΔC, suggesting that S2126 phosphorylation by CaMKIV is required for the dissociation of Cabin1 from MEF2. Together, these results support the notion that Ca2+/calmodulin may play a dual role to promote the complete dissociation of Cabin1 and MEF2D in response to TCR-initiated signaling: first by activating CaMKIV, which is necessary to phosphorylate Cabin1 on S2126 and promote binding of 14-3-3, and second by directly binding to Cabin1 at a nearby site in the C-terminus of Cabin1.
Discussion
Although CaMKIV is dispensable for the activation of naïve CD4+ T cells isolated from _Camk4_−/− mice (Anderson and Means, 2002), we have shown that downregulation of CaMKIV in peripheral human naïve T cells using RNA interference led to significant inhibition of TCR-mediated IL-2 transcription, suggesting that CaMKIV plays an essential role in TCR signaling in peripheral human T cells. This observation is consistent with previous findings in human Jurkat T cells and mouse thymocytes using both kinase-inactive CaMKIV and small molecule inhibitors of CaMKIV, such as KN62 (Anderson et al, 1997). It is also in agreement with our observation that CaMKIV inhibitors blocked TCR-mediated IL-2 production in primary human T cells (data not shown). Whereas the reason for the difference in dependence on CaMKIV between mouse and human T cells remains unknown, it seems possible that the lack of requirement for CaMKIV in naïve CD4+ T cells of the _Camk4_−/− mouse may be due to compensatory changes that favor the use of a different kinase in the Ca2+ signaling pathway in order to maintain T-cell homeostasis. Such compensatory mechanisms have been previously shown in chronic versus acute downregulation of other important cell regulatory proteins such as those involved in the Rb pathway (Sage et al, 2000). Alternatively, this may be due to the inability of murine Cabin1 to interact with 14-3-3 (see below).
One CaMKIV target in T cells that has been implicated in TCR-mediated immediate-early gene expression and IL-2 production is the CREB transcription factor (Sheng et al, 1991; Enslen et al, 1994; Matthews et al, 1994; Ho et al, 2000). In this paper, we identify the MEF2 corepressor Cabin1 as another substrate for CaMKIV that participates in TCR-initiated signal transduction. MEF2 is a unique transcription factor in its ability to both repress and activate gene expression and to integrate calcium signaling pathways in a variety of important physiological processes from cell division to differentiation to apoptosis. The calcium ‘switches' for MEF2 are encoded in all known transcriptional repressors of MEF2, which dissociate from MEF2 to enable binding by HATs in response to calcium signaling. Two types of calcium switching mechanisms have been elucidated and each appeared to operate independently to regulate a specific class of MEF2 repressors. While Cabin1, along with its associated corepressors mSin3 and HDAC1/2, is dissociated from MEF2 through competitive binding of Ca2+/calmodulin to Cabin1, all Class II HDACs are released from MEF2 through a 14-3-3-mediated nuclear export mechanism. This mechanism requires activation of kinases to phosphorylate the Class II HDACs and generate docking sites for 14-3-3. However, in the present study, we found that Cabin1-mediated repression of MEF2 is sensitive to perturbation by both types of calcium signaling mechanisms. On the one hand, Ca2+/calmodulin binds directly to Cabin1 and on the other hand, this complex also activates CaMKIV. In turn, CaMKIV phosphorylates Cabin1 on S2126 and generates a docking site for 14-3-3. Thus, direct Ca2+/calmodulin binding and CaMKIV-dependent nuclear export work in concert to dissociate Cabin1 from MEF2 and lead to activation of MEF2-dependent transcription.
We have shown that phosphorylation of Ser2126 of Cabin1 by CaMKIV is responsible for its binding to 14-3-3 and subsequent nuclear export, which is reminiscent of the regulation of nuclear export of Class II HDACs. However, there are significant differences between Cabin1 and Class II HDACs in the mechanisms of regulation of their nuclear export by kinases and 14-3-3. For example, it has been shown that HDAC4 forms a complex with 14-3-3 in the nucleus of undifferentiated muscle cells independent of CaMK activation (Zhao et al, 2001). The kinase(s) that creates 14-3-3-binding sites in HDAC4 remains to be identified. Activated CaMK appears to cause nuclear export of HDAC4 along with its associated proteins by phosphorylating residues that do not affect 14-3-3 binding. Although nuclear export of HDAC5 appears to be caused by 14-3-3 binding, at least in the heart the kinases responsible for generating the 14-3-3-binding sites appear to be PKC and PKD rather than the CaM kinases (Vega et al, 2004). In this study, however, we demonstrate that Cabin1 is directly phosphorylated by CaMKIV and this phosphorylation is necessary for the association between human Cabin1 and 14-3-3. We note that although CaMK consensus phosphorylation site centered around Ser2126 is conserved between mice and humans, the murine as well as rat ortholog of human Cabin1 lacks the 14-3-3 binding consensus. This raises the intriguing possibility that murine Cabin1 may not undergo the same nuclear export like human Cabin1, thus rendering murine CaMKIV inactive toward murine Cabin1 and MEF2 activation in response to calcium signal. It is possible that this difference in Cabin1 may also account for the difference in dependence on CaM kinases between murine and human naïve T cells.
