Average Risks of Breast and Ovarian Cancer Associated with BRCA1 or BRCA2 Mutations Detected in Case Series Unselected for Family History: A Combined Analysis of 22 Studies (original) (raw)
Abstract
Germline mutations in BRCA1 and BRCA2 confer high risks of breast and ovarian cancer, but the average magnitude of these risks is uncertain and may depend on the context. Estimates based on multiple-case families may be enriched for mutations of higher risk and/or other familial risk factors, whereas risk estimates from studies based on cases unselected for family history have been imprecise. We pooled pedigree data from 22 studies involving 8,139 index case patients unselected for family history with female (86%) or male (2%) breast cancer or epithelial ovarian cancer (12%), 500 of whom had been found to carry a germline mutation in BRCA1 or BRCA2. Breast and ovarian cancer incidence rates for mutation carriers were estimated using a modified segregation analysis, based on the occurrence of these cancers in the relatives of mutation-carrying index case patients. The average cumulative risks in _BRCA1-_mutation carriers by age 70 years were 65% (95% confidence interval 44%–78%) for breast cancer and 39% (18%–54%) for ovarian cancer. The corresponding estimates for BRCA2 were 45% (31%–56%) and 11% (2.4%–19%). Relative risks of breast cancer declined significantly with age for _BRCA1-_mutation carriers (P trend .0012) but not for _BRCA2-_mutation carriers. Risks in carriers were higher when based on index breast cancer cases diagnosed at <35 years of age. We found some evidence for a reduction in risk in women from earlier birth cohorts and for variation in risk by mutation position for both genes. The pattern of cancer risks was similar to those found in multiple-case families, but their absolute magnitudes were lower, particularly for BRCA2. The variation in risk by age at diagnosis of index case is consistent with the effects of other genes modifying cancer risk in carriers.
Introduction
Mutations in the breast and ovarian cancer–susceptibility genes BRCA1 (MIM 113705) (Miki et al. 1994) and BRCA2 (MIM 600185) (Wooster et al. 1995; Tavtigian et al. 1996) are found in a high proportion of multiple-case families with breast cancer, especially if they also include one or more case patients with ovarian cancer (Ford et al. 1994). Screening for mutations in these genes for predictive genetic testing has become widespread, with >750 protein-truncating mutations in these genes having been identified (see the Breast Cancer Information Core [BIC] Web site). Some women found to carry such mutations undergo prophylactic mastectomy and/or oophorectomy, because their cancer risk is extremely high. However, although it is very clear that mutations in these genes, segregating within these types of families, confer a substantial risk of both breast and ovarian cancer, the same may not apply to mutations detected in other settings, such as in families with less-extreme cancer histories or in incident cases, even those of early onset.
Several approaches have been used to estimate the average age-specific cumulative cancer risks, or penetrance, associated with mutations in BRCA1 and BRCA2. Early estimates applied the maximum-LOD-score (or linkage) method to multiple-case families collected for linkage studies for the identification of disease loci (Easton et al. 1993; Clerget-Darpoux 2001). Subsequent penetrance estimates have used the incidence of cancer in the relatives of mutation-carrying index case patients from case series unselected for family history. Analytically, these are the same method (i.e., a type of segregation analysis) applied with different corrections for family ascertainment. Both should give consistent estimates of penetrance, provided that the same penetrance function applies to all carriers. Different estimates will arise, however, either if the penetrance is mutation specific or if the penetrance is modified by other risk factors, genetic or environmental, that aggregate in families. Either of these phenomena would lead to a higher actual penetrance for mutations segregating in multiple-case families than for mutations segregating in the population as a whole. Some authors (e.g., Begg 2002) have described the penetrance estimates derived in this way as biased (Begg 2002). This is correct in the sense that they do not reflect the average risks to all carriers in the population. In practice, a counsellor is rarely interested in the risks to the “average” carrier. Virtually all genetic testing is conducted on women in families with multiple cases of the disease—the types of families from which the original penetrance estimates were derived. Some women are tested on the basis of weaker family histories or on the basis of having early-onset disease; risk estimates derived by studying the cancer incidence in relatives of population-based series of women with breast or ovarian cancer may then be more appropriate.
Published penetrance estimates are summarized in table 1. Breast and ovarian cancer risk estimates are generally higher in studies that are based on multiple-case families (Ford et al. 1994, 1998; Easton et al. 1995) than in those that are based on unselected series (Thorlacius et al. 1998; Hopper et al. 1999; Warner et al. 1999; Anglian Breast Cancer Study Group 2000). Another study, based on the family histories of 120 Ashkenazi Jewish volunteers in whom one of three different founder mutations common to this population had been identified, also reported lower penetrance estimates than reports based on multiple-case families (Struewing et al. 1997). These penetrance estimates are averages over the mutations segregating in the families in which mutations have been identified. There are, however, data to support the hypothesis of allelic risk heterogeneity, such that different mutations confer different risks. Specifically, BRCA2 mutations that occur in families with one or more cases of ovarian cancer tend to cluster in a central portion of the gene, termed the “ovarian cancer cluster region” (Gayther et al. 1997; Thompson and Easton 2001). A study of Breast Cancer Linkage Consortium (BCLC) families has shown that mutations in the ovarian cancer cluster region are associated with both a lower risk of breast cancer (relative risk [RR] 0.63) and a higher risk of ovarian cancer (RR = 1.9), as compared to mutations outside this region. Another study, based on probands with ovarian cancer, found that the _BRCA2-_associated breast cancer risk was associated only with mutations outside the ovarian cancer cluster region (Risch et al. 2001). Evidence for a genotype-phenotype correlation in BRCA1 has also been found. Gayther et al. (1995) have found that the risk of ovarian cancer relative to the risk of breast cancer was higher in families with protein-truncating mutations in the first two-thirds of the gene than in families with protein-truncating mutations in the last one-third of the gene. More recently, Thompson and Easton (2002) have found that mutations in a central region of BRCA1 were associated with a lower risk of breast cancer, and Risch et al. (2001) have reported that the risk of breast cancer increases with mutation position, from 5′ to 3′.
