Translocation of Cockayne syndrome group A protein to the nuclear matrix: Possible relevance to transcription-coupled DNA repair (original) (raw)
Abstract
Transcription-coupled repair (TCR) efficiently removes a variety of lesions from the transcribed strand of active genes. By allowing rapid resumption of RNA synthesis, the process is of major importance for cellular resistance to transcription-blocking genotoxic damage. Mutations in the Cockayne syndrome group A or B (CSA or CSB) gene result in defective TCR. However, the exact mechanism of TCR in mammalian cells remains to be elucidated. We found that CSA protein is rapidly translocated to the nuclear matrix after UV irradiation. The translocation of CSA was independent of Xeroderma pigmentosum group C, which is specific to the global genome repair subpathway of nucleotide excision repair (NER) and of the core NER factor Xeroderma pigmentosum group A but required the CSB protein. In UV-irradiated cells, CSA protein colocalized with the hyperphosphorylated form of RNA polymerase II, engaged in transcription elongation. The translocation of CSA was also induced by treatment of the cells with cisplatin or hydrogen peroxide, both of which produce damage that is subjected to TCR but not induced by treatment with dimethyl sulfate, which produces damage that is not subjected to TCR. The hydrogen peroxide-induced translocation of CSA was also CSB dependent. These findings establish a link between TCR and the nuclear matrix mediated by CSA.
Nucleotide excision repair (NER) is a versatile DNA repair system correcting a broad spectrum of DNA damage, including UV-induced cyclobutane pyrimidine dimers and (6–4) photoproducts as well as chemical carcinogen-induced lesions (1). The process of NER involves damage recognition, local opening of the DNA helix, dual incisions on both sides of the lesion, removal of the oligonucleotide containing the damage, gap-filling DNA synthesis, and ligation (2). There are two subpathways in NER (3). One is transcription-coupled repair (TCR), which efficiently removes the damage on the transcribed strand of transcriptionally active genes. The other is global genome repair (GGR), which occurs throughout the genome including the nontranscribed strand of active genes.
Xeroderma pigmentosum (XP) is an autosomal recessive disease characterized by hypersensitivity to sunlight and a high incidence of skin cancer on sun-exposed skin (1, 4). Cells from XP patients are hypersensitive to killing by UV irradiation. XP is classified into seven genetic complementation groups (XP-A to -G) and a variant form (XP-V) (1). The primary defect in XP-A to XP-G resides in NER and both TCR and GGR are defective in XP-A to XP-G except XP-C, in which only GGR is impaired (4). XP-V has normal NER activity but a defect in translesion DNA synthesis (5). Cockayne syndrome (CS) is an autosomal recessive disease that shows diverse clinical symptoms including photosensitivity, severe mental retardation, and developmental defects, but no predisposition to UV-induced skin cancer (6). CS is classified into two genetic complementation groups (CS-A and CS-B). XP-B patients and certain patients belonging to XP-D or XP-G show features of CS in addition to symptoms of XP (XP-B/CS, XP-D/CS, and XP-G/CS) (1,4). CS-A and CS-B cells are deficient in TCR but proficient in GGR (7,8). Moreover, it has been shown that oxidative damage, such as 8-oxoguanine and thymine glycol, on the transcribed strand is removed by TCR, and that TCR of oxidative damage is proficient in normal human, XP-A, XP-D, and XP-G cells, but deficient in CS-B, XP-B/CS, XP-D/CS, and XP-G/CS cells (9, 10). It has been reported also that TCR of oxidative damage is partially deficient in CS-A cells when compared with the cells from normal individual (10). From these results, it is suggested that TCR is a discrete pathway for the rapid removal of DNA damage that blocks transcription rather than a subpathway of NER, and that the CS-specific features result from defects in TCR of oxidative damage.
All of the XP and CS (XPA to XPG, XPV, CSA, and CSB) genes have been cloned (4, 11). The core reaction of global NER in humans has been reconstituted in vitro with purified proteins (12–14), whereas the molecular mechanism of TCR in NER has been resolved only in_Escherichia coli_. The transcription-repair coupling factor encoded by the mfd gene binds to and displaces an RNA polymerase arrested at a DNA lesion and then promotes removal of the damage by recruiting the UvrABC excinuclease (15). In human cells, CSA and CSB as well as XPB, XPD, XPG, hMSH2 (16, 17), hMLH1 (16, 17), BRCA1 (18), and XAB2 (19) are involved in TCR, but the exact mechanism of TCR remains to be elucidated. It has been reported that the CSA is a 44-kDa protein with five WD 40 repeats that appears to have the potential to interact with other proteins. It has been shown that the CSA protein interacts with XAB2, CSB, and the p44 subunit of transcription factor IIH (TFIIH) (19, 20). However, CSA neither binds to RNA polymerase II (RNAP II) (21) nor releases the stalled RNAP II elongation complex (22). Thus, the function of the CSA protein in TCR has been obscure. Here we report a function of CSA relevant to the mechanism of TCR: CSB-dependent translocation of the CSA protein to the nuclear matrix after DNA damage, which is known to be subjected to TCR.
Materials and Methods
Cell Lines.