Cabin1 represents a unique transcriptional repressor for MEF2, as all other known MEF2 repressors belong to the Class II HDAC family. Unlike Class II HDACs, Cabin1 does not possess intrinsic HDAC activity. Instead, it exerts its repression on MEF2 in large part by recruiting HDAC1 and 2 via another transcriptional corepressor mSin3 (Youn and Liu, 2000). In spite of these differences, there is conservation in the mechanism by which Cabin1 and Class II HDACs respond to calcium signaling. This leaves unanswered the question of whether the dissociation of Cabin1 from MEF2 through direct competitive binding of calmodulin that has been shown for Cabin1 may also be involved in the dissociation of Class II HDACs from MEF2. We have previously shown that HDAC4, like Cabin1, is also Ca2+/calmodulin-binding protein and the calmodulin-binding domain in HDAC4 overlaps its MEF2-binding domain (Youn et al, 2000b). By sequence comparison, the MEF2-binding domain and the putative calmodulin-binding domain are highly conserved among all Class II HDACs, suggesting that binding of calmodulin to cause dissociation of HDACs is a conserved mechanism among all Class II HDACs. Recently, it was reported that another member of the Class II HDAC family, HDAC5, binds to calmodulin and binding of calmodulin blocks the association of HDAC5 to MEF2 (Berger et al, 2003). It is thus clear that Class II HDACs, like Cabin1, are dissociated from MEF2 upon binding to calmodulin. We surmise that, like Cabin1, both calmodulin binding and kinase-induced nuclear export act in concert on all Class II HDACs to dissociate them from MEF2 in response to calcium-initiated signaling cascades.
Both Cabin1 and Class II HDACs have been shown to localize to either the nucleus or cytosol or both in a cell type-dependent manner (Lai et al, 1998; Sun et al, 1998; Grozinger and Schreiber, 2000; McKinsey et al, 2000a; Wang et al, 2000). It is likely that calcium-independent mechanisms exist that are responsible for the cytosolic localization of Cabin1 and Class II HDACs. As Cabin1 is a multidomain and multifunctional protein, its presence in the cytosolic compartment allows it to interact with its cytosolic partner or target proteins such as calcineurin. It remains unclear, however, whether the translocation of Cabin1 and its associated HDAC1/2 from the nucleus to cytosol plays any role in the context of MEF2 function during T-cell activation or thymocyte apoptosis. Nor is it known whether the nuclear export of Cabin1 and Class II HDACs simply serves to exclude these repressors from MEF2 and whether the newly translocated Class II HDACs continue to act on yet to be defined cytosolic substrate to facilitate muscle cell differentiation, thymocyte apoptosis or peripheral T-cell activation. The identification of additional cytosolic substrates for HDACs may help to shed light on this question.
We have previously shown that MEF2 serves as a site of signal integration of two independent calcium signaling modules, the calcineurin–NFAT module and the calmodulin–Cabin1/HDAC module (Youn et al, 2000a). In this study, we demonstrated that the CaMKIV signaling module is also integrated into the same signaling circuitry, and works in concert with calmodulin to dissociate MEF2 from its known repressors. Thus, MEF2 is a focal signal integration site for all three known calcium signaling modules to activate transcription of target genes in response to calcium signaling. Other than the aforementioned possibility of a cytosolic function for Cabin1 and Class II HDACs, the coexistence of two complementary modes of dissociation of repressors from MEF2 may ensure the complete removal of these repressors from MEF2 for its optimal transcriptional activation.
