Heterochromatin formation involves changes in histone modifications over multiple cell generations (original) (raw)
Abstract
Stable, epigenetic inactivation of gene expression by silencing complexes involves a specialized heterochromatin structure, but the kinetics and pathway by which euchromatin is converted to the stable heterochromatin state are poorly understood. Induction of heterochromatin in Saccharomyces cerevisiae by expression of the silencing protein Sir3 results in rapid loss of histone acetylation, whereas removal of euchromatic histone methylation occurs gradually through several cell generations. Unexpectedly, Sir3 binding and the degree of transcriptional repression gradually increase for 3–5 cell generations, even though the intracellular level of Sir3 remains constant. Strains lacking Sas2 histone acetylase or the histone methylases that modify lysines 4 (Set1) or 79 (Dot1) of H3 display accelerated Sir3 accumulation at HMR or its spreading away from the telomere, suggesting that these histone modifications exert distinct inhibitory effects on heterochromatin formation. These findings suggest an ordered pathway of heterochromatin assembly, consisting of an early phase, driven by active enzymatic removal of histone acetylation and resulting in incomplete transcriptional silencing, followed by a slower maturation phase, in which gradual loss of histone methylation enhances Sir association and silencing. Thus, the transition between euchromatin and heterochromatin is gradual and requires multiple cell division cycles.
Keywords: epigenetic transition, heterochromatin, histone methylation, Sir proteins, transcriptional silencing
Introduction
Epigenetically inheritable patterns of gene expression control important aspects of cell physiology, differentiation, and development. Eukaryotic genomes are composed of stable domains of euchromatin and heterochromatin that, respectively, are transcriptionally competent and silent. Heterochromatin accounts for diverse epigenetic phenomena, such as position effect variegation in Drosophila, X-chromosome inactivation in mammals, and telomeric and mating-type silencing in yeast. Despite notable difference among organisms and silencing systems, many functional and molecular aspects of heterochromatin are highly conserved (Moazed, 2001; Grewal and Moazed, 2003). Silent chromatin domains are compact, relatively inaccessible, and characterized by histone hypoacetylation and hypomethylation of lysines 4 and 79 of histone H3 (H3-K4 and H3-K79). The different proteins that mediate heterochromatin formation often possess enzymatic activities that covalently modify histones, and they interact with the modified histones, polymerize, and spread across large genomic regions. Spreading of heterochromatin is thus thought to occur through cycles of histone modification and binding, in which silencing complexes interact with the product of their own enzymatic activity (Moazed, 2001; Grewal and Moazed, 2003).
In Saccharomyces cerevisiae, heterochromatin is formed at the HMR and HML mating type loci and telomeric regions by products of the silent information regulator genes SIR2, SIR3, and SIR4, which form the Sir complex (Rusche et al, 2003). Heterochromatin formation is initiated at silencers that are composed of binding sites for sequence-specific DNA-binding proteins. These DNA-binding proteins (together with Sir1 at the mating type loci) recruit the Sir complex, which then spreads across the entire locus. Sir2 is an evolutionarily conserved NAD-dependent histone deacetylase (HDAC), whose enzymatic activity is important for silencing (Moazed, 2001). Histones at silenced loci are hypoacetylated at all tested lysine residues, and the Sir complex binds preferentially to hypoacetylated histones (Grunstein, 1998; Suka et al, 2001). Of particular importance for silencing is lysine 16 of histone H4 (H4-K16), which is a direct target for Sir2-mediated deacetylation (Grunstein, 1998; Suka et al, 2002; Rusche et al, 2003). Histone deacetylation by Sir2 is presumed to promote silencing by creating high-affinity binding sites for the spreading Sir complex.
Telomeric heterochromatin spreads from the chromosome end to a distance of several kb, and this domain can be enlarged by overexpression of Sir3 (Renauld et al, 1993; Hecht et al, 1996). The boundaries of telomeric heterochromatin domains are determined by several factors that limit Sir binding. Sas2, which acetylates the critical H4-K16, counteracts the histone deacetylation activity of Sir2 and thus blocks the spreading of heterochromatin (Kimura et al, 2002; Suka et al, 2002). H3 acetylases and other euchromatic components, the bromodomain protein Bdf1 and the histone variant H2A.Z, have similar effects (Kristjuhan et al, 2003; Ladurner et al, 2003; Meneghini et al, 2003). Methylated H3-K4 and H3-K79 are also marks associated with active chromatin (Bernstein et al, 2002; Ng et al, 2003a), and strains lacking the corresponding enzymes (Set1 and Dot1) compromise Sir binding and silencing at heterochromatin regions (Briggs et al, 2001; Ng et al, 2002; van Leeuwen et al, 2002). These histone methylases are thought to promote silencing indirectly, by preventing promiscuous binding of Sir proteins throughout the genome, thus concentrating the Sir proteins at their normal sites of action (van Leeuwen and Gottschling, 2002; van Leeuwen et al, 2002; Ng et al, 2003a; Santos-Rosa et al, 2004). While all these euchromatic factors display ‘antisilencing' properties, it is largely unknown how they impact the process of heterochromatin assembly.
Histone lysine methylation has emerged in recent years as an important mark associated with stable and transient transcriptional states, affecting both activation and silencing. For example, methylation of H3-K9 promotes heterochromatin formation in Schizosaccharomyces pombe and metazoans, while di-methylated H3-K4 and H3-K79 are universally associated with potentially active chromatin domains. Upon transcriptional induction, H3-K4 becomes tri-methylated at the active gene (Santos-Rosa et al, 2002; Ng et al, 2003b). While histone acetylation is highly dynamic and can be rapidly reversed by HDACs (Waterborg, 2001; Katan-Khaykovich and Struhl, 2002), histone methylation is stable in bulk chromatin, and transcriptionally induced H3-K4 tri-methylation persists to mark recently active genes after a transcriptional response has ended (Ng et al, 2003b).
The stability of histone methylation marks renders them particularly suitable for the propagation and inheritance of epigenetic states. Several mechanisms have been proposed to address the fate of such marks during transitions between epigenetic states, where histone methylation associated with the initial transcriptional state might counteract establishment of the new state. First, upon removal of a histone methylase, the modified histones can be slowly eliminated by dilution through replication cycles. Second, methylated histones can be removed through replication-independent histone exchange, as occurs during the act of transcriptional elongation that disrupts, and possibly evicts, histones from the DNA template (Ahmad and Henikoff, 2002; Saccani and Natoli, 2002; Ghosh and Harter, 2003; Janicki et al, 2004; Lee et al, 2004; Schwabish and Struhl, 2004). Third, histone methylation could be rapidly eliminated by cleavage of the histone tails (Jenuwein and Allis, 2001; Bannister et al, 2002) or active demethylation of specific lysine residues (Shi et al, 2004). Fourth, methylation marks may persist but no longer perform their function, due to removal of their interacting proteins or occurrence of additional modifications, such as phosphorylation, on nearby residues (Bannister et al, 2002; Fischle et al, 2003). The particular mechanism that counteracts histone methylation may depend on the specific methylation mark and circumstance, and it is likely to impact the nature of the transcriptional transition.
The intrinsic stability of epigenetic transcriptional states is important for the long-term maintenance of gene expression patterns; yet, transitions between such states can occur during development and cellular differentiation (Lyko and Paro, 1999; Heard, 2004; Su et al, 2004). In S. cerevisiae, subtelomeric silent chromatin is partially disrupted during the DNA damage response and reestablished following recovery (Martin et al, 1999; Mills et al, 1999), and its extent can be modulated in response to environmental conditions (Ai et al, 2002). Under normal growth conditions, even the relatively stable HML silencing is occasionally disrupted and re-established. At subtelomeric regions, where silencing is semistable, switches between silencing and activation occur more frequently (Pillus and Rine, 1989; Gottschling et al, 1990). The notion of heterochromatin domain formation through spreading and blocking of silencing proteins suggests a competition-based process, but a temporal dynamic view of heterochromatin formation is unknown.