The cell lines used in this study were HeLa cells and simian virus 40 immortalized human fibroblasts; MRC5SV (wild type), WI38VA13 (wild type), CS3BESV (CS-A), CS1ANSV (CS-B), CS1BESV (CS-B), XP2OSSV (XP-A), and XP4PASV (XP-C). Whole cell extracts were prepared according to published protocols (23, 24).
Antibodies.
Preparation and affinity purification of the rabbit polyclonal anti-CSA and anti-CSB antibodies have been described previously (24). Anti-hemagglutinin antigen (HA) antibody (3F10) was purchased from Roche Diagnostics. Anti-glucocorticoid receptor (E-20), anti-lamin B (C-20), and anti-XPA (FL-273) antibodies were from Santa Cruz Biotechnology. Anti-retinoblastoma antibody was from PharMingen. Anti-nuclear pore complex (mAb414), anti-RNAP II (8WG16 and H5) antibodies were from Berkeley Antibody. Anti-XPC antibody was provided by F. Hanaoka (Osaka University).
Generation of a Tagged CSA Construct.
The CSA cDNA (20) was obtained via reverse transcription–PCR from human granulocyte RNA. Primers encoding for CSA cDNA (underlined) flanked by _Eco_RI sites were used: 5′-GCTAGAATTCTAATGCTGGGGTTTTTGTCC-3′ and 5′-AGTGATGAAGAAGGATGAGAATTCTTGG-3′. The PCR product was cloned into pcDNA3 (Invitrogen), resulting in pcDNA3-CSA. Sequencing of the construct ruled out the presence of PCR-derived mistakes. For the generation of the tagged CSA construct, full length CSA cDNA mutated in the stop codon (TGA to TGG) was cloned in-frame and upstream of a sequence encoding six histidine residues (His6) followed by a HA epitope in a pBluescript backbone, yielding pBL-CSA-His6-HA. The part of the cDNA corresponding to the C terminus of His6-HA-tagged CSA was isolated by _Afl_II–_Xba_I digestion and exchanged with the corresponding sequence in the wild-type pcDNA3-CSA, resulting in pcDNA3-CSA-His6-HA. For simplicity, this construct is referred to as double-tagged CSA (dtCSA).
DNA Transfection and UV Survival.
CS-A fibroblasts (CS3BESV) were transfected with the pcDNA3-CSA or pcDNA3-dtCSA construct by using SuperFect transfection reagent (Qiagen, Chatsworth, CA). After selection with G418 (200 μg/ml), stable transfectants were selected for UV resistance by irradiation with 4 J/m2 of UV-C for 3 consecutive days. UV survival of the transfectants was assessed as previously described (24,25).
DNA Damage Induction and Fractionation of Cellular Proteins.
To examine the translocation of CSA protein induced by DNA damage, cells were incubated for a period after UV irradiation (5 J/m2 for NER-deficient cells and 20 J/m2 for NER-proficient cells). In some experiments, cells were treated with either 100 μM cisplatin [_cis_-diamminedichloroplatinum (II)] for 120 min, 10 mM hydrogen peroxide for 15 min followed by incubation for 60 min in the absence of hydrogen peroxide, or 150 μM dimethyl sulfate (DMS) for 30 min followed by incubation for 60 min in the absence of DMS. The dose of these chemical agents was based on previous reports (9, 26, 27).
The cells were harvested by trypsinization, and then cellular proteins were fractionated as described (28–30), with some modification. After being washed with PBS, cells were extracted in cytoskeleton (CSK) buffer [10 mM Pipes, pH 6.8/100 mM NaCl/300 mM sucrose/3 mM MgCl2], supplemented with 0.5% (vol/vol) Triton X-100/1 mM DTT/1 mM EGTA/1 mM PMSF/1 μg/ml of leupeptin/1 μg/ml of pepstatin at 4°C for 10 min. The cytoskeletal frameworks were separated from soluble proteins by centrifugation at 7,500 × g for 3 min (fraction 1). The pellet was washed twice with a solution containing 250 mM sucrose and 5 mM MgCl2 (fraction 2) and resuspended with 25 mM Tris⋅HCl, pH 7.4/250 mM sucrose/5 mM MgCl2/1 mM PMSF. Chromatin was solubilized by DNA digestion with 1 mg/ml of DNase I at 30°C for 1 h. The sample was centrifuged at 7,500 × g for 3 min (fraction 3). The pellet was washed three times with a low-salt buffer (10 mM Tris⋅HCl, pH 7.4/0.2 mM MgCl2/1 mM PMSF) (fraction 4), extracted consecutively with the low-salt buffer containing an increasing concentration of NaCl (0.3, 0.5, and 2.0 M) for 15 min, and centrifuged at 20,000 × g for 15 min (fraction 5, 6, and 7 respectively). The high-salt pellet was finally extracted with the low-salt buffer containing 1% (vol/vol) Triton X-100 for 15 min and centrifuged at 20,000 × g for 15 min (fraction 8). The remaining pellet was washed twice with the low-salt buffer and solubilized in SDS/PAGE loading buffer (fraction 9).
Immunofluorescence Microscopy.