Table 1.
Previously Published Breast and Ovarian Cancer Risks Associated with Mutations in BRCA1 and BRCA2
| Breast Cancer Risk(95% CI) at(%) | Ovarian Cancer Risk(95% CI) at(%) | ||||
|---|---|---|---|---|---|
| Family Ascertainmenta | Age 50 Years | Age 70 Years | Age 50 Years | Age 70 Years | Reference |
| BRCA1: | |||||
| Multiple-case families with Br/Ov | 73 (49–87) | 87 (72–95) | 29 (16–40) | 44 (28–56) | Ford et al. 1994 |
| Multiple-case families with Br/Ov | 51 | 85 | 23 | 63 | Easton et al. 1995 |
| Multiple-case families with Ov | 39 | 72 | 17 | 53 | Antoniou et al. 2000 |
| Hospital-based Ov cases | 34 (17–60) | 50 (26–82) | 21 (8–47) | 68 (36–94) | Antoniou et al. 2000 |
| Hospital-based Ashkenazi Jewish Br casesb | 60 | Warner et al. 1999 | |||
| Hospital-based Ashkenazi Jewish Br casesb | 46 (31–80) | Satagopan et al. 2001 | |||
| Population-based Br cases | 32 (2–62) | 47 (5–82) | 11 (1–74) | 36 (4–99) | Anglian Breast Cancer Study Group 2000 |
| Population-based Ov cases | 13 | 30 | 5.4 | 15 | Risch et al. 2001 |
| BRCA1 and BRCA2: | |||||
| Population-based Br cases | 10 (0–24) | 40 (16–64) | Hopper et al. 1999 | ||
| Unaffected Ashkenazi Jewish womenc | 33 (23–44) | 56 (40–73) | 7 (2–14) | 16 (6–28) | Struewing et al. 1997 |
| BRCA2: | |||||
| Multiple-case families with Br/Ov | 28 (9–44) | 84 (43–95) | .4 (0–1) | 27 (47) | Ford et al. 1998 |
| Multiple-case Ov families | 19 | 71 | 1 | 31 | Antoniou et al. 2000 |
| Hospital-based Ashkenazi Jewish Br casesd | 28 | Warner et al. 1999 | |||
| Hospital-based Ashkenazi Jewish Br casesd | 26 (14–50) | Satagopan et al. 2001 | |||
| Population-based Br casese | 17 (9–26) | 37 (22–54) | Thorlacius et al. 1998 | ||
| Population-based Br cases | 18 (2–32) | 56 (5–80) | 3 (0–19) | 10 (1–55) | Anglian Breast Cancer Study Group 2000 |
| Population-based Ov cases | 3.6 | 8.8 | 1.6 | 4.5 | Risch et al. 2001 |
Penetrance estimates based on multiple-case families may be inappropriate for the counselling of women without a strong family history of disease who have been found to carry a germline mutation in BRCA1 or BRCA2. Although estimates based on mutation testing in case series unselected for family history are more appropriate in this context, published estimates from individual studies have lacked precision, with most studies having identified a few dozen mutations at most. To improve the precision of penetrance estimates based on unselected case series, we have combined data from a large number of such studies into a formal meta-analysis. This combined data set has also allowed us to examine variations in penetrance by type of mutation, type and age at diagnosis of index case, birth cohort, and study center.
Subjects and Methods
Studies
Studies were eligible for this meta-analysis if all of the following criteria were met: (1) The study was based on mutation testing of a series of index cases either of female or male breast cancer or of invasive epithelial ovarian cancer. (2) Index cases were sampled independently of family history (although they may have been selected by age at diagnosis or ethnic group). (3) Index cases had been tested for BRCA1 and/or BRCA2 mutations by a systematic screen conducted independently of family history (mutation screening may have been of the entire coding sequence, of some part of the sequence, or of specific founder mutations). (4) Enumeration of at least all first-degree relatives of identified mutation carriers was available, along with ages at diagnosis of breast and ovarian cancers and ages at last observation.
Potentially eligible studies were identified by a literature search using Medline (National Library of Medicine) and by personal contact through the BCLC. We contacted 21 research groups, in total, that we believed to have data from one or more relevant studies, and we received data from 15. Participating investigators were asked to provide details on all recorded members of families in which the index case patient was found to have a BRCA1 or BRCA2 mutation. These details included date of birth, date of or age at diagnosis of any breast or ovarian cancer, and age at death or age at last observation. Data on cancers other than breast or ovarian cancer were sometimes also available but were not used in the present analysis. Investigators were also asked to provide details of each mutation identified.