Materials and methods
Molecular cloning
GFP-fused Cabin1 was subcloned into pEGFP-c2 mammalian expression vector (Clontech). pEGFP-Cabin1/S2126A was constructed using a site-directed mutagenesis kit (Stratagene). pSG-CaMKIV, pSG-CaMKIVΔC, pSG-CaMKIV/K75E and pCMV-CaMKII have been described previously (Chatila et al, 1996). To generate pBMN-CaMKIVΔC, the corresponding fragment with an N-terminal Flag tag was subcloned into _Bam_HI site of pBMN-GFP retroviral expression vector (Obigen). More detailed information on these plasmids is available upon request.
Metabolic labeling of cells
To label cellular proteins with 32P, cells were cultured in phosphate-free RPMI, supplemented with 3% dialyzed FBS. Cells were cultured in the appropriate medium for 15 min and then labeled with [32P]orthophosphate for 3 h. Typically, DO11.10 cells were labeled at 107 cells/ml in 3 ml of medium containing 0.5 mCi 32P.
In vitro kinase assay
CaMK assays were carried out based on protocols described previously (Chatila et al, 1996).
Fluorescent microscopy
Jurkat T cells were transfected with indicated plasmids, plated onto glass coverslips and treated as described. Cells were fixed in 4% paraformaldehyde for 30 min at room temperature, washed with PBS and then stained with DAPI (0.1 μg/ml) for 5 min at room temperature. The fluorescent images were recorded using a Nikon Microphot FXA Microscope. For statistic analysis, a total of 100 cells in a contiguous field were analyzed in each group.
Construction of siRNA expression vector
To eliminate the multiple cloning sites, the original pEGFP-c1 (Clontech) was cut by _Bgl_II and _Bam_HI, blunted by Klenow fragment and religated. The U6-siRNA cassette was released from pBS/U6 (kindly provided by Dr Y Shi) and inserted into the unique _Ase_I site of the modified pEGFP-c1 vector. In addition, _Bgl_II and _Sal_I were introduced into the U6-siRNA cassette for inserting the siRNA oligonucleotides. The resulting vector was denoted as pSS-U6-GFP, which contains GFP and neomycin resistance genes. To generate pSS-U6-siCaMKIV, two oligonucleotides were synthesized: oligo1 (sense, lower case denotes non-target sequence) 5′-gatccccGATGGCAACGAGGACATGAttcaag agaTCATGTCCTCGTTGCCATCTTTTTg-3′, oligo2 (antisense) 5′-tcgacAAAAAGATGGCAACGAGGACATGATCT CTTGAATCATGTCCTCGTTGCCATCGGG-3′. Oligo1 features a TTCAAGAGA loop situated between the sense and reverse complementary targeting sequences and a TTTTT terminator at the 3′ end. The two oligonucleotides were annealed and cloned into pSS-U6-GFP vector digested with _Bgl_II and _Sal_I. Positive clones containing the appropriate inserts were confirmed by DNA sequence. The control vector pSS-U6-siRL targeting Renilla luciferase (RL) was generated in a similar manner. The targeting sequence for RL is 5′-GTAGCGCGGTGTATTATAC-3′.
Construction of double-copy siRNA lentivirus vector
The double-copy siRNA construct was derived from FUGW (Qin et al, 2003; Tiscornia et al, 2003). The cassettes containing the U6 promoter were inserted into the 3′ U3 region of FUGW as follows. The _Bam_HI, _Eco_RI and _Xho_I sites in FUGW were separately eliminated by enzyme digestion, blunted with Klenow and religated. Then, one of the two _Kpn_I sites (at nucleotide 3856) was eliminated by partial enzyme digestion, blunting and religation. The modified FUGW was digested by _Bsp_EI, blunted using Klenow fragment and then further digested by _Kpn_I (cut at nucleotide 5355). The resulting lentivirus vector backbone was ligated to the siCaMKIV or siRL cassette released from pSS-U6-siCaMKIV or pSS-U6-siRL by digesting with _Kpn_I and _Sma_I. The resulting pFUP2-siCaMKIV and pFUP2-siRL vectors were confirmed by DNA sequencing.
Lentivirus production
Recombinant lentiviruses were generated using a three-plasmid system as described previous (Pan et al, 2004). Virus was harvested at 48 and 72 h after transfection and titer was determined based on percentages of GFP-positive Jurkat T cells after transduction with serially diluted viral supernatant. The titer, calculated as transducing units (TU)/ml of supernatant, was from 2 × 106 to 8 × 106 TU/ml. The virus-containing supernatant was concentrated using an Amicon Ultra Concentrator (Millipore) and stored at −80°C.