Cells grown on coverslips were washed twice with PBS and once with CSK buffer and incubated in CSK buffer supplemented with 0.5% (vol/vol) Triton X-100/0.5 mM PMSF/10 μg/ml of leupeptin (the Triton-extraction buffer) at 20°C for 20 min. In the indicated samples, DNase I (Roche Diagnostics; Grade II) was added to the above Triton-extraction buffer at a concentration of 0.2 mg/ml. Cells were washed twice with CSK buffer and fixed with 3% paraformaldehyde in PBS on ice for 20 min. Cells were then washed with PBS and treated sequentially with 50, 75, and 95% ethanol on ice for 5 min each. The samples were then blocked with PBS containing 5% normal goat serum (blocking buffer) at room temperature for 30 min and incubated in blocking buffer containing anti-HA (1 μg/ml) and antinuclear pore complex antibodies (0.5 μg/ml) at room temperature for 1 h. For double immunofluorescence labeling by using anti-HA and anti-RNAP II antibodies, the samples after incubation in blocking buffer were incubated in blocking buffer containing anti-HA antibody first and then in blocking buffer containing anti-RNAP II antibody (diluted 1:500). Cells were washed three times with PBS for 5 min, incubated with Alexa Fluor 488-conjugated anti-rat IgG antibody and Alexa Fluor 568-conjugated anti-mouse IgG antibody in PBS (diluted 1:500; Molecular Probes) at room temperature for 1 h, and washed three times with PBS. DNA staining was performed by incubating in TO-PRO-3 iodide in PBS (diluted 1:5000; Molecular Probes) at room temperature for 5 min and followed by three washes with PBS. The samples were examined with an MRC-1024 fluorescence microscopy system (Bio-Rad).
Results
UV Sensitivity and CSA Expression in the dtCSA/CS3BESV Cells.
We generated a cDNA construct that expresses CSA protein containing a His6 and a HA epitope at the C terminus (dtCSA) and transfected CS3BESV (CS-A) cells with it. To verify that the addition of the His6 and HA tags did not interfere with CSA function, we determined the relative UV sensitivity of the dtCSA-transfected CS3BESV (dtCSA/CS3BESV) cells. We found that the UV survival of the dtCSA/CS3BESV cells was similar to that of the MRC5SV (wild type) cells and the nontagged CSA-transfected CS3BESV (CSA/CS3BESV) cells (Fig. 6_A_, which is published as supporting information on the PNAS web site, www.pnas.org). Immunoblot analysis by using anti-CSA antibody indicated that the expression of dtCSA in the dtCSA/CS3BESV cells was similar to that of the endogenous CSA in HeLa cells, and anti-HA antibody was also able to detect dtCSA protein in the dtCSA/CS3BESV cells (Fig. 6_B_, which is published as supporting information on the PNAS web site).
Translocation of CSA Protein in UV-Irradiated Cells.
To gain an insight into the function of CSA protein, we examined whether UV-irradiation changes the subcellular localization of the protein. We fractionated cellular proteins from UV- and nonirradiated dtCSA/CS3BESV cells with serial extractions by using different concentrations of salt and detergent and analyzed the localization of dtCSA protein by immunoblotting with anti-HA antibody (Fig.1). In the nonirradiated cells, CSA protein was mainly detected in the fraction extracted with 0.5% Triton X-100 and in the wash fraction (lanes 1 and 2). Some of the CSA protein was detected in the DNase I-digested fraction (lane 3) and a very small amount in the fraction extracted with 0.3 M NaCl (lane 5). In the cells immediately after UV irradiation, the CSA protein was detected in the fractions extracted with salt (lanes 5–7) and 1% Triton X-100 (lane 8) besides the 0.5% Triton-soluble and DNase I-digested fractions (lanes 1–3) and was also detected in the remaining fraction resistant to high salt, DNase I, and high Triton, which we henceforth designate as “nuclear matrix” fraction (lane 9). Shortly after UV irradiation (15–30 min), this shift became more pronounced, whereas in the cells incubated for 60–180 min after UV irradiation, the CSA protein was detected mostly in the nuclear matrix fraction (lane 9) and in the 0.5% Triton-soluble fraction (lane 1). Essentially the same results were obtained from experiments with a lower dose (8 J/m2) of UV (data not shown). These results indicate that part of the CSA protein was translocated to the nuclear matrix fraction after UV irradiation.
Figure 1.
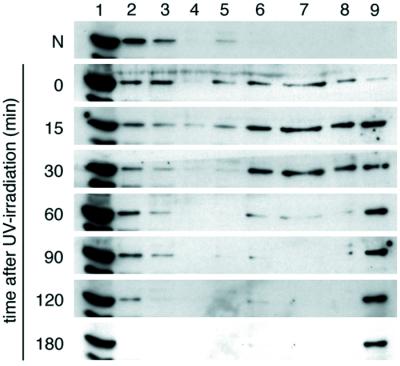
UV-induced translocation of CSA protein to the nuclear matrix in dtCSA/CS3BESV cells. Asynchronous cells were irradiated with 20 J/m2 of UV or nonirradiated and incubated for the indicated period, and then the cellular extracts were prepared as described in Materials and Methods. Proteins from each fraction with equivalent cell numbers were loaded for SDS/PAGE and analyzed by immunoblotting with anti-HA antibody. The lane numbers correspond to the fraction numbers.