Here we investigate the molecular events associated with heterochromatin assembly and spreading in S. cerevisiae, and the roles of histone modifications in these processes. Our results suggest that histone acetylation and methylation are removed in a temporally and mechanistically distinct manner, coinciding with the initiation and enhancement of Sir3 association with chromatin. Both histone modifications inhibit some aspect of heterochromatin formation, in that they control the rate of Sir3 association and spreading. These findings support a two-phase mechanism for the assembly of silent chromatin, driven by sequential changes in distinct histone modifications that limit Sir3 association. Unexpectedly, the transition between stable epigenetic states is gradual, taking 3–5 cell division cycles to complete.
Results
Complete transcriptional silencing and Sir3 association take several generations after Sir3 induction
To induce the formation of silent chromatin, we used a yeast strain in which HA-tagged Sir3 is expressed from the GAL10 promoter. Addition of 2% galactose to raffinose-grown cells causes a substantial induction of Sir3 expression after 1.5 h (Figure 1A), with no cytotoxic effect. Sir3 levels increase mildly (about two- to three-fold) up to 4.5 h, and show no obvious change afterwards. Transcriptional repression of HMRa1 is already evident after 1.5 h, and it is approximately 8-fold at 3 h (Figure 1B). At the 3-h time-point, the cells have undergone one cell division cycle, and, in this regard, efficient de novo silencing of HMRa1 requires passage through S-phase (Miller and Nasmyth, 1984) and a later M-phase event (Lau et al, 2002). Interestingly, RNA levels continue to decline throughout the time-course, reaching 71- and 217-fold repression after 7.5 and 15 h, respectively, even though Sir3 protein levels are unchanged. This continued decrease in RNA levels could reflect a decreasing, small subpopulation of cells that fail to initiate heterochromatin, or a gradual process of transcriptional inactivation over the whole population that takes several generations for complete silencing.
Figure 1.
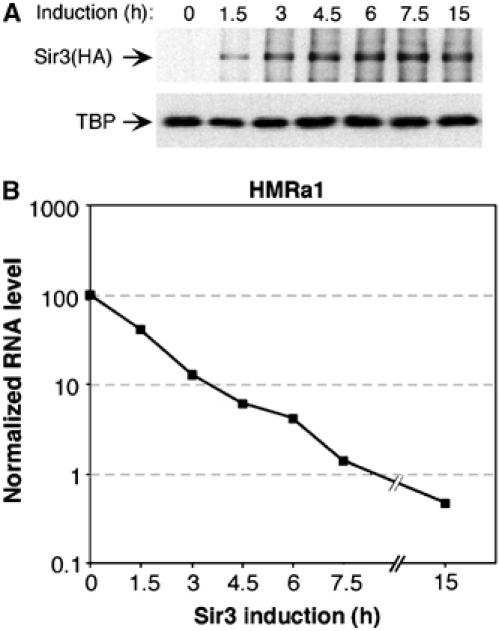
Transcriptional inactivation of HMRa1 following Sir3 induction. Expression of HA-tagged Sir3 from the GAL10 promoter was induced by treating raffinose-grown THC70 cells with 2% galactose for the indicated times. The average cell doubling time was 3.1 h. (A) Sir3 levels were monitored by Western blot analysis with an HA antibody. TBP served as a loading control. (B) HMRa1 RNA levels normalized to the DED1 control, averaged from two independent experiments.
To study the molecular events associated with heterochromatin formation, we used chromatin immunoprecipitation, focusing on two loci that are subject to silencing, the HMR mating type locus and the subtelomeric region of chromosome VI-R. At HMR, the a1 and a2 divergently transcribed genes are flanked by two silencers_, E_ and I, of which E is the stronger (Figure 2A). The telomeric silencer is defined by tandemly repeated Rap1 sites at the end of the chromosome, with the heterochromatic domain extending several kb away from the telomere.
Figure 2.
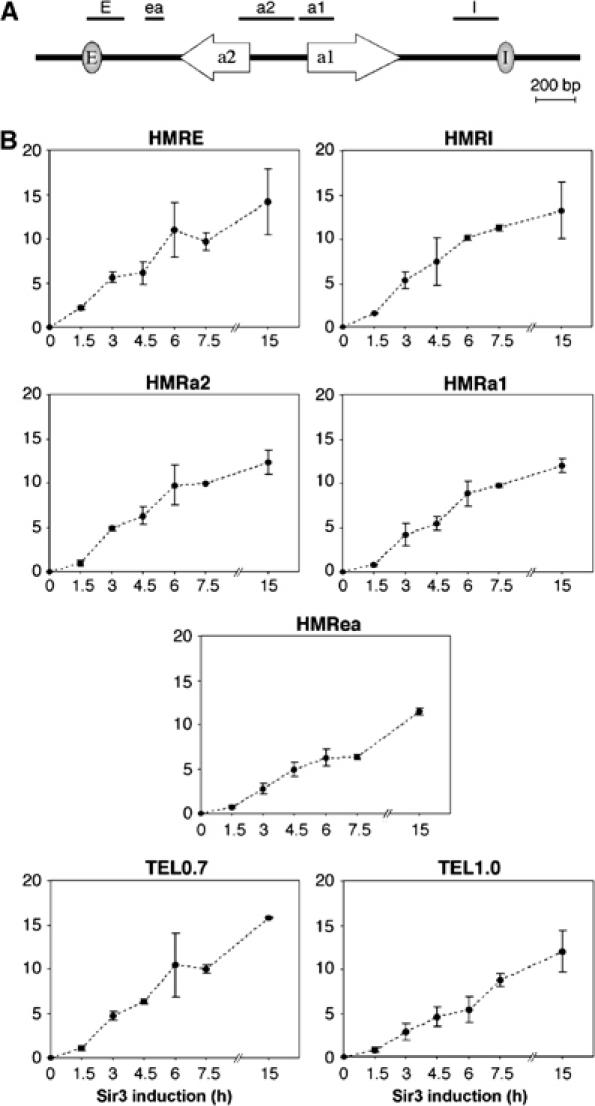
Sir3 association with silenced loci during heterochromatin assembly. (A) A diagram of the HMR locus, containing the divergently transcribed a1 and a2 genes, flanked by the E and I silencers. The positions of PCR products are shown above. (B) Sir3 association with the indicated genomic regions (TEL primer pairs are centered around 0.7 and 1 kb from the end of chromosome VI-R) in THC70 cells treated with 2% galactose for the indicated times. The level of Sir3 association at HMRa2 at 7.5 h was set as 10, and the average of two independent experiments is shown.
Sir3 binding (monitored with the HA-1 antibody) was detected at all loci at 1.5 h, whereas it is not detected prior to galactose induction (Figure 2B). Surprisingly, levels of Sir3 at all these heterochromatin loci increase throughout the entire 15-h time-course. This gradual increase in Sir3 association with heterochromatic loci occurs over several generations, even though intracellular Sir3 levels are essentially constant after the first generation following galactose induction. This observation suggests that the bulk of the population undergoes a gradual change in heterochromatin structure throughout the time-course. Furthermore, the continuous increase in Sir3 association up to 15 h roughly mirrors the continuous decline in transcription (Figure 1B), suggesting that incomplete transcriptional silencing is due to an intermediate state of heterochromatin.
Distinct kinetics of loss of H3 acetylation, H3-K79 methylation, and H3-K4 methylation during heterochromatin formation
Heterochromatic loci display low levels of H3 acetylation and methylation at H3-K4 and H3-K79 (see Supplementary Figure 1 for regional profiles of these modifications with different time-points). Upon Sir3 induction, H3 acetylation decreases substantially (two-fold) after 1.5 h, and it is largely eliminated by 3–4.5 h (Figure 3A). This rapid decrease in H3 acetylation is almost certainly due to histone deacetylation, with Sir2 histone deacetylase presumed to be the major enzymatic activity that is responsible.
Figure 3.