Isolation and culture of human primary T cells
Human peripheral blood mononuclear cells (PMBCs) were obtained from AllCells (Berkeley, CA). Naïve CD4+ T cells were purified by depletion of memory T cells as well as non-CD4+ cells, followed by positive selection with magnetic activated cell separation beads (Miltenyi Biotech). The purity of the CD4+/CD45RA+ T cells was more than 95% as judged by FACS analysis using CD45RA-FITC and CD4-PE staining. Cells were cultured in RPMI 1640 supplemented with 10% fetal bovine serum at a density of 1 × 106 cells/ml in the presence of human recombinant IL-7 at 5 ng/ml (BD Pharmingen).
Transduction of human CD4+ naïve T cells
Cells (5 × 105) were mixed with viral supernatants in the presence of polybrene (8 μg/ml) in a 5 ml tube, followed by addition of 10 mM HEPES and centrifugation at 2000 g for 3 h at 37°C. After incubation with viral supernatant for 10 h, cells were washed and incubated in fresh culture medium containing IL-7 for 12–16 h, followed by another cycle of trandsduction. Cells were washed and plated at the density of 1 × 106 cells/ml in the presence of IL-7 as a T-cell survival factor. A fraction of cells (1 × 105) from each group was analyzed by FACS to determine the efficiency of transduction (∼90%) by monitoring GFP expression 60 h after transduction. The remaining cells were stimulated with either plate-bound anti-CD3 (5 μg/ml) and soluble anti-CD28 (2 μg/ml; BD Pharmingen) or control IgG for 6 h. Supernatants were collected for IL-2 ELISA assay using a human IL-2 ELISA kit II (BD Pharmingen, CA). The cells were harvested for RT–PCR and Western blot analysis.
CHIP assay
CHIP assays were carried out as described previously (Pan et al, 2004).
RT–PCR
The Titan One Tune RT–PCR system (Roche Biochemicals) was used to detect IL-2 or GAPDH mRNA according to the manufacturer's instructions. The sequences of the IL-2 primers are 5′-GATTGCACTAATTCTTGCACTTGTCA-3′ (sense) and 5′-CGTTGATATTGCTGATTAAGTCCCTG-3′ (antisense). The GAPDH primers are 5′-TCCACCACCCTGTTGCTGTA-3′ (sense) and 5′-ACCACAGTCCATGCCATCAC-3′ (antisense).
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Acknowledgments
This work was supported in part by a start-up fund from Johns Hopkins School of Medicine, the eck Center (JOL) and NIH grants HD-07503 and GM-33976 (ARM).
References
- Anderson KA, Means AR (2002) Defective signaling in a subpopulation of CD4(+) T cells in the absence of Ca(2+)/calmodulin-dependent protein kinase IV. Mol Cell Biol 22: 23–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KA, Ribar TJ, Illario, M, Means AR (1997) Defective survival and activation of thymocytes in transgenic mice expressing a catalytically inactive form of Ca2+/calmodulin-dependent protein kinase IV. Mol Endocrinol 11: 725–737 [DOI] [PubMed] [Google Scholar]
- Berger I, Bieniossek C, Schaffitzel C, Hassler M, Santelli E, Richmond TJ (2003) Direct interaction of Ca2+/calmodulin inhibits histone deacetylase 5 repressor core binding to myocyte enhancer factor 2. J Biol Chem 278: 17625–17635 [DOI] [PubMed] [Google Scholar]
- Blaeser F, Ho N, Prywes R, Chatila TA (2000) Ca2+-dependent gene expression mediated by MEF2 transcription factors. J Biol Chem 275: 197–209 [DOI] [PubMed] [Google Scholar]
- Chatila T, Anderson KA, Ho N, Means AR (1996) A unique phosphorylation-dependent mechanism for the activation of Ca2+/calmodulin-dependent protein kinase type IV/GR. J Biol Chem 271: 21542–21548 [DOI] [PubMed] [Google Scholar]
- Crabtree GR, Clipstone NA (1994) Signal transmission between the plasma membrane and nucleus of T lymphocytes. Annu Rev Biochem 63: 1045–1083 [DOI] [PubMed] [Google Scholar]
- Enslen H, Sun P, Brickey D, Soderling SH, Klamo E, Soderling TR (1994) Characterization of Ca2+/calmodulin-dependent protein kinase IV. Role in transcriptional regulation. J Biol Chem 269: 15520–15527 [PubMed] [Google Scholar]