Details of the studies included are given in table 2. Of the 22 studies included, 16 were conducted by ascertainment of female breast cancer index cases, 2 were conducted by ascertainment of male breast cancer cases, and 4 were conducted by ascertainment of ovarian cancer cases. Of the studies based on breast cancer index cases, 10 restricted ascertainment by age at diagnosis; in 9 of these, the upper limit for age at diagnosis was ⩽50 years. None of the studies based on either male breast cancer index cases or ovarian cancer index cases imposed a restriction on age at diagnosis. Sixteen of the studies ascertained cases through a population-based cancer registry, whereas the remainder were based on unselected, hospital-based series. The recruitment method for the studies varied widely. Most studies obtained a blood sample and family-history data simultaneously, but some studies collected a blood sample first and then retrospectively obtained family-history data on mutation carriers whereas others obtained family history data first and collected a blood sample later. Furthermore, in some studies, not all available blood samples were analyzed.
Table 2.
Description of Studies Included in the Present Analysis
| No. of Patients | No. of Carriersof Mutations in | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Population and Index Casea (Years of Study) | Age atDiagnosis(years) | Eligible | Enrolled | Tested | Sourceb | Gene(s)Tested | Method(s)/Mutation(s)Analyzedc | BRCA1 | BRCA2 | Reference(s) |
| Australia: | ||||||||||
| Br (1992–95) | <40 | 639 | 467 | 388 | P | BRCA1/BRCA2 | PTT, sequencing | 7 | 9 | Southey et al. 1999 |
| Canada: | ||||||||||
| Ov (1995–96) | Any | 1,024 | 649d | 649 | P | BRCA1/BRCA2 | PTT, DGGE, ASO | 39 | 21 | Risch et al. 2001 |
| Canada/United States/Israel: | ||||||||||
| Ov (1995–96) | Any | 254 | 213 | 208 | H | BRCA1/BRCA2 | 185delAG, 5382insC, 6174delT | 57 | 29 | Moslehi et al. 2000 |
| Canada/United States: | ||||||||||
| Br (1996–98) | Any | 700 | 457 | 412e | H | BRCA1/BRCA2 | 185delAG, 5382insC, 6174delT | 27 | 13 | Warner et al. 1999 |
| Finland: | ||||||||||
| Br (1997–99) | Any | 1,262 | 1,035 | 1035 | P | BRCA1/BRCA2 | ASO or RFLPf | 4 | 14 | Syrjäkoski et al. 2000 |
| Hungary: | ||||||||||
| Br (1996–98) | Any | 900 | 739 | 739 | H | BRCA1/BRCA2 | HA, sequencing | 14g | 3 | Van Der Looij et al. 2000 |
| Hong Kong: | ||||||||||
| Br (1992–93) | Any | 130 | 130 | 120 | H | BRCA1 | SSCP | 6 | … | Tang et al. 1999 |
| Iceland: | ||||||||||
| Br (1955–96) | Any | 1,214 | 541 | 541 | P | BRCA2 | 999del5 | … | 56 | Thorlacius et al. 1998 |
| MBr (1955–96) | Any | 34 | 34 | 34 | P | BRCA2 | 999del5 | … | 13 | Thorlacius et al. 1998 |
| Italy: | ||||||||||
| Br (1991–99) | <36 | 122 | 74 | 61 | H | BRCA1/BRCA2 | PTT, SSCP, sequencing | 15 | 9 | De Benedetti et al. 1998; unpublished data |
| Poland: | ||||||||||
| Br (1999–2000) | <50 | 381 | 362 | 362 | H | BRCA1 | T300G, 4158delA, 5382insC | 27 | … | Unpublished data |
| Ov (1999–2000) | Any | 166 | 159 | 159 | H | BRCA1 | T300G, 4158delA, 5382insC | 21 | … | Unpublished data |
| Sweden: | ||||||||||
| Br (1990–95) | <41 | 262 | 250 | 234 | P | BRCA1/BRCA2 | PTT, SSCP, DHPLC | 16 | 5 | Loman et al. 2001 |
| United Kingdom: | ||||||||||
| Br (1991–96) | <55 | 2,017 | 1,486 | 1,443 | P | BRCA1/BRCA2 | HA | 8 | 15 | Anglian Breast Cancer Study Group 2000 |
| Br (1996–99) | <40 | 314 | 190 | 92 | P | BRCA1/BRCA2 | HA | 2 | 3 | Unpublished data |
| MBr (1971–99) | Any | 166 | 110 | 88 | P | BRCA1/BRCA2 | HA | 0 | 5 | Basham et al. 2002 |
| Br (1992–95) | <40 | 172 | 171 | 171 | P | BRCA1 | HA, SSCP | 11 | … | Eccles et al. 1998 |
| Br (1980–97) | <30 | 126 | 99 | 99 | P | BRCA1/BRCA2 | Sequencing | 9 | 7 | Unpublished data |
| Br (1982–85) | <36 | 1,049 | 755 | 256 | P | BRCA1/BRCA2 | HA | 9 | 6 | Peto et al. 1999 |
| Br (1988–89) | 36–45 | 838 | 644 | 363 | P | BRCA1/BRCA2 | HA | 7 | 7 | Peto et al. 1999 |
| United States: | ||||||||||
| Br (1994–95) | Any | 1671 | 1,310 | 687 | P | BRCA1/BRCA2 | HA, SSCP | 7 | 4 | Anton-Culver et al. 2000 |
| MBr (1980–94) | Any | 67 | 54 | 54 | P | BRCA1/BRCA2 | HA, SSCP | 0 | 2 | Friedman et al. 1997 |
| Ov (1994–95) | Any | 144 | 120 | 116 | P | BRCA1 | HA, SSCP | 3 | … | Anton-Culver et al. 2000 |
Mutations were included in the present analysis if they were “pathogenic” according to the generally accepted criteria (see the BIC Web site)—that is, frameshift or nonsense mutations, splice-site mutations predicted to cause aberrant splicing, large deletions or duplications, and missense mutations classified as such by BIC. In practice, the last group included only mutations in the ring-finger domain of BRCA1. In-frame deletions and known polymorphisms or “unclassified variants” were not included. A variety of mutation-screening techniques was used by the studies. Of the studies, 14 screened for mutations in both genes, 6 screened for mutations in BRCA1 only, and 2 screened for mutations in BRCA2 only. Six studies investigated specific founder mutations (in the Ashkenazi Jewish, Icelandic, and Polish populations), whereas the remaining 16 studies screened the coding sequence of either or both genes.