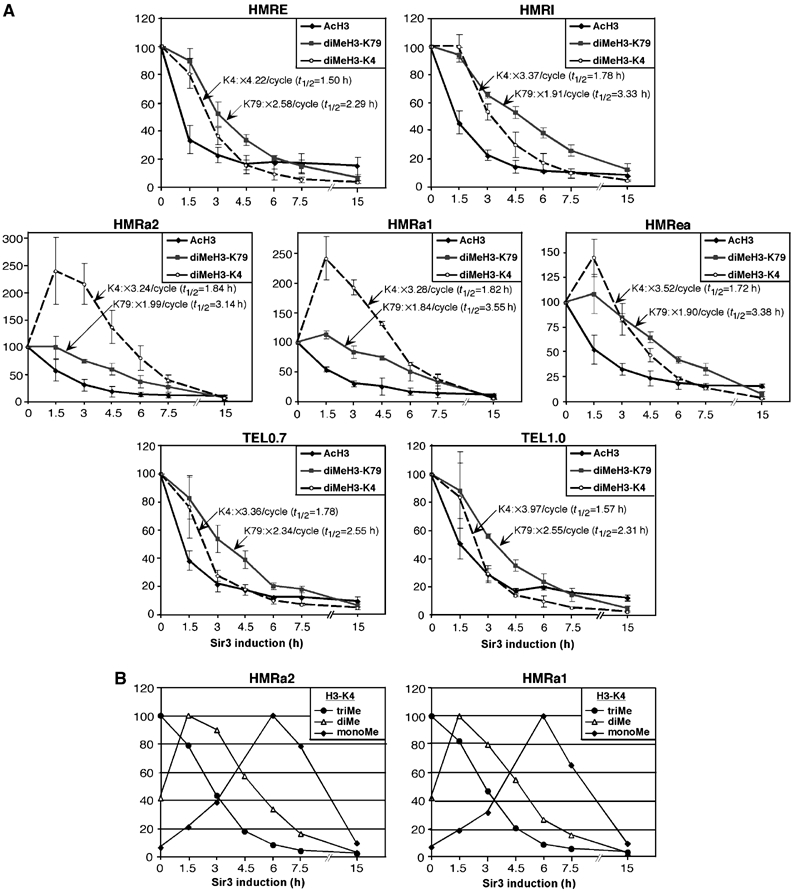
Dynamics of H3 acetylation and methylation during heterochromatin assembly. Levels of H3 acetylation (AcH3), H3-K79 di-methylation (diMeH3-K79) and H3-K4 di- (diMeH3-K4), tri-, and mono-methylation after induction of Sir3 expression. For each histone modification, the initial (A) or maximal (B) level was set to 100. The exponentially declining phase of each H3 methylation graph was used to calculate the half-life of histone methylation on chromatin (_t_1/2) and the modification's decline per 3.1 h replication cycle. The results represent the average of three (A) or two (B) independent experiments.
In contrast to the rapid deacetylation of H3, levels of H3-K79 di-methylation decrease much more slowly. Levels of H3-K79 di-methylation are essentially unchanged 1.5 h after induction, and they decline gradually throughout the time-course, with an average half-life of 2.9 h at the exponentially declining phase of each curve (Figure 3A). As the average cell-doubling time in these experiments is around 3.1 h, levels of di-methylated H3-K79 decrease on average 2.2-fold per cell cycle (ranging between 1.9- and 2.6-fold depending on the locus). These results are consistent with di-methylated H3-K79 being removed primarily by two-fold dilutions through replication cycles.
Loss of H3-K4 di-methylation during heterochromatin formation occurs with kinetics that are distinct from those of both H3 acetylation and di-methylated H3-K79 (Figure 3A). At the HMR silencers and subtelomeric regions (but not the HMRa1/a2 region; see below), di-methylated H3-K4 declines exponentially at a fairly constant rate throughout the time-course, long after H3 is completely deacetylated. However, loss of di-methylated H3-K4 is more rapid than loss of di-methylated H3-K79, with the average half-life of H3-K4 di-methylation being 1.7 h, which corresponds to a 3.6-fold decrease per cell cycle (range between 3.2- and 4.2-fold). The different persistence times of H3-K4 and H3-K79 methylation marks strongly suggest a mechanistic difference in their removal from chromatin. Whereas replication-mediated dilution can largely account for the loss of di-methylated H3-K79, the more rapid removal of H3-K4 methylation requires a replication-independent component that is specific for H3-K4 modification.
In contrast to the results at the HMR silencer and subtelomeric regions, a 2.4-fold increase in di-methylated H3-K4 is observed around the HMRa1/a2 genes at the early time-points of Sir3 induction, after which methylation levels gradually decline throughout the time-course. To explore the basis for this unexpected initial increase in H3-K4 di-methylation, we examined H3-K4 mono- and tri-methylation. Tri-methylation of H3-K4 is maximal prior to induction and then displays a gradual, continuous decline throughout the entire time-course, with an average half-life of 1.4 h, and a 4.6-fold decrease per cell cycle (Figure 3B). In contrast, mono-methylation of H3-K4 keeps increasing until later times (around 6 h), reaching low levels again only at 15 h. The time-course of the three H3-K4 methylation marks, showing consecutive peaks of tri-, di-, mono-, and finally no methylation, supports the notion that heterochromatin is assembled through a gradual process that takes multiple cell divisions.
Replication-dependent and -independent removal of euchromatic histone modifications
Heterochromatin formation in S. cerevisiae depends on an unknown S-phase event that is not DNA replication (Miller and Nasmyth, 1984; Kirchmaier and Rine, 2001; Li et al, 2001). To address the mechanism by which histone methylation marks are removed during heterochromatin assembly, and specifically the role of DNA replication, we used an experimental system that uncouples the progression through S-phase from replication (Kirchmaier and Rine, 2001). In the strain used, a derivative of HMR containing a synthetic silencer with Gal4-binding sites is flanked by two target sites for the FLP recombinase. FLP induction results in the formation of an extrachromosomal ring that lacks any origin of DNA replication (Figure 4A). By first inducing ring formation and then expressing a Gal4–Sir1 fusion protein, heterochromatin assembles at an extrachromosomal HMR locus that is stable throughout the cell cycle but does not replicate (Figure 4B).
Figure 4.
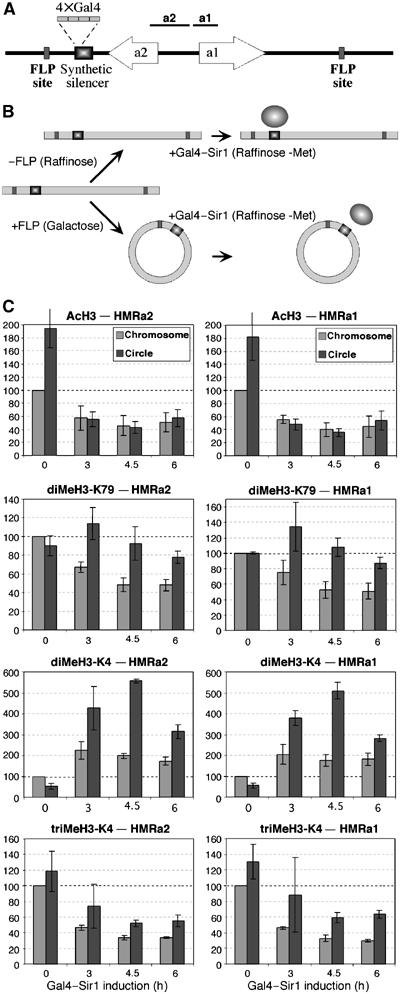
Replication-dependent and -independent changes in histone modifications during heterochromatin assembly via a synthetic silencer. (A) A synthetic derivative of the HMR silencer (contains Rap1 and Abf1 sites and four copies of the Gal4-binding site) directs heterochromatin formation upon expression of a Gal4–Sir1 fusion. Two FLP target sites flank HMR, and FLP induction results in excision of HMR from the chromosome, to form a nonreplicating DNA ring. The positions of PCR products are shown above the a1 and a2 genes. (B) JRY7131 cells were grown in raffinose (control) or galactose to induce FLP, resulting in HMR excision and ring formation. Both cultures were subsequently washed and grown in raffinose media lacking methionine to induce Gal4–Sir1. (C) Changes in histone modifications at the replicating chromosomal (chromosome) and nonreplicating ring-borne (circle) HMR locus following Gal4–Sir1 induction. The chromosomal modification level at time 0 was set to 100, and the average of three independent experiments is shown.