- Esau C, Boes M, Youn HD, Tatterson L, Liu JO, Chen J (2001) Deletion of calcineurin and myocyte enhancer factor 2 (MEF2) binding domain of Cabin1 results in enhanced cytokine gene expression in T cells. J Exp Med 194: 1449–1459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelfand EW, Cheung RK, Grinstein S, Mills GB (1986) Characterization of the role for calcium influx in mitogen-induced triggering of human T cells. Identification of calcium-dependent and calcium-independent signals. Eur J Immunol 16: 907–912 [DOI] [PubMed] [Google Scholar]
- Grozinger CM, Schreiber SL (2000) Regulation of histone deacetylase 4 and 5 and transcriptional activity by 14-3-3-dependent cellular localization. Proc Natl Acad Sci USA 97: 7835–7840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho N, Liauw JA, Blaeser F, Wei F, Hanissian S, Muglia LM, Wozniak DF, Nardi A, Arvin KL, Holtzman DM, Linden DJ, Zhuo M, Muglia LJ, Chatila TA (2000) Impaired synaptic plasticity and cAMP response element-binding protein activation in Ca2+/calmodulin-dependent protein kinase type IV/Gr-deficient mice. J Neurosci 20: 6459–6472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hook SS, Means AR (2001) Ca(2+)/CaM-dependent kinases: from activation to function. Annu Rev Pharmacol Toxicol 41: 471–505 [DOI] [PubMed] [Google Scholar]
- Imboden JB, Weiss A, Stobo JD (1985) The antigen receptor on a human T cell line initiates activation by increasing cytoplasmic free calcium. J Immunol 134: 663–665 [PubMed] [Google Scholar]
- Kasler HG, Victoria J, Duramad O, Winoto A (2000) ERK5 is a novel type of mitogen-activated protein kinase containing a transcriptional activation domain. Mol Cell Biol 20: 8382–8389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai MM, Burnett PE, Wolosker H, Blackshaw S, Snyder SH (1998) Cain, a novel physiologic protein inhibitor of calcineurin. J Biol Chem 273: 18325–18331 [DOI] [PubMed] [Google Scholar]
- Lewis RS (2001) Calcium signaling mechanisms in T lymphocytes. Annu Rev Immunol 19: 497–521 [DOI] [PubMed] [Google Scholar]
- Li X, Song S, Liu Y, Ko SH, Kao HY (2004) Phosphorylation of the histone deacetylase 7 modulates its stability and association with 14-3-3 proteins. J Biol Chem 279: 34201–34208 [DOI] [PubMed] [Google Scholar]
- Liu J (1993) FK506 and cyclosporin, molecular probes for studying intracellular signal transduction. Immunol Today 14: 290–295 [DOI] [PubMed] [Google Scholar]
- Matthews RP, Guthrie CR, Wailes LM, Zhao X, Means AR, McKnight GS (1994) Calcium/calmodulin-dependent protein kinase types II and IV differentially regulate CREB-dependent gene expression. Mol Cell Biol 14: 6107–6116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinsey TA, Zhang CL, Lu J, Olson EN (2000a) Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature 408: 106–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinsey TA, Zhang CL, Olson EN (2000b) Activation of the myocyte enhancer factor-2 transcription factor by calcium/calmodulin-dependent protein kinase-stimulated binding of 14-3-3 to histone deacetylase 5. Proc Natl Acad Sci USA 97: 14400–14405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinsey TA, Zhang CL, Olson EN (2002) MEF2: a calcium-dependent regulator of cell division, differentiation and death. Trends Biochem Sci 27: 40–47 [DOI] [PubMed] [Google Scholar]
- Meller N, Liu YC, Collins TL, Bonnefoy-Berard N, Baier G, Isakov N, Altman A (1996) Direct interaction between protein kinase C theta (PKC theta) and 14-3-3 tau in T cells: 14-3-3 overexpression results in inhibition of PKC theta translocation and function. Mol Cell Biol 16: 5782–5791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miska EA, Karlsson C, Langley E, Nielsen SJ, Pines J, Kouzarides T (1999) HDAC4 deacetylase associates with and represses the MEF2 transcription factor. EMBO J 18: 5099–5107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan F, Ye Z, Cheng L, Liu JO (2004) Myocyte enhancer factor 2 mediates calcium-dependent transcription of the interleukin-2 gene in T lymphocytes: a calcium signaling module that is distinct from but collaborates with the nuclear factor of activated T cells (NFAT). J Biol Chem 279: 14477–14480 [DOI] [PubMed] [Google Scholar]