Statistical Methods
_Kaplan-Meier estimation.—_The cumulative probabilities of breast and ovarian cancer in mothers and sisters of BRCA1- and _BRCA2-_mutation carriers were estimated using the Kaplan-Meier product-limit method, using the program Stata (version 7). For this analysis, censoring age was the age at breast cancer diagnosis, age at ovarian cancer diagnosis, age at last follow-up, or age 70 years, whichever occurred first. SEs and confidence limits were obtained using Greenwood’s formula.
_Penetrance estimation.—_We used the information on disease occurrence in relatives of mutation-positive index case patients to estimate age-specific breast and ovarian cancer incidences in mutation carriers by maximum likelihood, using modified segregation analyses implemented in Mendel (Lange et al. 1988; Antoniou et al. 2001). This is essentially the same methodology and software as that used for penetrance analysis in multiple-case families (but with a different ascertainment correction). Relatives were assumed to be followed from age 20 years and to be censored at the age at first cancer diagnosis, at the age at death, at the age at last follow-up, or at age 70 years, whichever came first. Information on mutation status in relatives was incorporated when available. Females born before 1890 were excluded from the analyses. Individuals with no age information (608 females from entire data set) or no year of birth (53 females) were censored at age 0 years. To correct for ascertainment, we maximized the conditional likelihood of the pedigree given the phenotypic and genotypic information of the index case.
The main analyses were based on the fitting of fixed age-specific incidence rates for carriers. Initially, these rates were assumed to be independent of country of origin or year of birth, but we then explored variation in rates according to these covariates. Breast cancer incidence in carriers was assumed to follow 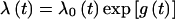 , where λ0(t) is the background incidence for England and Wales (1973–77) and _exp_[g(t)] is the age-specific RR of breast cancer in carriers as compared to population rates. The ovarian cancer incidences were assumed to follow a similar model. Conditional on the genotype, the probability of developing breast cancer was assumed to be independent of the probability of developing ovarian cancer. We estimated the age-specific log(RR) parameters g(t) for five age groups: 20–29, 30–39, 40–49, 50–59, and 60–69 years. We then fitted models with carrier incidences parameterized in terms of rate ratios relative to country-, age-, and period-specific incidences. In all analyses, cancer incidences in noncarriers were assumed to follow country- and cohort-specific rates (Waterhouse et al. 1976, 1982; Muir et al. 1987; Parkin et al. 1992, 1997).
, where λ0(t) is the background incidence for England and Wales (1973–77) and _exp_[g(t)] is the age-specific RR of breast cancer in carriers as compared to population rates. The ovarian cancer incidences were assumed to follow a similar model. Conditional on the genotype, the probability of developing breast cancer was assumed to be independent of the probability of developing ovarian cancer. We estimated the age-specific log(RR) parameters g(t) for five age groups: 20–29, 30–39, 40–49, 50–59, and 60–69 years. We then fitted models with carrier incidences parameterized in terms of rate ratios relative to country-, age-, and period-specific incidences. In all analyses, cancer incidences in noncarriers were assumed to follow country- and cohort-specific rates (Waterhouse et al. 1976, 1982; Muir et al. 1987; Parkin et al. 1992, 1997).
To test for differences in incidences among different subgroups, we fitted models in which we added a subgroup-specific log(RR) parameter. For example, to test for differences among centers, we fitted models including the five age-specific log(RR) estimates (for all centers) but also allowed an additional center-specific log(RR) (constant over age). A likelihood-ratio test was then used to test for heterogeneity of risk among centers. Similar tests were used to explore variations in incidences by year of birth, type of mutation, and type and age of index case patient. Trend tests were used to test whether the log(RR) estimates increased or decreased significantly with age.