Expression of Gal4–Sir1 in the absence of FLP causes a decrease in H3 acetylation (two-fold), di-methylated H3-K79 (two-fold), and tri-methylated H3-K4 (three-fold), whereas levels of di-methylated H3-K4 initially increase and then slightly decrease (Figure 4C). All these effects are similar to those observed at the early times of heterochromatin formation via Sir3 induction (Figure 3). The smaller decreases in euchromatic histone modifications upon Gal4–Sir1 induction are consistent with the reduced silencing capacity of the Gal4-based silencer as compared to more efficient natural silencers (Kirchmaier and Rine, 2001; Li et al, 2001).
Induction of FLP resulted in efficient excision and ring formation, as verified by PCR (data not shown). At the ring-borne HMR, H3 acetylation levels decline upon Gal4–Sir1 induction to a level comparable to that of the chromosomal locus (Figure 4C), confirming that loss of H3 acetylation is independent of DNA replication. By contrast, di-methylated H3-K79 levels are significantly higher at the ring-borne HMR, as compared to the chromosomal locus. The ring-borne HMR shows no reduction at 4.5 h, and only a 15–20% reduction at 6 h, whereas the chromosomal locus shows a two-fold decrease at 4.5 h. These results are consistent with the kinetic analysis (Figure 3), and they strongly suggest that DNA replication plays a major role in the removal of di-methylated H3-K79 marks during heterochromatin formation.
The dynamics of di-methylated H3-K4 also differ between the chromosomal and extrachromosomal HMR locus (Figure 4C). In both cases, methylation increased upon Gal4–Sir1 induction, yet a higher increase occurred at the ring-borne HMR, and was followed by a substantial decrease. The levels of tri-methylated H3-K4 declined significantly at both the chromosomal and the extrachromosomal locus, indicating replication-independent removal. The higher tri-methylated H3-K4 levels at the ring-borne HMR following induction may also suggest a role for replication in tri-methylated H3-K4 removal. Altogether, these experiments suggest that di-methylated H3-K79 is removed primarily through replication, while H3-K4 methylation loss is mediated by replication-dependent and -independent processes, consistent with the distinct dynamics of these methylation marks upon Sir3 expression.
H3 methylation delays Sir3 accumulation at HMR
To address whether and how the process of heterochromatin assembly is impacted by the relatively persistent histone methylation marks, we examined the kinetics of Sir3 association in strains lacking Dot1 (H3-K79 methylase), Set1 (H3-K4 methylase), or Sas2 (H4-K16 acetylase that counteracts Sir2-mediated deacetylation and the spreading of heterochromatin). The strains grew at comparable rates during a 6-h galactose induction, and had roughly comparable levels of Sir3 expression (Figure 5B; there is perhaps a small decrease in the set1 strain at later times and a slightly higher level in the sas2 strain at certain times). As expected, transcriptional analysis of a natural heterochromatic gene (Yfr055W, located ∼5 kb from the end of chromosome 6R) shows weakened silencing in the dot1 strain, enhanced silencing in the sas2 strain, and marginally increased transcription in the set1 strain (Supplementary Figure 2).
Figure 5.
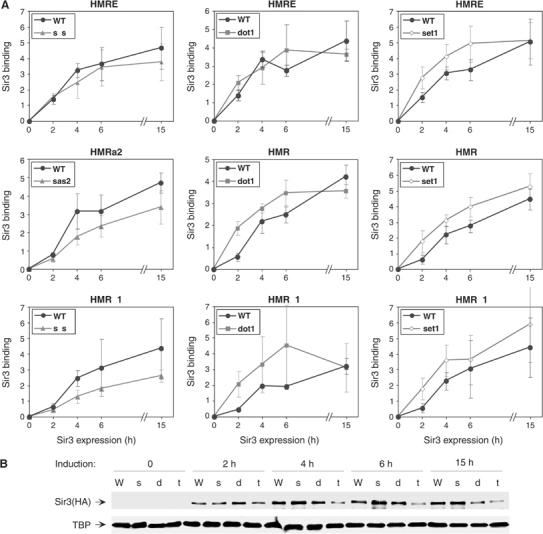
Effects of histone-modifying enzymes on Sir3 association at HMR. (A) Sir3 association at the indicated loci in wild-type (THC70; W), sas2 (s), dot1 (d), or set1 (t) cells induced for Sir3 expression for the indicated times. The level of Sir3 binding in a wild-type strain at TEL 0.27 at 15 h (see Figure 7) was set as 5. The average of three independent experiments is shown. (B) Western blot analysis of HA-Sir3 levels, using TBP as a loading control.
As shown above, Sir3 binding at HMRa1/a2 in the wild-type strain is relatively low at the 2 h time-point, and increases substantially afterwards (Figure 5A). At the HMRE silencer, the delay in Sir3 binding is smaller. Deletion of SAS2 does not relieve the delay in Sir3 association or enhance Sir3 binding at HMR, but rather causes a slight decrease in Sir3 association (Figure 5A, left panels). By contrast, Sir3 accumulation at HMR is significantly faster in the dot1 strain (Figure 5A, middle panels), with a three- to four-fold enhancement being evident at HMRa1/a2 2 h after induction. A similar enhancing effect, albeit less pronounced, is observed in the set1 deletion strain (three-fold increase in HMRa1/a2 binding at 2 h, Figure 5A, right panels). These results suggest that persistent euchromatic histone methylation marks, generated by Dot1 and Set1, delay the accumulation of silencing proteins at HMR.
Histone modifications affect the kinetics of Sir3 spreading to subtelomeric regions
To study the effects of histone modifications on Sir3 spreading away from a silencer, we first determined the Sir3-binding profiles at the subtelomeric region of chromosome VI-R after a 15-h induction (Figure 6A). In the wild-type strain, binding is maximal near the telomere and gradually decreased over distance. Sir3 binding remained relatively high up to 10 kb, and then significantly dropped around 15–17 kb. Sir3 association in this strain extends further than in strains expressing SIR3 from its own promoter (Hecht et al, 1996), probably due to higher induced Sir3 levels. As expected from the role of Sas2 in limiting the spread of telomeric silencing (Kimura et al, 2002; Suka et al, 2002), Sir3 binding in the sas2 strain is enhanced at positions more than 5 kb from the telomere. Also, as expected (Ng et al, 2002; van Leeuwen et al, 2002), loss of Dot1 does not affect Sir3 binding at the telomere, but it does reduce Sir3 association at telomere-distal positions. In our strain, loss of Set1 has a minimal effect on Sir3 association at telomeric loci.
Figure 6.
