RNA interference in mammalian cells using siRNAs synthesized with T7 RNA polymerase (original) (raw)
Abstract
Methods that allow the specific silencing of a desired gene are invaluable tools for research. One of these is based on RNA interference (RNAi), a process by which double-stranded RNA (dsRNA) specifically suppresses the expression of a target mRNA. Recently, it has been reported that RNAi also works in mammalian cells if small interfering RNAs (siRNAs) are used to avoid activation of the interferon system by long dsRNA. Thus, RNAi could become a major tool for reverse genetics in mammalian systems. However, the high cost and the limited availability of the short synthetic RNAs and the lack of certainty that a designed siRNA will work present major drawbacks of the siRNA technology. Here we present an alternative method to obtain cheap and large amounts of siRNAs using T7 RNA polymerase. With multiple transfection procedures, including calcium phosphate co-precipitation, we demonstrate silencing of both exogenous and endogenous genes.
INTRODUCTION
Over the last few years, RNA interference (RNAi) has been recognized as a major mechanism of post-transcriptional gene silencing in the nematode Caenorhabditis elegans and the fruitfly Drosophila, as well as in plants (1). This phenomenon is based on double-stranded RNA (dsRNA) that triggers the silencing of gene expression in a sequence-specific manner. According to the prevailing model, the injected or transfected dsRNA is processed into small RNAs (guide RNAs or small interfering RNAs, siRNAs) of 21–25 nt, depending on the species (2,3). The siRNAs probably associate with a multicomponent nuclease, identified in Drosophila and called RISC (RNA-induced silencing complex), and guide this enzyme for sequence-specific degradation of the mRNA (4). In mammalian cells, the interferon-mediated antiviral response to long dsRNA that leads to the shutdown of protein synthesis precludes the use of RNAi (5). To bypass this non-specific effect, short interfering dsRNAs of 21 nt (which do not activate the antiviral response) have been used instead (6). The function of several endogenous genes has recently been investigated with this technique in mammalian cells (7,8). This new approach requires the chemical synthesis of short RNAs involving high cost without a guarantee that the purchased siRNA will be effective in silencing (6). To alleviate these problems, we present a simple alternative to obtain large amounts of short interfering RNAs with T7 RNA polymerase-directed in vitro transcription. The obtained siRNAs promote silencing in different mammalian cells of both exogenous and endogenous genes.
MATERIALS AND METHODS
T7 siRNA synthesis
Desalted DNA oligonucleotides were ordered from Microsynth (Switzerland). (i) T7, 5′-TAATACGACTCACTATAG-3′. (ii) GFP as in Caplen et al. (9): sense, 5′-ATGAACTTCAGGGTCAGCTTGCTATAGTGAGTCGTAT-TA-3′; antisense, 5′-CGGCAAGCTGACCCTGAAGTTCTATAGTGAGTCGTATTA-3′. (iii) PKR nucleotides 931–949 relative to the start codon: sense, 5′-AAGATCAAGTTTTGCCAATGCTATAGTGAGTCGTATTA-3′; antisense, 5′-AAGCATTGGCAAAACTTGATCTATAGTGAGTCGTAT-TA-3′. The oligonucleotide-directed production of small RNA transcripts with T7 RNA polymerase has been described (10). For each transcription reaction, 1 nmol of each oligonucleotide was annealed in 50 µl of TE buffer (10 mM Tris–HCl pH 8.0, and 1 mM EDTA) by heating at 95°C; after 2 min, the heating block was switched off and allowed to cool down slowly to obtain dsDNA. Transcription was performed in 50 µl of transcription mix: 1× T7 transcription buffer (40 mM Tris–HCl pH 7.9, 6 mM MgCl2, 10 mM DTT, 10 mM NaCl and 2 mM spermidine) 1 mM rNTPs, 0.1 U yeast pyrophosphatase (Sigma), 40 U RnaseOUT (Life Technologies) and 100 U T7 RNA polymerase (Fermentas) containing 200 pmol of the dsDNA as template. After incubation at 37°C for 2 h, 1 U RNase free-DNase (Promega) was added at 37°C for 15 min. Sense and antisense 21-nt RNAs generated in separate reactions were annealed by mixing both crude transcription reactions, heating at 95°C for 5 min followed by 1 h at 37°C to obtain ‘T7 RNA polymerase synthesized small interfering double-stranded RNA’ (T7 siRNA). The mixture (100 µl) was then adjusted to 0.2 M sodium acetate pH 5.2, and precipitated with 2.5 vol ethanol. After centrifugation, the pellet was washed once with 70% ethanol, dried, and resuspended in 50 µl of water.