It has been reported that the XPG protein is redistributed in the nucleus after UV irradiation (31) and the XPA protein associates more tightly with genomic DNA after UV irradiation (29). Therefore, we examined whether the subcellular localization of other DNA repair proteins changes after UV irradiation. (Fig. 7, which is published as supporting information on the PNAS web site). The XPA protein was detected only in the 0.5% Triton-soluble fraction in the nonirradiated cells, whereas it was also detected in the 0.3 M NaCl fraction in the UV-irradiated cells. These results were consistent with the previously described finding (29). It has been reported that the CSB protein interacts with the in vitro translated CSA protein (20) but not with CSA protein in vivo (24). In our present experimental conditions, the subcellular translocation of CSB protein was not evident after UV irradiation. In addition, the subcellular localization of XPC protein did not change. Although part of RNAP II was detected in the nuclear matrix fraction, the subcellular localization was not influenced by UV irradiation. Because RNAP II is involved in transcription as well as in DNA repair, the majority located in the nuclear matrix fraction may be involved in transcription. Thus, among the repair proteins tested in this study, CSA protein was the only one that was clearly translocated to the nuclear matrix fraction after UV irradiation.
Our fractionation procedures were validated by comparison with the distribution of glucocorticoid receptor, Rb protein, and lamin B (data not shown). The localization of these proteins was consistent with the one in a previous report (30).
Immunofluorescence Analysis of CSA Protein in the UV-Irradiated Cells.
The localization of CSA protein in the UV-irradiated cells was also examined by immunofluorescence analysis. An asynchronous population of dtCSA/CS3BESV cells was irradiated with UV. Two hours after UV irradiation, cells were permeabilized with a buffer containing 0.5% Triton X-100 before fixation, and the subcellular localization of CSA protein was monitored by immunofluorescence by using anti-HA rat monoclonal antibody (Fig. 2A). The entire cell population displayed strong nuclear staining for CSA protein that was resistant to the Triton extraction, and the staining patterns of CSA protein were not homogeneous in nuclei, with a diffuse nucleoplasmic staining and foci. On the other hand, the nonirradiated cells did not show any immunofluorescence in their nuclei. When cells were not extracted by Triton before fixation, CSA protein was uniformly stained in the nuclei, and the intensity of the CSA staining was the same in both UV and nonirradiated cells (data not shown). To exclude the possibility that the CSA protein may associate tightly to DNA after UV irradiation, the UV-irradiated dtCSA/CS3BESV cells were treated with DNase I in the presence of 0.5% Triton X-100 (Fig.2B). No significant difference in the staining of CSA protein was detected between DNase I-treated and untreated cells, although DNA was completely digested in the treated cells as shown by no reactivity to TO-PRO-3 iodide. On the other hand, nuclear pores in the nuclear envelope were preserved during DNase I treatment, as shown by reactivity to antinuclear pore complex antibody, verifying the integrity of the nuclear matrix even after DNase I treatment. These results indicate that the CSA protein is associated with nuclear matrix but not directly with DNA after UV irradiation.
Figure 2.

Immunofluorescence staining of CSA protein in dtCSA/CS3BESV cells after UV irradiation. (A) Asynchronous dtCSA/CS3BESV cells were irradiated with 20 J/m2 of UV (+UV) or nonirradiated (−UV) and treated with the Triton-extraction buffer before fixation. The CSA protein was detected with anti-HA rat monoclonal antibody and visualized with Alexa Fluor 488-conjugated anti-rat IgG antibody (green). DNA was shown by TO-PRO-3 staining (dark-red). (B) Asynchronous dtCSA/CS3BESV cells were irradiated with 20 J/m2 of UV and treated with Triton-extraction buffer supplemented with or without 0.2 mg/ml of DNase I before fixation. The CSA protein was detected as described. Nuclear pore complex was detected with antinuclear pore complex antibody and visualized by Alexa Fluor 568-conjugated anti-mouse IgG antibody (red).
CSA Protein Was Not Translocated in CSB Cells After UV Irradiation.
We then examined the translocation of CSA protein after UV irradiation in NER-deficient cells (Fig.3A). The XP2OSSV (XP-A) cells are deficient in both TCR and GGR. The XP4PASV (XP-C) cells are selectively deficient in GGR, whereas the CS1ANSV and CS1BESV (both are CS-B) cells are selectively defective in TCR. The CSA protein was translocated to the nuclear matrix fraction 60 min after UV irradiation in the XP-A and the XP-C cells as well as in the normal human cells (WI38VA13). In normal cells, the CSA protein was detected in the nuclear matrix fraction even 12 h after UV irradiation (data not shown). In contrast, the translocation of the CSA protein did not occur in two different CS-B cell lines (Fig. 3A and data not shown). The CSA protein was not detected in the nuclear matrix fraction even 180 min post UV incubation in CS-B cells (Fig. 3B and data not shown). However, the CS-B cells stably expressing double-tagged CSB cDNA (dtCSB/CS1ANSV), which were proficient in TCR (24), resumed normal CSA translocation to the nuclear matrix fraction after UV irradiation. These results indicate that the UV-induced translocation of the CSA protein to the nuclear matrix fraction depends on the CSB protein.