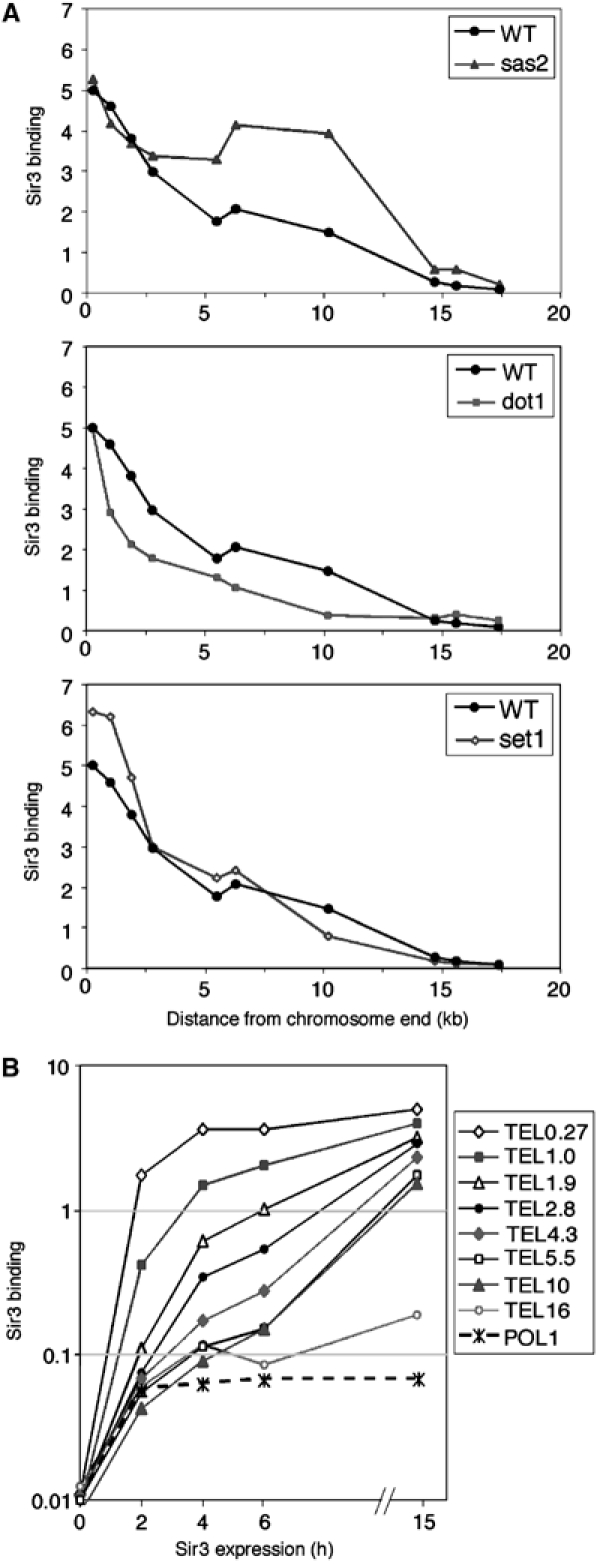
Sir3 association with subtelomeric chromatin in wild-type and mutant strains. (A) Sir3 association at the indicated subtelomeric loci of chromosome VI-R in wild-type (THC70; WT), sas2, dot1, or set1 cells treated with 2% galactose for 15 h. The level of Sir3 binding in a wild-type strain at the telomeric-most position was set as 5. The average of two independent experiments in shown. (B) Sir3 association at the indicated subtelomeric region (TEL primer names indicate distances in kb from the end of chromosome VI-R) at the indicated times after Sir3 induction. The POL1 coding region served as control for nonspecific Sir3 association with chromatin, and the average of five independent experiments is shown.
Analysis of Sir3-binding kinetics in the wild-type strain shows a dramatic difference between different subtelomeric positions (Figure 6B). Near the telomere, significant Sir3 binding occurs early on, reaching 75% of the final level by 4 h. As the distance from the telomere increases, Sir3 association is progressively slower. At 5.5 and 10 kb, binding is modest during the first 6 h, reaching only ∼10% of the final level. Substantial Sir3 association with these telomere-distal regions thus required more than two generations.
In the sas2 strain, Sir3 association is dramatically enhanced at genomic regions 4–10 kb from the telomere between 2–6 h after induction, whereas Sir3 binding at the telomere (0.27 kb) is largely similar to that of the wild-type strain (Figure 7A). To address whether this effect might be due to the slightly increased Sir3 expression in the sas2 strain, we modified Sir3 expression levels by reducing galactose concentrations such that Sir3 levels were comparable between the wild-type and sas2 strains and produced a Sir3-binding profile resembling that of natural Sir3 strains (Supplementary Figure 3). Under these conditions, the kinetics of Sir3 binding at the 0.27 kb position is comparable in wild-type and sas2 strains, whereas the sas2 strain displays markedly higher Sir3 association at the 4 and 6 h time-points in regions 2.8–10 kb from the telomere. Thus, loss of Sas2 greatly accelerates Sir3 spreading to distal positions, suggesting that Sas2-mediated histone acetylation is a major factor in controlling the rate of Sir3 spreading.
Figure 7.
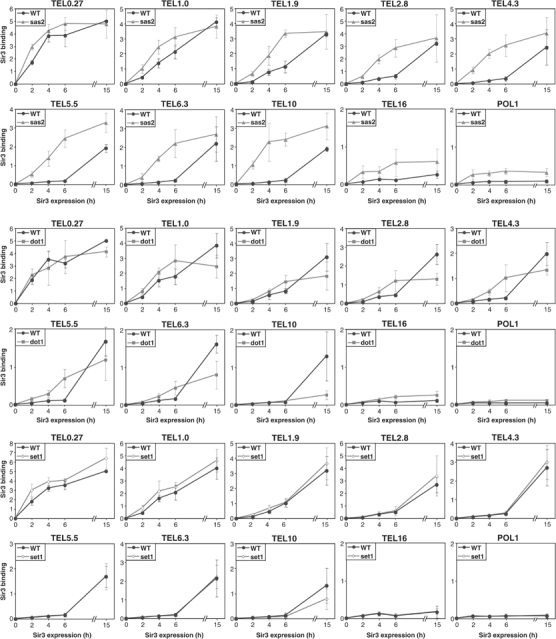
Effects of Sas2 (A), Dot1 (B), and Set1 (C) on the kinetics of Sir3 association with subtelomeric chromatin. ChIP samples from the experiments shown in Figure 5 were analyzed for Sir3 binding using primer pairs to different positions within the subtelomeric region of chromosome VI-R. ‘TEL' primer names indicate distances in kb from the chromosome end. The POL1 coding region served as control for nonspecific Sir3 association.
A significant, but less dramatic, increase in Sir3-binding kinetics is observed in the dot1 strain (Figure 7B). The dot1 effect is evident at 2.8–6.3 kb and most prominent at 4.3 and 5.5 kb, where the 6-h Sir3 binding was enhanced five- to six-fold. The accelerated kinetics of Sir3 spreading in the dot1 strain does not, however, result in higher steady-state binding; indeed, the steady-state level of Sir3 binding is lower in dot1 strains. In contrast to Sas2 and Dot1, loss of Set1 does not alter Sir3 spreading kinetics significantly (Figure 7C). Thus, both Sas2-mediated histone acetylation and Dot1-mediated H3-K79 methylation delay Sir3 spreading and heterochromatin formation, with Sas2 playing the major role.
Discussion
Heterochromatin formation is a gradual process, taking multiple cell generations
The dynamics of heterochromatin formation and the role of various histone modifications in the transition from an active to a fully silenced state are poorly understood. Here we follow this process temporally, by inducing heterochromatin rapidly and efficiently using a galactose-regulated Sir3 gene. Although Sir3 overexpression alters the balance between the cellular concentrations of the different silencing proteins, thus possibly affecting some quantitative parameters of heterochromatin assembly, the basic mechanistic aspects of this process are likely to be the same. In particular, heterochromatin containing overexpressed Sir3 is affected by mutations of histone-modifying enzymes in a manner similar to that of natural strains.
Our results show that the transition from an active to a fully silenced state is surprisingly slow, taking multiple cell generations. Specifically, the degree of transcriptional silencing and the level of Sir3 association progressively increase throughout the 15-h time-course, which corresponds to five generations. In addition, the kinetics of various forms of H3-K4 methylation suggest a gradual loss of Set1 action throughout the process of heterochromatin formation. Thus, although euchromatin and heterochromatin represent stable epigenetic states, the gradual and progressive transition between these stable states suggests the existence of metastable, intermediate states of chromatin. As discussed below, our results suggest that the need to remove different histone modifications defines the dynamic nature of heterochromatin formation.
Early and late events in heterochromatin assembly involving distinct histone modifications
Our results suggest that the pathway of heterochromatin assembly consists of two phases, controlled by distinct histone modifications (Figure 8). The ‘initiation' phase is characterized by moderate association of Sir proteins, rapid histone deacetylation by Sir2 (and possibly other HDACs), and a minimal change in histone methylation. This initial state of ‘partial heterochromatin' results in significant, but incomplete, transcriptional silencing. We suggest that this initial phase of heterochromatin formation limits the access or activity of histone methylases. In the subsequent ‘maturation' phase, H3-K79 and H3-K4 methylation is slowly lost throughout the time-course (at least three generations) by silencing-independent mechanisms (including DNA replication), thereby allowing progressive enhancements of Sir3 binding. This increased association of Sir proteins is required for the final heterochromatic state in which transcription in the region is completely silenced.
Figure 8.