To construct CIs for the log(RR) estimates, we assumed that the parameters were asymptotically normally distributed with the covariance matrix given by inverting the information matrix. Cumulative risk or penetrance and 95% CIs were calculated from the cumulative incidence λ(t), where 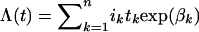 , where i k is the incidence in noncarriers in the k_th age band of length t k and β_k is the ln(RR) in the _k_th age band. The variance of the cumulative risk is given by the expression
, where i k is the incidence in noncarriers in the k_th age band of length t k and β_k is the ln(RR) in the _k_th age band. The variance of the cumulative risk is given by the expression

and the cumulative risk F(t) is then given by F(t)=1-_exp_[-Λ(t)], with a 95% CI of  . Uncertainty in RRs for factors with more than two categories (e.g., center) is presented as floating CIs (Easton et al. 1991).
. Uncertainty in RRs for factors with more than two categories (e.g., center) is presented as floating CIs (Easton et al. 1991).
Results
The 22 studies included in the present analysis screened a total of 6,965 female breast cancer cases, 176 male breast cancer cases, and 998 ovarian cancer cases and identified 289 BRCA1- and 221 _BRCA2-_mutation carriers (table 2). Table 2 also shows the number of individuals eligible for each study, the number enrolled, and the number of samples analyzed. However, estimation of a response rate that is comparable across all studies is not possible, because of the variety of protocols used in recruitment and data gathering (see the “Subjects and Methods” section). Family-history data were not available for 12 mutation carriers, leaving 280 families of _BRCA1-_mutation carriers and 218 families of _BRCA2-_mutation carriers in the present analysis. Among the first-degree relatives of _BRCA1-_mutation–positive index case patients, 125 breast cancers and 41 ovarian cancers were identified, and, among the first-degree relatives of _BRCA2-_mutation–positive index case patients, 87 breast cancers and 13 ovarian cancers were identified.
Kaplan-Meier Estimates
Figure 1 shows the age-specific cumulative probabilities of breast and ovarian cancer in mothers and sisters of _BRCA1-_mutation carriers, using Kaplan-Meier estimation. The estimated cumulative risks of breast cancer by age 70 years were 29% (95% CI 23%–35%) in mothers and 42% (95% CI 30%–56%) in sisters; the corresponding cumulative risks of ovarian cancer were 15% (95% CI 10%–21%) in mothers and 14% (95% CI 7.5%–24%) in sisters. Although the estimated breast cancer risks are higher in sisters than mothers at all ages, the difference in risks was not statistically significant (log-rank _P_=.056 for breast cancer; log-rank _P_=.75 for ovarian cancer).
Figure 1.

Kaplan-Meier cumulative breast (upper lines) and ovarian (lower lines) cancer probability in sisters (thick lines) and mothers (thin lines) of _BRCA1-_mutation–carrying index case patients.
Figure 2 shows the corresponding Kaplan-Meier estimates for mothers and sisters of _BRCA2-_mutation carriers. The cumulative risks of breast cancer by age 70 years were 19% (95% CI 14%–26%) in mothers and 25% (95% CI 18%–34%) in sisters, whereas the cumulative risks of ovarian cancer were 5.1% (95% CI 2.7%–9.6%) in mothers and 4.5% (95% CI 1.7%–12%) in sisters. Again, the differences in risks between mothers and sisters were not statistically significant for either cancer (_P_=.12 for breast cancer; _P_=.53 for ovarian cancer).
Figure 2.

Kaplan-Meier cumulative breast (upper lines) and ovarian (lower lines) cancer probability in sisters (thick lines) and mothers (thin lines) of _BRCA2-_mutation–carrying index case patients.
Average Penetrance Estimates
For the main analysis, we assumed that the age-specific incidences were the same for all mutation carriers and that incidences in noncarriers were country and birth-cohort specific. The RRs of breast and ovarian cancer in BRCA1- and _BRCA2-_mutation carriers, compared to population rates for England and Wales in 1973–77, are shown in table 3. For BRCA1, the breast cancer RR increased to 33 in the 30–39-years age group and decreased with age thereafter (P trend .012), whereas the ovarian cancer RR estimates showed no apparent trend with age. The estimated breast cancer RR to _BRCA2-_mutation carriers was 19 in the 20–29-years age group and fell to ∼10 in older-age groups (P trend .98). Ovarian cancer RRs for _BRCA2-_mutation carriers were only estimated for ages ⩾40 years, because there were no ovarian cancer cases diagnosed at <40 years of age in the first-degree relatives of _BRCA2-_mutation carriers. The RR increased to a maximum of 19 in the 50–59-years age group and then decreased.
Table 3.
RRs of Breast and Ovarian Cancer in Mutation Carriers
| RRa (95% CI) of Cancerfor Carriers of Mutations in | ||||
|---|---|---|---|---|
| BRCA1 | BRCA2 | |||
| Age Group | BreastCancer | OvarianCancer | BreastCancer | OvarianCancer |
| 20–29 years | 17 (4.2–71) | 1.0 | 19 (4.5–81) | 1.0 |
| 30–39 years | 33 (23–49) | 49 (21–111) | 16 (9.3–29) | 1.0 |
| 40–49 years | 32 (24–43) | 68 (42–111) | 9.9 (6.1–16) | 6.3 (1.4–28) |
| 50–59 years | 18 (11–30) | 31 (14–66) | 12 (7.4–19) | 19 (9.0–41) |
| 60–69 years | 14 (6.3–31) | 50 (22–114) | 11 (6.3–20) | 8.4 (2.2–32) |
The corresponding age-specific incidences are shown in table 4, and the cumulative cancer risks (penetrances) are shown in figures 3 and 4. The breast cancer incidence in _BRCA1-_mutation carriers increased with age up to age 45–49 years but remained roughly constant thereafter. The ovarian cancer rates were low below age 30 years and rose steeply with age thereafter, to ∼2% per annum, only slightly less than the breast cancer rates. The breast cancer incidence in _BRCA2-_mutation carriers increased progressively with age, whereas the ovarian cancer incidence increased up to age 55–59 years and then decreased slightly. The cumulative breast cancer risk by age 70 years in _BRCA1-_mutation carriers was estimated to be 65% (95% CI 51%–75%), and the ovarian cancer risk was estimated to be 39% (95% CI 22%–51%). For _BRCA2-_mutation carriers, the cumulative breast cancer risk by age 70 years was estimated to be 45% (95% CI 33%–54%), and that for ovarian cancer was estimated to be 11% (95% CI 4.1%–18%).