Figure 3.

Translocation of CSA protein to the nuclear matrix in DNA repair-deficient cell lines following UV-irradiation. (A) The XP-A (XP2OSSV), XP-C (XP4PASV), and CS-B (CS1ANSV) cells, which are deficient in NER, were irradiated with 5 J/m2 of UV, whereas the wild-type (WI38VA13) cells and the CS1ANSV cells expressing the dtCSB construct (dtCSB/CS1ANSV), which are proficient in NER, were irradiated with 20 J/m2 of UV. Each fraction was prepared from cells that were incubated for 60 min after UV irradiation or from nonirradiated cells and then immunoblotted with anti-CSA antibody. The lane numbers correspond to the fraction numbers. (B) The nuclear matrix fraction was prepared from UV-irradiated cells after incubation for the period indicated, and the translocation of CSA protein to the nuclear matrix was examined by immunoblotting with anti-CSA antibody.
Colocalization of CSA Protein with the Hyperphosphorylated Form of RNAP II in the UV-Irradiated Cells.
It has been reported that the hyperphosphorylated form of RNAP II, which is essential for transcriptional elongation, interacts with the nuclear matrix (32–34). Furthermore, NER may also occur in association with the nuclear matrix (35–38). We therefore examined whether RNAP II and CSA protein were colocalized in the nuclear matrix (Fig.4). The dtCSA/CS3BESV cells were incubated for 120 min after UV irradiation and permeabilized with a buffer containing Triton X-100 and DNase I before fixation. The localization of RNAP II and CSA protein in same cells was monitored with anti-RNAP II monoclonal antibody [hypophosphorylated form (IIa) and hyperphosphorylated form (IIo) detected by 8WG16, and IIo detected by H5] and anti-HA rat monoclonal antibody, respectively. CSA protein was almost completely colocalized with RNAP II foci detected by H5 but only partially colocalized with RNAP II foci detected by 8WG16. These results suggest that CSA protein cooperates with the hyperphosphorylated form of RNAP II at the nuclear matrix in the process of TCR.
Figure 4.

CSA protein was colocalized with the hyperphosphorylated form of RNAP II in the nuclear matrix after UV irradiation. Asynchronous dtCSA/CS3BESV cells were irradiated with 20 J/m2 of UV and treated with Triton X-100 and DNase I before fixation. CSA protein was detected with anti-HA rat monoclonal antibody and visualized with Alexa Fluor 488-conjugated anti-rat IgG antibody (green;Top). RNAP II was detected with anti-RNAP II monoclonal antibodies (IIo by H5, IIa + IIo by 8WG16) and visualized with Alexa Fluor 568-conjugated anti-mouse IgG antibody (red;Middle). Merged images are displayed (Bottom).
Translocation of CSA Protein to the Nuclear Matrix by Other DNA Damage That Is Subjected to TCR.
To further elucidate the relationship between translocation of CSA protein to the nuclear matrix and the TCR process, we examined the effect of other DNA-damaging agents such as cisplatin, hydrogen peroxide, and DMS on the translocation of CSA protein. Intrastrand crosslinks induced by cisplatin and thymine glycol induced by hydrogen peroxide are known to be removed by TCR similar to UV damage (9, 27). In contrast, DMS-induced lesions (7-methylguanine and 3-methyladenine) are efficiently repaired by global base excision repair (BER) and not subjected to TCR (26). As shown in Fig.5A, CSA protein was translocated to the nuclear matrix fraction on treatment of the cells with cisplatin or hydrogen peroxide but not with DMS. Moreover, the translocation of CSA protein did not occur in the hydrogen peroxide-treated CS-B cells but did occur in XP-A, XP-C, and dtCSB/CS1ANSV cells (Fig. 5B). Thus the hydrogen peroxide-induced translocation of CSA protein to the nuclear matrix fraction also depended on the CSB protein. All these results indicate that the translocation of CSA protein to the nuclear matrix fraction occurs in connection with TCR.
Figure 5.

Translocation of CSA protein to the nuclear matrix by chemical agents whose damage is subjected to TCR. (A) The dtCSA/CS3BESV cells were irradiated with 20 J/m2 of UV and incubated for 60 min or treated (incubated) with DNA damaging agents (100 μM cisplatin; 10 mM hydrogen peroxide; 150 μM DMS). Each fraction was prepared from cells and then immunoblotted with anti-HA antibody. The lane numbers correspond to the fraction numbers. (B) XP-A (XP2OSSV), XP-C (XP4PASV), CS-B (CS1ANSV), wild-type (WI38VA13) cells, and CS1ANSV cells expressing the dtCSB construct (dtCSB/CS1ANSV) were treated with 10 mM hydrogen peroxide and then immunoblotted with anti-CSA antibody. The lane numbers correspond to the fraction numbers.