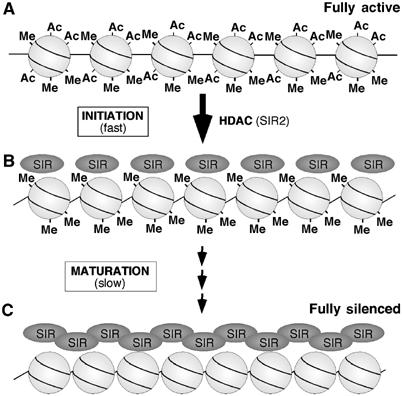
A two-phase mechanism for the ordered assembly of heterochromatin, driven by sequential changes in histone modifications. (A) Euchromatic regions have an open, accessible chromatin structure characterized by histone acetylation, and methylation at H3-K4 and H3-K79. (B) During the initiation phase of heterochromatin assembly, rapid histone deacetylation by Sir2 (and possibly other enzymes) generates moderate-affinity binding sites for the Sir complex, thus promoting the initial association of Sir proteins with chromatin. This generates a heterochromatic-like structure that partially inhibits transcription and the activity of histone methylases. This intermediate chromatin state, however, still retains histone methylation marks, due to their stable nature, and these prevent further binding of Sir proteins. (C) During the following maturation phase, the relatively slow and gradual removal of histone methylation allows further accumulation of Sir proteins, resulting in a complete heterochromatic structure that fully silences transcription.
The correlations between changes in histone modifications and heterochromatin formation, as defined by Sir3 binding and transcriptional silencing, suggest that the former may be controlling the latter. This idea is strongly supported by our observations that euchromatic histone modifications exert distinct inhibitory effects on the rate of heterochromatin formation. Loss of Dot1 or Set1, but not Sas2, accelerates the accumulation of Sir3 at HMR. Loss of Sas2, and to a lesser extent Dot1, dramatically accelerates Sir3 spreading to telomere-distal regions. Collectively, our results suggest that the sequential loss of multiple euchromatic marks may be a driving force in the formation of silent chromatin. Although the stability of the silenced state has not been directly assayed here, we speculate that this notable characteristic of heterochromatin develops during the ‘maturation' phase, with the progressive hypomethylation of histones. As a possible analogy, transcriptional inactivation of certain mammalian genes is initially reversible, and becomes epigenetically silenced at later stages of chromatin assembly (Wutz and Jaenisch, 2000; Su et al, 2004). A combination of histone modifications with distinct dynamic properties, which can be sequentially eliminated through active and passive mechanisms, may thus mediate an ordered, gradual transition between stable epigenetic states.
Potential molecular mechanisms underlying the dynamics of histone methylation
Unlike the rapid loss of histone acetylation via the action of Sir2 and possibly other histone deacetylases, loss of H3-K79 and H3-K4 methylation in the course of heterochromatin formation is gradual and relatively slow due to the relative stability of these modifications. The dynamics of the different H3-K4 methylation marks at HMRa1/a2 reveal consecutive peaks of tri-, di-, and mono-methylation, with the latter decreasing substantially only after more than three generations. Importantly, the decline of a given methylation mark correlates with rising levels of the mark with one fewer methyl groups. This observation cannot be explained simply by passive dilution through DNA replication. Instead, the observation suggests that, during heterochromatin assembly, histones are exchanged and Set1 action on newly deposited histones is gradually reduced to favor tri-, then di-, then mono-methylation, with complete inhibition occurring at later times. The progressive association of Sir proteins is likely to progressively restrict access of Set1 to chromatin, and, in this view, the shift between the different methylated H3-K4's represents another indication that the transition between euchromatin and heterochromatin is gradual and takes multiple cell generations. An alternative mechanism, which we consider less likely, is direct conversion of tri- to di- to mono-methylation by an unknown histone demethylase. As indicated by the slow and gradual changes in H3-K4 methylation marks, such a hypothetical histone demethylase would be inefficient in the context of heterochromatin assembly.
Unlike the case for H3-K4 methylation, both the kinetic analysis and synthetic silencer experiments suggest that replication-coupled histone deposition serves as the major (though not exclusive) removal mechanism for H3-K79 methylation. We consider four possibilities for why H3-K4 and H3-K79 methylation is lost with different kinetics during heterochromatin formation. First, replication-independent histone exchange might preferentially occur on H3-K4-methylated nucleosomes. Second, histone tail cleavage (Jenuwein and Allis, 2001) would preferentially remove H3-K4 methylation, although such a mechanism would have to be coupled with re-methylation of newly deposited histones to account for the consecutive peaks of different H3-K4 methylated forms. Third, the more rapid disappearance of H3-K4 methylation might be due to an H3-K4-specific histone demethylase, and such an enzyme has been described recently in mammalian cells (Shi et al, 2004). Fourth, the extent of histone exchange, inferred from the pattern of H3-K4 methylation, might be masked by efficient H3-K79 methylation of newly deposited histones. In this regard, >90% of H3 is methylated at K79 (considering all three forms), whereas only 35% is methylated at K4 (van Leeuwen and Gottschling, 2002).
Spreading kinetics of heterochromatin
In various eukaryotes, the formation of heterochromatin domains involves spreading of silencing proteins away from their nucleation centers. Described in dynamic terms, but experimentally studied in static systems, spreading of budding yeast heterochromatin involves a competition between the opposing enzymatic activities of Sir2 and Sas2, which create a gradient of H4-K16 acetylation across the subtelomeric region (Kimura et al, 2002; Suka et al, 2002). Our kinetic analysis indicates that spreading of Sir3 from a silencer is a surprisingly slow process, lasting several cell generations. This slow rate of spreading is not an inherent kinetic property of the Sir proteins, because loss of Sas2 (and to a lesser extent Dot1) accelerates the rate of spreading. Instead, the properties of euchromatin, the substrate for heterochromatin formation, determine the rate of Sir spreading. Euchromatin may thus restrict the invasion of heterochromatin through a kinetic inhibition, where the need to counteract Sas2-mediated acetylation (and to a lesser extent Dot1-mediated methylation) slows down the advancing Sir complex. In a simple model of heterochromatin formation, the extent of a silent domain would be determined by a balance between the spreading rate of individual Sir molecules and their stability on chromatin, either of which may be influenced by histone modifications. In any event, our results suggest a functional link between the spreading kinetics of heterochromatin and the steady-state genomic partition into active and inactive regions. We note that kinetic inhibition on Sir spreading is more likely to be effective at subtelomeric regions, with undefined heterochromatin–euchromatin boundaries, than at the HMR locus, which contains discrete boundary elements (Rusche et al, 2003).
Chromatin dynamics in the establishment and maintenance of epigenetic states
Various euchromatic marks have been implicated in inhibiting the binding of silencing proteins; yet, the functional relationship between them and why multiple euchromatic marks are needed have remained unclear. The euchromatic histone modification pattern includes both a dynamic (acetylation) and a relatively stable (methylation) component with ‘antisilencing' properties. As a consequence of their dynamic properties, the sequential removal of histone acetylation and methylation seems to promote different phases of heterochromatin formation, which together mediate an ordered transition between stable epigenetic states. In particular, this transition involves an intermediate state(s), in which Sir protein association has not reached the level of the final state and transcriptional silencing is significant, but incomplete. The pathway of silent chromatin assembly thus exemplifies how the distinct dynamic nature of histone acetylation and methylation can be used to drive and control a complex chromatin process.
Although histone acetylases and methylases both control the genomic distribution of silencing proteins, these enzymes affect heterochromatin in a different manner. Whereas Sas2 limits Sir3 accumulation at telomere-distal regions, the most notable effect of Dot1 is to increase the steady-state level of Sir3 at some of these same regions. Considering these phenotypes, and the distinct characteristics of the corresponding histone modifications, it is possible that the two euchromatic enzymes fulfill different, partially overlapping roles, which together maintain the proper partition of the genome into active and silent chromatin. Sas2-mediated histone acetylation may be primarily responsible for limiting the linear spreading of silencing proteins from their nucleation centers, whereas histone methylation marks generated by Dot1 (and Set1) may function mainly to prevent promiscuous binding and titration of Sir proteins throughout the genome. The use of reversible histone acetylation to control the spreading of heterochromatin generates a dynamic boundary that can limit the extent of heterochromatin domains, yet can be overcome to allow silent chromatin assembly at the appropriate location and circumstance. On the other hand, the more stable histone methylation marks would be ideal for preventing the binding of silencing proteins throughout the euchromatic genome at all times. A combination of reversible and irreversible histone modifications may thus provide the required stability to epigenetic chromatin states, while maintaining enough flexibility to allow properly orchestrated epigenetic transitions.