Table 4.
Estimated Breast and Ovarian Cancer Incidence (%) in Mutation Carriers
| Estimated Cancer Incidencefor Carriers of Mutations in | ||||
|---|---|---|---|---|
| BRCA1 | BRCA2 | |||
| Age Group | BreastCancer | OvarianCancer | BreastCancer | OvarianCancer |
| 20–24 years | .02 | .001 | .02 | .001 |
| 25–29 years | .11 | .002 | .12 | .002 |
| 30–34 years | .74 | .18 | .36 | .004 |
| 35–39 years | 1.59 | .28 | .78 | .01 |
| 40–44 years | 2.92 | .87 | .91 | .08 |
| 45–49 years | 4.28 | 1.49 | 1.34 | .14 |
| 50–54 years | 2.65 | .96 | 1.76 | .60 |
| 55–59 years | 3.01 | 1.19 | 2.00 | .75 |
| 60–64 years | 2.70 | 2.26 | 2.17 | .38 |
| 65–69 years | 2.96 | 2.49 | 2.38 | .42 |
Figure 3.

Cumulative risk of breast (♦) and ovarian (▪) cancer in _BRCA1-_mutation carriers.
Figure 4.

Cumulative risk of breast (♦) and ovarian (▪) cancer in _BRCA2-_mutation carriers.
Analysis by Center
We investigated potential heterogeneity of risk among centers by fitting models with additional center-specific RR parameters. For BRCA1, we grouped the U.K., Canadian, Polish, and other centers (43, 124, 48, and 65 families, respectively). RRs by center are shown in table 5. The estimated cancer risks were somewhat lower in the Canadian and Polish families than in the U.K. and other families, but there was no significant evidence of heterogeneity (_P_=.32).
Table 5.
RRs for Mutation Carriers, as Compared to Baseline, for Models Allowing for Center, Type-of-Index-Case, and Year-of-Birth Effects[Note]
| RR (95% Floated CI) for Carriers of Mutations in | ||||
|---|---|---|---|---|
| BRCA1 (_P_=.32) | BRCA2 (_P_=.13) | |||
| Center | Breast Cancer | Ovarian Cancer | Breast Cancer | Ovarian Cancer |
| United Kingdoma | 1.0 (.55–1.8) | 1.0 (.41–2.4) | 1.0 (.69–1.4) | 1.0 (.33–3.1) |
| Canada | .51 (.36–.71) | .34 (.19–.59) | .53 (.30–1.0) | 3.1 (1.3–7.1) |
| Poland | .47 (.28–.78) | .40 (.18–.90) | … | … |
| Iceland | … | … | 1.0 (.69–1.4) | 1.0 (.33–3.1) |
| Other | .69 (.44–1.1) | .52 (.25–1.1) | 1.4 (.77–2.6) | 4.2 (1.2–14) |
| RR (95% Floated CI) for Carriers of Mutations in | ||||
| BRCA1 (_P_=.015) | BRCA2 (_P_=.01) | |||
| Type of Index Case | Breast Cancer | Ovarian Cancer | Breast Cancer | Ovarian Cancer |
| Breast cancer at ⩾35 yearsa | 1.0 (.68–1.5) | 1.0 (.53–1.9) | 1.0 (.68–1.5) | 1.0 (.42–2.4) |
| Breast cancer at <35 years | 2.2 (1.4–3.3) | 1.8 (.82–4.0) | 1.2 (.57–2.5) | 14 (3.8–53) |
| Male breast cancer | … | … | 1.3 (.65–2.7) | 1.8 (.92–3.6) |
| Ovarian cancer | .83 (.59–1.2) | 1.1 (.69–1.8) | .45 (.23–.89) | 6.5 (2.1–21) |
| RR (95% Floated CI) for Carriers of Mutations in | ||||
| BRCA1 (_P_=.00001) | BRCA2 (_P_=.16) | |||
| Year of Birth | Breast Cancer | Ovarian Cancer | Breast Cancer | Ovarian Cancer |
| Before 1920a | 1.0 (.53–1.9) | 1.0 (.27–3.7) | 1.0 (.60–1.7) | … |
| 1920–39 | 2.7 (1.9–3.8) | 12 (7.3–18) | 1.2 (.71–1.9) | … |
| 1940–59 | 3.7 (2.6–5.1) | 9.5 (4.9–19) | 2.1 (1.2–3.6) | … |
| 1960 onward | 7.7 (2.6–23) | … | 4.8 (1.0–23) | … |
For BRCA2, we grouped the U.K., Canadian, Icelandic, and other centers (44, 63, 69, and 42 families, respectively). Again, there was no significant evidence of heterogeneity (P = .13). The estimated breast cancer risks for Canadian centers were lower than for the U.K. and Icelandic centers (RR = 0.53), and the ovarian cancer risk was higher (RR = 3.1). There was also some suggestion of higher cancer risks for “other centers” as compared to the U.K. center (RR = 1.4 for breast cancer, and RR = 4.2 for ovarian cancer).