- Qin XF, An DS, Chen IS, Baltimore D (2003) Inhibiting HIV-1 infection in human T cells by lentiviral-mediated delivery of small interfering RNA against CCR5. Proc Natl Acad Sci USA 100: 183–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao A, Luo C, Hogan PG (1997) Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol 15: 707–747 [DOI] [PubMed] [Google Scholar]
- Rittinger K, Budman J, Xu J, Volinia S, Cantley LC, Smerdon SJ, Gamblin SJ, Yaffe MB (1999) Structural analysis of 14-3-3 phosphopeptide complexes identifies a dual role for the nuclear export signal of 14-3-3 in ligand binding. Mol Cell 4: 153–166 [DOI] [PubMed] [Google Scholar]
- Sage J, Mulligan GJ, Attardi LD, Miller A, Chen S, Williams B, Theodorou E, Jacks T (2000) Targeted disruption of the three Rb-related genes leads to loss of G(1) control and immortalization. Genes Dev 14: 3037–3050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Thompson MA, Greenberg ME (1991) CREB: a Ca(2+)-regulated transcription factor phosphorylated by calmodulin-dependent kinases. Science 252: 1427–1430 [DOI] [PubMed] [Google Scholar]
- Sparrow DB, Miska EA, Langley E, Reynaud-Deonauth S, Kotecha S, Towers N, Spohr G, Kouzarides T, Mohun TJ (1999) MEF-2 function is modified by a novel co-repressor, MITR. EMBO J 18: 5085–5098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Youn H-D, Loh C, Stolow M, He W, Liu JO (1998) Cabin 1, a negative regulator for calcineurin signaling in T lymphocytes. Immunity 8: 703–711 [DOI] [PubMed] [Google Scholar]
- Tiscornia G, Singer O, Ikawa M, Verma IM (2003) A general method for gene knockdown in mice by using lentiviral vectors expressing small interfering RNA. Proc Natl Acad Sci USA 100: 1844–1848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truneh A, Albert F, Golstein P, Schmitt-Verhulst AM (1985) Early steps of lymphocyte activation bypassed by synergy between calcium ionophores and phorbol ester. Nature 313: 318–320 [DOI] [PubMed] [Google Scholar]
- Vega RB, Harrison BC, Meadows E, Roberts CR, Papst PJ, Olson EN, McKinsey TA (2004) Protein kinases C and D mediate agonist-dependent cardiac hypertrophy through nuclear export of histone deacetylase 5. Mol Cell Biol 24: 8374–8385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang AH, Kruhlak MJ, Wu J, Bertos NR, Vezmar M, Posner BI, Bazett-Jones DP, Yang XJ (2000) Regulation of histone deacetylase 4 by binding of 14-3-3 proteins. Mol Cell Biol 20: 6904–6912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RR, Kwon YG, Taing M, Lawrence DS, Edelman AM (1998) Definition of optimal substrate recognition motifs of Ca2+–calmodulin-dependent protein kinases IV and II reveals shared and distinctive features. J Biol Chem 273: 3166–3172 [DOI] [PubMed] [Google Scholar]
- Yang CC, Ornatsky OI, McDermott JC, Cruz TF, Prody CA (1998) Interaction of myocyte enhancer factor 2 (MEF2) with a mitogen-activated protein kinase, ERK5/BMK1. Nucleic Acids Res 26: 4771–4777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn HD, Chatila TA, Liu JO (2000a) Integration of calcineurin and MEF2 signals by the coactivator p300 during T-cell apoptosis. EMBO J 19: 4323–4331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn HD, Grozinger CM, Liu JO (2000b) Calcium regulates transcriptional repression of myocyte enhancer factor 2 by histone deacetylase 4. J Biol Chem 275: 22563–22567 [DOI] [PubMed] [Google Scholar]
- Youn HD, Liu JO (2000) Cabin1 represses MEF2-dependent Nur77 expression and T cell apoptosis by controlling association of histone deacetylases and acetylases with MEF2. Immunity 13: 85–94 [DOI] [PubMed] [Google Scholar]
- Youn HD, Sun L, Prywes R, Liu JO (1999) Apoptosis of T cells mediated by Ca(2+)-induced release of the transcription factor MEF2. Science 286: 790–793 [DOI] [PubMed] [Google Scholar]
- Zhao X, Ito A, Kane CD, Liao TS, Bolger TA, Lemrow SM, Means AR, Yao TP (2001) The modular nature of histone deacetylase HDAC4 confers phosphorylation-dependent intracellular trafficking. J Biol Chem 276: 35042–35048 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3