Cell culture
Human HeLa and HEK293T cells were grown at 37°C in Dulbecco’s modified Eagle’s medium (DMEM) (Life Technologies) supplemented with 5% fetal calf serum, glutamine, penicillin and streptomycin. Cells were seeded into 6-well plates in 2.5 ml of medium 2–3 h prior to transfection by the calcium phosphate co-precipitation technique. Unless noted, 2 µl of T7 siRNA plus 1 µg/well of plasmids pEGFP-C1 (Clontech) and pcDNA3-Luc (11) in 90 µl of water were prepared in a tube to which 30 µl of 1 M CaCl2 was added. The calcium/DNA/siRNA solution was mixed quickly with 120 µl of 2× phosphate solution (140 mM NaCl, 1.5 mM Na2HPO4, 50 mM HEPES, pH 7.02) and the precipitate was immediately added to the wells. For silencing of an endogenous gene, 2 µl of T7 siRNA plus 2 µg of vector pcDNA3 were transfected by the calcium phosphate co-precipitation technique. For liposome-mediated transfection, cells were seeded into 6-well plates 4–5 h prior to treatment. Co-transfection of plasmids and T7 siRNAs was carried out with Lipofectamine (Life Technologies) as directed by the manufacturer for adherent cell lines. Unless noted otherwise, 2 µl of T7 siRNA and 0.5 µg of pEGFP-C1 formulated into liposomes were applied per well. The final volume was 1 ml/well. Cells were harvested and lysed 24 and 40 h later for GFP and PKR analysis, respectively.
Western blotting
Cells were lysed in 10 mM Tris–HCl pH 7.5, 150 mM NaCl, 0.1% SDS, 1% Triton X-100, 0.5% deoxycholate, 5% glycerol, protease inhibitors and 1 mM DTT. Equal amounts of total proteins were separated on a 10% polyacrylamide gel and transferred to nitrocellulose. The proteins were revealed either with anti-GFP (Clontech), anti-PKR (Santa-Cruz), anti-luciferase (12) or anti-Hsp90 (a kind gift from Dr David Toft, Mayo Clinic) antibodies.
RESULTS AND DISCUSSION
To generate siRNAs in vitro, we designed the strategy presented in Figure 1. An 18mer oligonucleotide encompassing the T7 promoter is annealed to a 38mer (39mer) oligonucleotide with the complementary sequence of the T7 promoter downstream of the target sequence preceded by two additional nucleotides (reading the sequence 5′→3′). The transcribed sequence is 19 nt (20 nt) plus 2 nt, which can be any nucleotides in the case of the sense RNA but must be complementary nucleotides in the antisense RNA, since it has been shown that the antisense RNA of the siRNA guides target recognition (13). It is noteworthy that T7 RNA polymerase can transcribe a template where only the promoter is double stranded (10). The last guanosine of the T7 promoter is the first ribonucleotide that is incorporated into the RNA by the T7 RNA polymerase during transcription and, therefore, all siRNAs designed by this method will start with a G. Thus, the design of T7 siRNA requires that the sequence starts with a G and has a C at position 19 (position 20) to allow annealing with the complementary RNA, which also starts with a G (see Fig. 1). This G-N17 (N18)-C rule does not restrict the T7 siRNA design since this sequence is frequently found in any gene (on average about five times in a random sequence of 100 bp). To obtain dsRNA for RNAi, only two DNA oligonucleotides corresponding to the sense and antisense sequences of the target gene have to be ordered. The T7 promoter oligonucleotide is invariant and common to any target gene. Following transcription reactions, sense and antisense transcripts are annealed and ethanol precipitated, yielding what we refer to as T7 siRNAs. The integrity of the transcripts was checked on a Nusieve agarose gel (data not shown).
Figure 1.
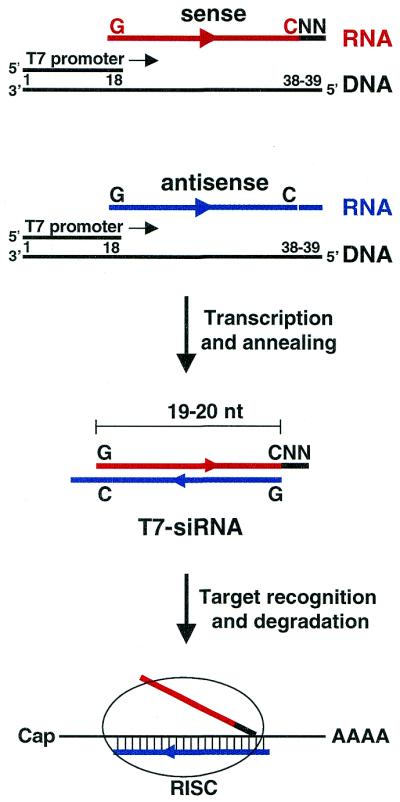
Strategy to generate T7 siRNAs (see Results and Discussion for details). The sequence of the gene of interest is shown in red (sense) or blue (antisense), while the two unrelated nucleotides are in black. RISC stands for the RNA-induced silencing complex that targets the mRNA for cleavage.