Discussion
We found that CSA protein was rapidly translocated to the nuclear matrix after UV irradiation (Figs. 1 and 2) and that the translocation was independent of XPC and XPA but required CSB protein (Fig. 3). In mammalian cells, the XPC/hHR23B complex serves as a damage sensor to initiate GGR and recruits the core NER machinery to the lesion (39,40). The DNA around the lesion is then opened in an ATP-dependent manner by the general transcription factor TFIIH, which contains XPB and XPD helicases. Open complex formation also depends on the presence of XPA and RPA (41). The XPA protein verifies the damage in an open DNA conformation and is crucial for proper orientation of the remainder of the NER machinery. These results indicate that the UV-induced translocation of CSA protein to the nuclear matrix fraction is relevant neither to the GGR-specific step(s) nor to the core NER reactions. On the other hand, the genetic and cell biological evidence indicates that the CSA and CSB proteins are involved in the TCR-specific step(s), although the exact mechanism of TCR in mammalian cells is unknown as yet. Our results therefore suggested that the UV-induced translocation of CSA protein to nuclear matrix is specifically related to the TCR process.
It has been reported that oxidative DNA damage such as thymine glycol and 8-oxoguanine is removed by TCR that does not involve NER (9). To verify that the translocation of CSA protein to the nuclear matrix is also related to TCR of oxidative damage, we examined the effect of hydrogen peroxide on the translocation of CSA as well as cisplatin and DMS, which produce damage that is subjected to NER and BER, respectively. It has been shown that the damage on the transcribed strand induced by cisplatin or hydrogen peroxide is removed by TCR, but the damage by DMS is not (9, 26, 27). We found that the translocation of CSA was induced by treatment of the cells with cisplatin or hydrogen peroxide but not by treatment with DMS (Fig. 5A). In addition, the hydrogen peroxide-induced translocation of CSA was also CSB-dependent (Fig. 5B). These results strongly suggest that the translocation of CSA to the nuclear matrix is relevant not only to TCR in NER but also to TCR of oxidative DNA damage. The mechanisms of the two TCR processes are expected to be much the same. Presumably, the elongating RNAP II complex that encounters a lesion detects the DNA damage early in the TCR pathway. The actual repair proteins involved in either NER or BER are then recruited, and the lesion is repaired. The transcription is resumed when the lesion has been repaired. Consistent with this model, CSA protein was colocalized with the hyperphosphorylated form of RNAP II, which is engaged in transcription elongation in UV-irradiated cells (Fig. 4).
Our results revealed that the CSA protein was translocated to the nuclear matrix, whereas most of the CSB protein was detected in the 0.5% Triton-soluble and DNase I-digested fractions and not in the nuclear matrix fraction following DNA damage (Fig. 7, which is published as supporting information on the PNAS web site). Although this finding does not indicate a direct involvement of CSB in the recruitment of CSA to the nuclear matrix on DNA damage, it does not exclude the possibility of transient interactions or an indirect role. On the other hand, the CSB protein interacts with RNAP II and can stimulate transcriptional elongation (42). In addition, it counteracts TFIIS-induced transcript shortening (42) but does not dissociate the stalled RNAP II from DNA (22). It has been shown that CSB interacts with a ternary complex containing DNA, RNA and stalled RNAP II in a manner that requires ATP hydrolysis, and that the quaternary complex can recruit TFIIH (21, 43). These results suggest that in the TCR subpathway, the CSB protein interacts with a stalled RNAP II elongation complex and plays an important role in recruiting TFIIH to the site of a DNA lesion. Moreover, CSB is able to remodel chromatin structure at the expense of ATP hydrolysis (44). Thus the CSB protein may induce conformational changes in the stalled RNAP II complex or in the surrounding nucleosome structure during TCR. The conformational changes in the stalled RNAP II complex and/or the alterations of nucleosome structure induced by CSB may allow the CSA protein to translocate to the nuclear matrix, where transcription elongation and TCR occur. It may also make the DNA lesion on the transcribed strand accessible to actual repair factors.
The nuclear matrix is thought to play an important role in chromatin organization by providing the axis where complex arrays of chromatin loops are anchored. Additionally, the nuclear matrix is implicated in nuclear metabolism including DNA replication, transcription, and mRNA splicing. As for repair, enrichment of UV-induced repair patches in nuclear matrix fractions has been reported. Interestingly, this phenomenon was enhanced in TCR-proficient XP-C cells and abolished in TCR-deficient CS-B fibroblasts (45). These findings indicate that TCR takes place in the nuclear matrix, consistent with the present results.
In conclusion, our findings provide evidence that the involvement of CSA in TCR depends on CSB, and that CSA acts at a stage before actual repair factors, highlight a direct link between TCR and the nuclear matrix, and suggest that the protein complex required for TCR is not preassembled but may rapidly form when transcription elongation has been arrested.
Supplementary Material
Supporting Figures
Acknowledgments
We thank Dr. F. Hanaoka (Osaka University) for anti-XPC antibody and Drs. Y. Nakatsu and W. Vermeulen for helpful suggestions and critical reading of the manuscript. We also thank Mr. Vincent van den Boom (Erasmus University) for providing the anti-CSA antibody. This work was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan, Core Research for Evolutional Science and Technology (CREST) of Japan Science and Technology (JST), the European Community (DNA repair disorders and DNage contract QLRT 1999–02002), and the National Institutes of Health (Contract AG17242–02).