Lastly, the competition between heterochromatic and euchromatic states, which defines the gradual nature of the transition, is also likely to be affected by ongoing transcriptional activity. Significantly, H3-K4 tri-methylation occurs around the 5′ end of active genes due to targeting of Set1 methylase (Santos-Rosa et al, 2002; Ng et al, 2003b). The relative resistance of active genes to the invasion of heterochromatin, as observed at HMRa1/a2, may be due to transcription-coupled generation of histone methylation marks. In this regard, the first few generations of heterochromatin assembly are characterized by reduced, but still ongoing, transcription. The process by which active genes become stably silenced would thus represent a continuous, dynamic competition, where the initial active state counteracts establishment of the new silenced state through the generation of targeted and persistent euchromatic histone modifications.
Materials and methods
DNAs and yeast strains
THC70 was a gift from Mark Gartenberg (Cheng and Gartenberg, 2000). The THC70-derived sas2 (YKY7), dot1 (YKY10), and set1 (YKY8) strains were constructed by PCR-based gene replacement of the wild-type loci with _loxP_-_LEU2_-loxP (Gueldener et al, 2002). JRY7131 was a gift from Jasper Rine (Kirchmaier and Rine, 2001). THC70 and its derivatives were grown in YP medium containing 2% raffinose, and Sir3 expression was induced by treating the cells with galactose. JRY7131 was initially grown in synthetic complete media lacking histidine (for plasmid selection) and containing 2% raffinose and 100 μM methionine. Induction of FLP and Gal4–Sir1 was carried out by growing cells in media containing 2% galactose or lacking methionine, respectively.
Transcriptional analysis
HMRa1 mRNA levels were determined with respect to DED1 mRNA levels by reverse transcriptase, quantitative PCR in real time, as described (Proft and Struhl, 2002).
Chromatin immunoprecipitation
Chromatin immunoprecipitation was performed essentially as described (Kuras and Struhl, 1999; Aparicio et al, 2004), with modifications. Insoluble chromatin was pelleted by spinning for 20 min in a microfuge, followed by resuspension and sonication. The resulting chromatin solutions were cleared by spinning for 30 min in a microfuge. For the experiments shown in Figure 4 and Supplementary Figure 3, crosslinked whole-cell extracts were used, by omitting the above 20-min spin. Immunoprecipitations were carried out in 150 mM NaCl, using antibodies against the following: di-acetylated H3 (lysines 9 and 14), di-methylated H3-K79, di-methylated H3-K4 (all from Upstate Biotechnology), tri-methylated H3-K4 (AbCam), mono-methylated H3-K4 (AbCam), and the HA1 epitope (F7, Santa Cruz). Quantitative PCR analyses were performed in real time using an Applied Biosystems 7700 sequence detector. IP efficiency was calculated as the ratio between the amounts of IP PCR product and input PCR product. For analysis of histone modifications, the IP efficiency of the tested locus was normalized with respect to that of a control locus (POL1 coding region or ACT1 promoter). Error bars represent standard deviations.
The statistical significance of the difference between each pair of histone modifications in Figure 3A was examined by the unpaired _t_-test. For the relevant time-points where differences are claimed, _P_-values are typically <0.05 for individual time-points and <10−4 for the combination of relevant time-points. H3 acetylation levels are lower than the corresponding H3-K79 di-methylation levels from 1.5 to 6 h, with _P_-values (from the four time-points combined) of 2 × 10−4 for TEL1.0 and below 10−4 for each of the other six loci. H3 acetylation levels are lower than the corresponding H3-K4 di-methylation levels at HMRE, HMRI, and the TEL regions at 1.5 h and at HMRE and HMRI at 3 h, with a _P_-value (combined) below 10−4. H3-K4 di-methylation levels are lower than the corresponding H3-K79 di-methylation levels from 3 to 7.5 h, with _P_-values (combined) of 2 × 10−4 for HMRE and below 10−4 for HMRI, _TEL_0.7, and _TEL_1.0. In the synthetic silencer experiments (Figure 4C), the levels of all histone methylation marks are higher at the extrachromosomal than at the chromosomal HMR from 3 to 6 h, with _P_-values (from the three time-points combined) of <10−4 for both HMRa1 and HMRa2.
Western blotting
Electrophoretically separated proteins (from crosslinked or non-crosslinked cell extracts) were probed with monoclonal anti-HA (12CA5) or polyclonal anti-TBP antibodies.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure Legends
Acknowledgments
We thank Marc Gartenberg, Ann Kirchmaier, and Jasper Rine for strains and plasmids, and Huck Hui Ng for fruitful discussions. This work was supported by an EMBO postdoctoral fellowship to YK-K and by research grant GM53720 to KS from the National Institutes of Health.
References
- Ahmad K, Henikoff S (2002) The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly. Mol Cell 9: 1191–1200 [DOI] [PubMed] [Google Scholar]
- Ai W, Bertram PG, Tsang CK, Chan TF, Zheng XF (2002) Regulation of subtelomeric silencing during stress response. Mol Cell 10: 1295–1305 [DOI] [PubMed] [Google Scholar]
- Aparicio OM, Geisberg JV, Struhl K (2004) Chromatin immunoprecipitation for determining the association of proteins with specific genomic sequences in vivo. In Current Protocols in Molecular Biology, Ausubel FA, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K (eds), pp 21.3.1–21.3.17. New York: John Wiley & Sons [DOI] [PubMed] [Google Scholar]
- Bannister AJ, Schneider R, Kouzarides T (2002) Histone methylation. Dynamic or static? Cell 109: 801–806 [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Humphrey EL, Erlich RL, Schneider R, Bouman P, Liu JS, Kouzarides T, Schreiber SL (2002) Methylation of histone H3 Lys4 in coding regions of active genes. Proc Natl Acad Sci USA 99: 8695–8700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs SD, Bryk M, Strahl BD, Cheung WL, Davie JK, Dent SY, Winston F, Allis CD (2001) Histone H3 lysine 4 methylation is mediated by Set1 and required for cell growth and rDNA silencing in Saccharomyces cerevisiae. Genes Dev 15: 3286–3295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng TH, Gartenberg MR (2000) Yeast heterochromatin is a dynamic structure that requires silencers continuously. Genes Dev 14: 452–463 [PMC free article] [PubMed] [Google Scholar]
- Fischle W, Wang Y, Allis CD (2003) Binary switches and modification cassettes in histone biology and beyond. Nature 425: 475–479 [DOI] [PubMed] [Google Scholar]
- Ghosh MK, Harter ML (2003) A viral mechanism for remodeling chromatin structure in G0 cells. Mol Cell 12: 255–260 [DOI] [PubMed] [Google Scholar]
- Gottschling DE, Aparicio OM, Billington BL, Zakian VA (1990) Position effect at S. cerevisiae telomeres: reversible repression of pol II transcription. Cell 63: 751–762 [DOI] [PubMed] [Google Scholar]
- Grewal SI, Moazed D (2003) Heterochromatin and epigenetic control of gene expression. Science 301: 798–802 [DOI] [PubMed] [Google Scholar]
- Grunstein M (1998) Yeast heterochromatin: regulation of its assembly and inheritance by histones. Cell 93: 325–328 [DOI] [PubMed] [Google Scholar]
- Gueldener U, Heinisch J, Koehler GJ, Voss D, Hegemann JH (2002) A second set of loxP marker cassettes for Cre-mediated multiple gene knockouts in budding yeast. Nucl Acids Res 30: e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heard E (2004) Recent advances in X-chromosome inactivation. Curr Opin Cell Biol 16: 247–255 [DOI] [PubMed] [Google Scholar]