Since our goal was to perform siRNA-mediated silencing with T7 siRNAs, we first selected as a target GFP, which has already been used to study RNAi. The plasmid pEGFP-C1 was transfected together with a 22 nt T7 siRNA into HeLa cells with lipofectamine. Cells were harvested 24 h later and the levels of the exogenously expressed protein were monitored by immunoblot analysis. As shown in Figure 2A, GFP protein levels dropped in the presence of T7 siRNAi targeted to GFP. An unrelated T7 siRNA targeted against the kinase PKR (see below) did not affect GFP levels. Numerous different approaches have been developed to facilitate the transfer of foreign genes into cells, among them the calcium phosphate co-precipitation technique. We thus decided to compare lipofectamine with calcium phosphate. As shown in Figure 2B, GFP expression was also reduced following transfection of T7 siRNAs by the calcium phosphate co-precipitation technique. In the same experiment, the expression of firefly luciferase was not affected by any of the T7 siRNAs, indicating that silencing triggered by T7 siRNAs in mammalian cells is specific, as reported previously for chemically synthesized siRNAs (6,9). Caplen and colleagues used siRNAs of different lengths and showed that both 21- and 22-nt siRNAs work well to silence GFP (9). We have noticed that 22-nt-long T7 siRNAs may be more efficient for silencing transfected genes than 21 nt T7 siRNAs (data not shown). We conclude that T7 siRNAs can be introduced into cells and mediate their silencing effects with the non-liposome based calcium phosphate co-precipitation method.
Figure 2.
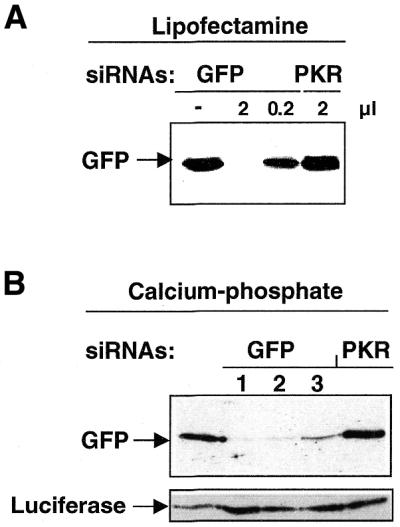
T7 siRNAs silence transfected genes in mammalian cells. (A) Plasmid pEGFP-C1 and decreasing amounts of siRNAs were co-transfected using a lipofectamine-based protocol. GFP was revealed with an anti-GFP antibody. T7 siRNA against PKR was transfected as a specificity control. (B) T7 siRNAs directed against GFP (1–3 represent siRNAs from three independent transcription reactions) or PKR were transfected using the calcium phosphate co-precipitation technique into HeLa cells with the plasmids pEGFP-C1 and pcDNA3-Luc. GFP and luciferase were detected with anti-GFP and anti-luciferase polyclonal antibodies, respectively.
To complete our study, we tested T7 siRNA on an endogenous gene, the gene for the interferon-induced protein kinase PKR (14). For this target, the T7 siRNA was 21 nt long, as described recently for other endogenous targets (6,7). In this case, the main challenge for a successful RNAi experiment is to achieve a high transfection efficiency. Using either lipofectamine or calcium phosphate co-precipitation, the transfection efficiency reached almost 80% for HeLa and 293T cells (as judged by transfection of GFP; data not shown). As presented in Figure 3, T7 siRNA targeted against human PKR, transfected into 293T by the calcium phosphate co-precipitation technique, was able to down-regulate endogenous PKR 48 h after the beginning of transfection. The silencing was almost completely effective with 2 µl of T7 siRNA. The residual PKR protein observed by immunoblot may reflect the 10–20% untransfected cells. The T7 siRNA was specific since no effect was observed on the unrelated protein Hsp90 (Fig. 3).
Figure 3.
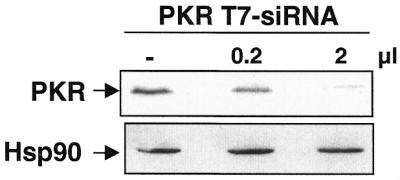
T7 siRNA silences an endogenous gene in mammalian cells. Aliquots of 0.2 or 2 µl of T7 siRNA directed against PKR were transfected using the calcium phosphate co-precipitation technique into 293T cells. Forty hours after transfection, cells were harvested and equal amounts of proteins were loaded on a 10% polyacrylamide gel. PKR was detected using a polyclonal antibody against PKR. Hsp90 was revealed as a specificity and loading control.