Abbreviations
CS
Cockayne syndrome
CSA
CS group A
dtCSA
double-tagged CSA
TCR
transcription-coupled repair
NER
nucleotide excision repair
BER
base excision repair
RNAP II
RNA polymerase II
XP
Xeroderma pigmentosum
GGR
global genome repair
HA
hemagglutinin antigen
DMS
dimethyl sulfate
CSK
cytoskeleton
RNAP II
RNA polymerase II
TFIIH
transcription factor IIH
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Friedberg E C, Walker G C, Siede W. DNA Repair and Mutagenesis. Washington, DC: Am. Soc. Microbiol.; 1995. [Google Scholar]
- 2.de Laat W L, Jaspers N G J, Hoeijmakers J H J. Genes Dev. 1999;13:768–785. doi: 10.1101/gad.13.7.768. [DOI] [PubMed] [Google Scholar]
- 3.Hanawalt P C, Spivak G. In: Advances in DNA Damage and Repair. Dizdaroglu M, Karakaya A E, editors. New York: Kluwer Academic/Plenum; 1999. pp. 169–179. [Google Scholar]
- 4.Bootsma D, Kreamer K H, Cleaver J E, Hoeijmakers J H J. In: The Genetic Basis of Human Cancer. Vogelstein B, Kinzler K W, editors. New York: McGraw–Hill; 1998. pp. 245–274. [Google Scholar]
- 5.Cordonnier A M, Fuchs P P. Mutat Res. 1999;435:111–119. doi: 10.1016/s0921-8777(99)00047-6. [DOI] [PubMed] [Google Scholar]
- 6.Nance M A, Berry S A. Am J Med Genet. 1992;42:68–84. doi: 10.1002/ajmg.1320420115. [DOI] [PubMed] [Google Scholar]
- 7.Venema J, Mullenders L H, Natarajan A T, van Zeeland A A, Mayne L V. Proc Natl Acad Sci USA. 1990;87:4707–4711. doi: 10.1073/pnas.87.12.4707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Hoffen A, Natarajan A T, Mayne L V, van Zeeland A A, Mullenders L H, Venema J. Nucleic Acids Res. 1993;21:5890–5895. doi: 10.1093/nar/21.25.5890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Le Page F, Kwoh E E, Avrutskaya A, Gentil A, Leadon S A, Sarasin A, Cooper P K. Cell. 2000;101:159–171. doi: 10.1016/s0092-8674(00)80827-2. [DOI] [PubMed] [Google Scholar]
- 10.Leadon S A, Cooper P K. Proc Natl Acad Sci USA. 1993;90:10499–10503. doi: 10.1073/pnas.90.22.10499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Masutani C, Kusumoto R, Yamada A, Dohmae N, Yokoi M, Yuasa M, Araki M, Iwai S, Takio K, Hanaoka F. Nature (London) 1999;399:700–704. doi: 10.1038/21447. [DOI] [PubMed] [Google Scholar]
- 12.Aboussekhra A, Biggerstaff M, Shivji M K, Vilpo J A, Moncollin V, Podust V N, Protic M, Hubscher U, Egly J M, Wood R D. Cell. 1995;80:859–868. doi: 10.1016/0092-8674(95)90289-9. [DOI] [PubMed] [Google Scholar]
- 13.Mu D, Park C H, Matsunaga T, Hsu D S, Readon J T, Sancar A. J Biol Chem. 1995;270:2415–2418. doi: 10.1074/jbc.270.6.2415. [DOI] [PubMed] [Google Scholar]
- 14.Araujo S J, Tirode F, Coin F, Pospiech H, Syvaoja J E, Stucki M, Hubscher U, Egly J M, Wood R D. Genes Dev. 2000;14:349–359. [PMC free article] [PubMed] [Google Scholar]
- 15.Selby C P, Sancar A. Science. 1993;260:53–58. doi: 10.1126/science.8465200. [DOI] [PubMed] [Google Scholar]
- 16.Mellon I, Rajpal D K, Koi M, Boland C R, Champe G N. Science. 1996;272:557–560. doi: 10.1126/science.272.5261.557. [DOI] [PubMed] [Google Scholar]
- 17.Leadon S A, Avrutskaya A V. Cancer Res. 1997;57:3784–3791. [PubMed] [Google Scholar]
- 18.Gowen L C, Avrutskaya A V, Latour A M, Koller B H, Leadon S A. Science. 1998;281:1009–1012. doi: 10.1126/science.281.5379.1009. [DOI] [PubMed] [Google Scholar]
- 19.Nakatsu Y, Asahina H, Citterio E, Rademarkers S, Vermeulen W, Kamiuchi S, Yeo J P, Khaw M C, Saijo M, Kodo N, et al. J Biol Chem. 2000;10:34931–34937. doi: 10.1074/jbc.M004936200. [DOI] [PubMed] [Google Scholar]
- 20.Henning K A, Li L, Iyer N, McDaniel L D, Reagan M S, Legerski R, Schultz R A, Stefanini M, Lehmann A R, Mayne L V, Friedberg E C. Cell. 1995;82:555–564. doi: 10.1016/0092-8674(95)90028-4. [DOI] [PubMed] [Google Scholar]
- 21.Tantin D. J Biol Chem. 1998;273:27794–27799. doi: 10.1074/jbc.273.43.27794. [DOI] [PubMed] [Google Scholar]