Effect of Year of Birth
We investigated the effect of birth cohort on breast and ovarian cancer risks by fitting models with additional parameters for birth cohorts (before 1920, 1920–39, 1940–59, and from 1960 onward for breast cancer; before 1920, 1920–39, and from 1940 onward for ovarian cancer). For _BRCA1-_mutation carriers, we found higher risks for both breast and ovarian cancer (_P_=.011 and _P_=.0013, respectively) in more-recent birth cohorts (table 5). The RR of breast cancer in _BRCA2-_mutation carriers also increased with more-recent birth cohort (table 5), but not significantly (_P_=.16). There were too few ovarian cancer cases among the first-degree relatives of the index case patients to assess cohort effects on ovarian cancer risk in _BRCA2-_mutation carriers.
Effect of Type of Index Case
One hundred seventeen families were ascertained through a _BRCA1-_mutation–carrying index case patient with ovarian cancer, and 163 families were ascertained through a _BRCA1-_mutation–carrying index case patient with breast cancer. We fitted models, adding an RR parameter for type of index case (table 5). The breast cancer risk for _BRCA1-_mutation carriers ascertained through an ovarian cancer index case was lower than that for carriers ascertained through a breast cancer index case (RR = 0.60 [95% CI 0.38–0.94]; cumulative risk by age 70 years 56% vs. 72%). The ovarian cancer risks, however, did not differ significantly by type of index case (RR = 0.86 [95% CI 0.42–1.8]).
Families ascertained through a breast cancer case were subdivided further by age at diagnosis of the index case. The breast and ovarian cancer risk estimates were higher in the families of the early-onset index cases, although only the breast cancer effect was significant (table 5) (breast cancer RR = 2.2 [95% CI 1.4–3.3]; ovarian cancer RR = 1.8 [95% CI 0.82–4.0]). On the basis of this analysis, the breast cancer risk for _BRCA1-_mutation carriers for families ascertained through early-onset index cases was estimated to be 87% (95% CI 67%–95%) by age 70 years, and the ovarian cancer risk was estimated to be 51% (95% CI 9.1%–73%), compared to 61% (41%–74%) and 32% (11%–49%) for families ascertained through an older index case patient. The risk estimates for older index case patients with breast cancer were comparable to those for ovarian cancer index case patients (54% and 36%).
Fifty _BRCA2-_mutation–positive families were ascertained through an ovarian cancer index case, 148 were ascertained through a female breast cancer index case, and 20 were ascertained through a male breast cancer index case. Of the female index case patients, 46 received a diagnosis at <35 years of age, and 102 received a diagnosis at ⩾35 years of age. The estimated breast cancer risk in carriers ascertained through a breast cancer index case was higher than in those ascertained through an ovarian cancer index case (RR = 0.42 [95% CI 0.20–0.88]). Conversely, the ovarian cancer risk was higher in the families ascertained through an ovarian cancer index case (RR = 2.4 [95% CI 0.74–8.1]). There was no evidence of a difference in risk according to whether the index case patient with breast cancer was a male or a female who received a diagnosis at ⩾35 years of age (RR = 1.3 [95% CI 0.65–2.7]). Among carriers ascertained through a female breast cancer index case, there was no significant difference in the breast cancer risks according to whether the index case was diagnosed at <35 years of age or at a later age (RR = 1.2 [95% CI 0.57–2.5]; cumulative risks by age 70 years 55% [16%–76%] vs. 49% [32%–61%]), but there was some evidence of higher ovarian cancer risk for families ascertained through early-onset breast cancer cases (RR = 13 [95% CI 2.4–70]; cumulative risks by age 70 years 35% [0.61%] vs. 3% [0–7%]).
Since ascertainment criteria varied by center, we also fitted models in which RRs for center and type of index case were fitted simultaneously. Under this model, the breast cancer risk for _BRCA1-_mutation carriers ascertained through index cases diagnosed as breast cancer at <35 years of age remained higher than that for carriers whose diagnosis was given at later ages (RR = 2.2 [95% CI 1.2–4.2]). Some suggestion of a higher ovarian cancer risk among carriers ascertained through ovarian cancer index cases, not evident from the univariate analysis, emerged when center was taken into account (RR = 1.9 [95% CI 0.67–5.2]). We were unable to fit the effects of index case and center for BRCA2. These effects were confounded because all the ovarian cancer index cases were from one center (Canada).