In conclusion, we report here a new protocol for the synthesis of siRNAs by T7 RNA polymerase and their transfer into cells. The main advantage of this technique is its simplicity and its extremely low cost compared with the current prices for synthetic RNA oligonucleotides. The amount of T7 siRNA required for each sample is small and represents ∼1–5% of the synthesized short RNA. We estimate that the average yield of T7 siRNA per transcription reaction is 1–5 µg (per 200 pmol template) and that silencing experiments can be carried out with 40–200 ng of T7 siRNA (in 2 µl; see Figs 2 and 3). But even 10 times less was sufficient for a noticeable decrease in GFP expression (Fig. 2A, lane 0.2). The demonstration that RNAi also works with the calcium phosphate technique further contributes to making this powerful technology widely available and applicable. With our approach, RNA interference in mammalian cells may become as easy as in C.elegans or Drosophila, where long dsRNAs synthesized by T7 RNA polymerase can be used.
Acknowledgments
ACKNOWLEDGEMENTS
We thank David Toft for the Hsp90 antibody. We are grateful to Katharina Strub and Laurent Huck for experimental advice. This work was supported by the Swiss National Science Foundation, Krebs Forschung Schweiz and the Canton de Genève.
REFERENCES
- 1.Carthew R.W. (2001) Gene silencing by double-stranded RNA. Curr. Opin. Cell Biol., 13, 244–248. [DOI] [PubMed] [Google Scholar]
- 2.Hammond S.M., Bernstein,E., Beach,D. and Hannon,G.J. (2000) An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature, 404, 293–296. [DOI] [PubMed] [Google Scholar]
- 3.Zamore P.D., Tuschl,T., Sharp,P.A. and Bartel,D.P. (2000) RNAi: double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell, 101, 25–33. [DOI] [PubMed] [Google Scholar]
- 4.Bernstein E., Caudy,A.A., Hammond,S.M. and Hannon,G.J. (2001) Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature, 409, 363–366. [DOI] [PubMed] [Google Scholar]
- 5.Williams B.R. (1999) PKR; a sentinel kinase for cellular stress. Oncogene, 18, 6112–6120. [DOI] [PubMed] [Google Scholar]
- 6.Elbashir S.M., Harborth,J., Lendeckel,W., Yalcin,A., Weber,K. and Tuschl,T. (2001) Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature, 411, 494–498. [DOI] [PubMed] [Google Scholar]
- 7.Harborth J., Elbashir,S.M., Bechert,K., Tuschl,T. and Weber,K. (2001) Identification of essential genes in cultured mammalian cells using small interfering RNAs. J. Cell Sci., 114, 4557–4565. [DOI] [PubMed] [Google Scholar]
- 8.Garrus J.E., von Schwedler,U.K., Pornillos,O.W., Morham,S.G., Zavitz,K.H., Wang,H.E., Wettstein,D.A., Stray,K.M., Cote,M., Rich,R.L., Myszka,D.G. and Sundquist,W.I. (2001) Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell, 107, 55–65. [DOI] [PubMed] [Google Scholar]
- 9.Caplen N.J., Parrish,S., Imani,F., Fire,A. and Morgan,R.A. (2001) Specific inhibition of gene expression by small double-stranded RNAs in invertebrate and vertebrate systems. Proc. Natl Acad. Sci. USA, 98, 9742–9747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Milligan J.F. and Uhlenbeck,O.C. (1989) Synthesis of small RNAs using T7 RNA polymerase. Methods Enzymol., 180, 51–62. [DOI] [PubMed] [Google Scholar]
- 11.Donzé O., Jagus,R., Koromilas,A.E., Hershey,J.W. and Sonenberg,N. (1995) Abrogation of translation initiation factor eIF-2 phosphorylation causes malignant transformation of NIH 3T3 cells. EMBO J., 14, 3828–3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Donzé O. and Picard,D. (1999) Hsp90 binds and regulates Gcn2, the ligand-inducible kinase of the alpha subunit of eukaryotic translation initiation factor 2. Mol. Cell. Biol., 19, 8422–8432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lipardi C., Wei,Q. and Paterson,B.M. (2001) RNAi as random degradative PCR: siRNA primers convert mRNA into dsRNAs that are degraded to generate new siRNAs. Cell, 107, 297–307. [DOI] [PubMed] [Google Scholar]
- 14.Donzé O., Abbas-Terki,T. and Picard,D. (2001) The Hsp90 chaperone complex is both a facilitator and a repressor of the dsRNA-dependent kinase PKR. EMBO J., 20, 3771–3780. [DOI] [PMC free article] [PubMed] [Google Scholar]