- 22.Selby C P, Sancar A. Proc Natl Acad Sci USA. 1997;94:11205–11209. doi: 10.1073/pnas.94.21.11205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Manley J L, Fire A, Samuels M, Sharp P A. Methods Enzymol. 1983;101:568–582. doi: 10.1016/0076-6879(83)01038-1. [DOI] [PubMed] [Google Scholar]
- 24.van Gool A J, Citterio E, Rademakers S, van Os R, Vermeulen W, Constantinou A, Egly J M, Bootsma D, Hoeijmakers J H J. EMBO J. 1997;16:5955–5965. doi: 10.1093/emboj/16.19.5955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sijbers A M, v&er Spek P J, Odijk H, van den Berg J, van Duin M, Westerveld A, Jaspers N G J, Bootsma D, Hoeijmakers J H J. Nucleic Acids Res. 1996;24:3370–3380. doi: 10.1093/nar/24.17.3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scicchitano D A, Hanawalt P C. Proc Natl Acad Sci USA. 1989;86:3050–3054. doi: 10.1073/pnas.86.9.3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.May A, Nairn R S, Okumoto D S, Wassermann K, Stevnsner T, Jones J C, Bohr V A. J Biol Chem. 1993;268:1650–1657. [PubMed] [Google Scholar]
- 28.He D C, Nickerson J A, Penman S. J Cell Biol. 1990;110:569–580. doi: 10.1083/jcb.110.3.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Otrin V R, McLenigan M, Takao M, Levine A S, Protic M. J Cell Sci. 1997;110:1159–1168. doi: 10.1242/jcs.110.10.1159. [DOI] [PubMed] [Google Scholar]
- 30.Reyes J C, Muchardt C, Yaniv M. J Cell Biol. 1997;137:263–274. doi: 10.1083/jcb.137.2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park M S, Knauf J A, Pendergrass S H, Coulon C H, Strniste G F, Marrone B L, MacInnes M A. Proc Natl Acad Sci USA. 1996;93:8368–8373. doi: 10.1073/pnas.93.16.8368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mortillaro M J, Blencowe B J, Wei X, Nakayasu H, Du L, Warren S L, Sharp P A, Berezney R. Proc Natl Acad Sci USA. 1996;93:8253–8257. doi: 10.1073/pnas.93.16.8253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Patturajan M, Wei X, Berezney R, Corden J L. Mol Cell Biol. 1998;18:2406–2415. doi: 10.1128/mcb.18.4.2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wei X, Somanathan S, Samarabandu J, Berezney R. J Cell Biol. 1999;146:543–548. doi: 10.1083/jcb.146.3.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McCready S J, Cook P R. J Cell Sci. 1984;70:189–196. doi: 10.1242/jcs.70.1.189. [DOI] [PubMed] [Google Scholar]
- 36.Harless J, Hewitt R R. Mutat Res. 1987;183:177–184. doi: 10.1016/0167-8817(87)90060-5. [DOI] [PubMed] [Google Scholar]
- 37.Mullenders L H F, van Zeeland A A, Natarajan A T. J Cell Sci Suppl. 1987;6:243–262. doi: 10.1242/jcs.1984.supplement_6.17. [DOI] [PubMed] [Google Scholar]
- 38.Koehler D R, Hanawalt P C. Nucleic Acids Res. 1996;24:2877–2884. doi: 10.1093/nar/24.15.2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sugasawa K, Ng J M Y, Masutani C, Iwai S, van der Spek P J, Eker A P M, Hanaoka F, Bootsma D, Hoeijmakers J H J. Mol Cell. 1998;2:223–232. doi: 10.1016/s1097-2765(00)80132-x. [DOI] [PubMed] [Google Scholar]
- 40.Yokoi M, Masutani C, Maekawa T, Sugasawa K, Ohkuma Y, Hanaoka F. J Biol Chem. 2000;275:9870–9875. doi: 10.1074/jbc.275.13.9870. [DOI] [PubMed] [Google Scholar]
- 41.Evans E, Moggs J G, Hwang J R, Egly J M, Wood R D. EMBO J. 1997;16:6559–6573. doi: 10.1093/emboj/16.21.6559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Selby C P, Sancar A. J Biol Chem. 1997;272:1885–1890. doi: 10.1074/jbc.272.3.1885. [DOI] [PubMed] [Google Scholar]
- 43.Tantin D, Kansal A, Carey M. Mol Cell Biol. 1997;17:6803–6814. doi: 10.1128/mcb.17.12.6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Citterio E, van den Boom V, Schnitzler G, Kanaar R, Bonte E, Kingston R E, Hoeijmakers J H J, Vermeulen W. Mol Cell Biol. 2000;20:7643–7653. doi: 10.1128/mcb.20.20.7643-7653.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mullenders L H, van Kesteren van Leeuwen A C, van Zeeland A A, Natarajan A T. Nucleic Acids Res. 1988;16:10607–10622. doi: 10.1093/nar/16.22.10607. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Figures