- Hecht A, Strahl-Bolsinger S, Grunstein M (1996) Spreading of transcriptional repressor SIR3 from telomeric heterochromatin. Nature 383: 92–96 [DOI] [PubMed] [Google Scholar]
- Janicki SM, Tsukamoto T, Salghetti SE, Tansey WP, Sachidanandam R, Prasanth KV, Ried T, Shav-Tai Y, Bertrand E, Singer RH, Spector DL (2004) From silencing to gene expression: real-time analysis in single cells. Cell 116: 683–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD (2001) Translating the histone code. Science 293: 1074–1080 [DOI] [PubMed] [Google Scholar]
- Katan-Khaykovich Y, Struhl K (2002) Dynamics of global histone acetylation and deacetylation in vivo: rapid restoration of normal histone acetylation status upon removal of activators and repressors. Genes Dev 16: 743–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura A, Umehara T, Horikoshi M (2002) Chromosomal gradient of histone acetylation established by Sas2 and Sir2 functions as a shield against gene silencing. Nat Genet 32: 370–377 [DOI] [PubMed] [Google Scholar]
- Kirchmaier AL, Rine J (2001) DNA replication-independent silencing in S. cerevisiae. Science 291: 646–650 [DOI] [PubMed] [Google Scholar]
- Kristjuhan A, Wittschieben BO, Walker J, Roberts D, Cairns BR, Svejstrup JQ (2003) Spreading of Sir3 protein in cells with severe histone H3 hypoacetylation. Proc Natl Acad Sci USA 100: 7551–7556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuras L, Struhl K (1999) Binding of TBP to promoters in vivo is stimulated by activators and requires Pol II holoenzyme. Nature 399: 609–612 [DOI] [PubMed] [Google Scholar]
- Ladurner AG, Inouye C, Jain R, Tjian R (2003) Bromodomains mediate an acetyl-histone encoded antisilencing function at heterochromatin boundaries. Mol Cell 11: 365–376 [DOI] [PubMed] [Google Scholar]
- Lau A, Blitzblau H, Bell SP (2002) Cell-cycle control of the establishment of mating-type silencing in S. cerevisiae. Genes Dev 16: 2935–2945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CK, Shibata Y, Rao B, Strahl BD, Lieb JD (2004) Evidence for nucleosome depletion at active regulatory regions genome-wide. Nat Genet 36: 900–905 [DOI] [PubMed] [Google Scholar]
- Li YC, Cheng TH, Gartenberg MR (2001) Establishment of transcriptional silencing in the absence of DNA replication. Science 291: 650–653 [DOI] [PubMed] [Google Scholar]
- Lyko F, Paro R (1999) Chromosomal elements conferring epigenetic inheritance. BioEssays 21: 824–832 [DOI] [PubMed] [Google Scholar]
- Martin SG, Laroche T, Suka N, Grunstein M, Gasser SM (1999) Relocalization of telomeric Ku and SIR proteins in response to DNA strand breaks in yeast. Cell 97: 621–633 [DOI] [PubMed] [Google Scholar]
- Meneghini MD, Wu M, Madhani HD (2003) Conserved histone variant H2A.Z protects euchromatin from the ectopic spread of heterochromatin. Cell 112: 725–736 [DOI] [PubMed] [Google Scholar]
- Miller AM, Nasmyth KA (1984) Role of DNA replication in the repression of silent mating type loci in yeast. Nature 312: 247–251 [DOI] [PubMed] [Google Scholar]
- Mills KD, Sinclair DA, Guarente L (1999) MEC1-dependent redistribution of the Sir3 silencing protein from telomeres to DNA double-strand breaks. Cell 97: 609–620 [DOI] [PubMed] [Google Scholar]
- Moazed D (2001) Common themes in mechanisms of gene silencing. Mol Cell 8: 489–498 [DOI] [PubMed] [Google Scholar]
- Ng HH, Ciccone DN, Morshead KB, Oettinger MA, Struhl K (2003a) Lysine-79 of histone H3 is hypomethylated at silenced loci in yeast and mammalian cells: a potential mechanism for position-effect variegation. Proc Natl Acad Sci USA 100: 1820–1825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng HH, Feng Q, Wang H, Erdjument-Bromage H, Tempst P, Zhang Y, Struhl K (2002) Lysine methylation within the globular domain of histone H3 by Dot1 is important for telomeric silencing and Sir protein association. Genes Dev 16: 1518–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng HH, Robert F, Young RA, Struhl K (2003b) Targeted recruitment of Set1 histone methylase by elongating Pol II provides a localized mark and memory of recent transcriptional activity. Mol Cell 11: 709–719 [DOI] [PubMed] [Google Scholar]
- Pillus L, Rine J (1989) Epigenetic inheritance of transcriptional states in S. cerevisiae. Cell 59: 637–647 [DOI] [PubMed] [Google Scholar]
- Proft M, Struhl K (2002) Hog1 kinase converts the Sko1-Cyc8-Tup1 repressor complex into an activator that recruits SAGA and SWI/SNF in response to osmotic stress. Mol Cells 9: 1307–1317 [DOI] [PubMed] [Google Scholar]
- Renauld H, Aparicio OM, Zierath PD, Billington BL, Chablani SK, Gottschling DE (1993) Silent domains are assembled continuously from the telomere and are defined by promoter distance and strength, and by SIR3 dosage. Genes Dev 7: 1133–1145 [DOI] [PubMed] [Google Scholar]
- Rusche LN, Kirchmaier AL, Rine J (2003) The establishment, inheritance, and function of silenced chromatin in Saccharomyces cerevisiae. Annu Rev Biochem 72: 481–516 [DOI] [PubMed] [Google Scholar]
- Saccani S, Natoli G (2002) Dynamic changes in histone H3 Lys9 methylation occurring at tightly regulated inducible inflammatory genes. Genes Dev 16: 2219–2224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Rosa H, Bannister AJ, Dehe PM, Geli V, Kouzarides T (2004) Methylation of H3 lysine 4 at euchromatin promotes Sir3 association with heterochromatin. J Biol Chem 279: 47506–47512 [DOI] [PubMed] [Google Scholar]
- Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NC, Schreiber SL, Mellor J, Kouzarides T (2002) Active genes are tri-methylated at K4 of histone H3. Nature 419: 407–411 [DOI] [PubMed] [Google Scholar]
- Schwabish MA, Struhl K (2004) Evidence for eviction and rapid deposition of histones upon transcriptional elongation by RNA polymerase II. Mol Cell Biol 24: 10111–10117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y (2004) Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119: 941–953 [DOI] [PubMed] [Google Scholar]
- Su RC, Brown KE, Saaber S, Fisher AG, Merkenschlager M, Smale ST (2004) Dynamic assembly of silent chromatin during thymocyte maturation. Nat Genet 36: 502–506 [DOI] [PubMed] [Google Scholar]
- Suka N, Luo K, Grunstein M (2002) Sir2 and Sas2 opposingly regulate acetylation of yeast histone H4 lysine 16 and spreading of heterochromatin. Nat Genet 32: 378–383 [DOI] [PubMed] [Google Scholar]
- Suka N, Suka Y, Carmen AA, Wu J, Grunstein M (2001) Highly specific antibodies determine histone acetylation in yeast heterochromatin and euchromatin. Mol Cell 8: 473–479 [DOI] [PubMed] [Google Scholar]
- van Leeuwen F, Gafken PR, Gottschling DE (2002) Dot1p modulates silencing in yeast by methylation of the nucleosome core. Cell 109: 745–756 [DOI] [PubMed] [Google Scholar]
- van Leeuwen F, Gottschling DE (2002) Genome-wide histone modifications: gaining specificity by preventing promiscuity. Curr Opin Cell Biol 14: 756–762 [DOI] [PubMed] [Google Scholar]
- Waterborg JH (2001) Dynamics of histone acetylation in Saccharomyces cerevisiae. Biochemistry 40: 2599–2605 [DOI] [PubMed] [Google Scholar]
- Wutz A, Jaenisch R (2000) A shift from reversible to irreversible X inactivation is triggered during ES cell differentiation. Mol Cell 5: 695–705 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure Legends