We also fitted models allowing for both type of index case and year of birth. The estimated effects that type of index case had on both breast and ovarian cancer risk were similar to those estimated previously. Thus, the RR based on breast cancer cases diagnosed at <35 years of age, relative to those diagnosed at later ages, was 1.9 (95% CI 1.1–3.3), whereas the RR for mutation carriers ascertained through an ovarian cancer index case was 0.84 (95% CI 0.51–1.37). Adjustment for type of index case did not materially affect the year of birth effect: the estimated RRs for mutation carriers born in the 1920–39, 1940–59, and 1960-onward cohorts were 1.8 (95% CI 0.88–3.6), 2.5 (1.2–5.3), and 4.9 (1.4–18), respectively. We fitted similar models for BRCA2, but the RRs for both the type-of-index-case effect and the birth-cohort effect were similar to those when each effect was considered individually.
Center- and Cohort-Specific Incidence Models
The apparent variation in incidence by center and birth cohort may reflect variations in population-specific incidence rates. We therefore also performed analyses in which we estimated the age-specific RRs in carriers relative to population- and cohort-specific incidence rates. The RRs estimated in these models were very similar to those estimated in the analyses that assumed a constant background incidence rate (table 6). We then fitted models allowing for heterogeneity between center and birth cohort. The RR estimates by center were very similar to those for the fixed incidences models, with no significant evidence for heterogeneity of risk by center for either gene.
Table 6.
RR Estimates for Mutation Carriers, Based on Country- and Cohort-Specific Background Rates
| RR Estimates (95% Floated CI)for Carriers of Mutations in | ||||
|---|---|---|---|---|
| BRCA1 | BRCA2 | |||
| Age Group | BreastCancer | OvarianCancer | BreastCancer | OvarianCancer |
| 20–29 years | 18 (4.4–75) | 1.0 | 19 (4.4–82) | 1.00 |
| 30–39 years | 36 (25–52) | 38 (17–88) | 16 (9.3–29) | 1.00 |
| 40–49 years | 31 (25–52) | 61 (38–99) | 9.5 (5.9–15) | 6.3 (1.4–28) |
| 50–59 years | 16 (9.6–27) | 30 (14–65) | 11 (6.6–17) | 19 (9.1–41) |
| 60–69 years | 11 (5.0–25) | 48 (22–109) | 9.2 (5.1–17) | 7.3 (1.8–30) |
The cohort effects were slightly less marked than the fixed incidence rate model. For BRCA1, the breast cancer RR estimates for the three later birth cohorts, relative to the before-1920 cohort, were as follows: 1920–39, 2.6 (95% CI 1.3–5.2); 1940–59, 3.1 (95% CI 1.5–6.5); and 1960 onward, 6.2 (95% CI 1.7–22.1). The ovarian cancer RRs for the 1920–39 and 1940–59 birth cohorts, compared to the before-1920 cohort, were estimated as 9.8 (95% CI 2.6–36.6) and 7.6 (95% CI 1.9–30.8), respectively. There were no ovarian cancer cases in relatives born after 1960, so no RR parameter was estimated for this cohort. Both effects remained highly significant. For BRCA2, the corresponding breast cancer RRs were 0.94 (95% CI 0.49–1.8), 1.4 (95% CI 0.65–3.0), and 3.6 (95% CI 0.65–19), respectively. As with the fixed incidence rate model, the cohort effects were not significant for _BRCA2-_mutation carriers (_P_=.26). There were too few ovarian cancer cases among the first-degree relatives of the index case patients to assess cohort effects on ovarian cancer risk in _BRCA2-_mutation carriers.
Effect of Mutation Position
We investigated the possibility of allelic heterogeneity in risk by classifying mutations according to their position and by fitting models comparable to the previous BCLC analyses (Thompson and Easton 2001). Families with the BRCA1 C61G missense mutation were excluded from the present analysis. BRCA1 mutations were categorized into three groups as defined previously: nucleotides 1–2400 (137 index cases), 2401–4184 (55 index cases), and 4185 onward (88 index cases). The RR of breast cancer for mutations in the central region as compared to that for mutations in the 5′ region was estimated to be 0.93, and that for mutations in the 3′ region was estimated to be 1.4; the corresponding risks for ovarian cancer were 1.8 and 1.1. These models did not fit significantly better than the null model.
BRCA2 mutations were divided into those within the ovarian cancer cluster region (nucleotides 3059–6629 [97 index cases]) and those outside the ovarian cancer cluster region (mutations at all other nucleotides [121 index cases]). We fitted models in which the breast and ovarian cancer risks were allowed to vary between the two regions. The estimated breast cancer risk was lower among carriers of mutations in the ovarian cancer cluster region as compared to mutations outside the ovarian cancer cluster region (RR = 0.57 [95% CI 0.32–1.0]). The corresponding ovarian cancer RR was 2.1 (95% CI 0.62–7.0).
Discussion
In this meta-analysis, we have used data from 22 studies that have tested patients with breast or ovarian cancer who were unselected for family history of germline mutations in BRCA1 and/or BRCA2 as a basis for the estimation of breast and ovarian cancer incidences and cumulative risks in mutation carriers. We are aware of a few studies that could not be included in the present analysis, and, undoubtedly, there are other such studies ongoing. Nevertheless, this overview represents the large majority of the available data, and, especially given the costs of such studies, it seems unlikely that much greater precision will be available in the near future from studies of this design.
The major perceived strength of this approach, as compared with the linkage approach based on multiple-case families, is that it produces estimates that are less susceptible to the effects of other familial risk factors and mutation-specific differences in risk. Although this is true, it is important to note that the families that we have analyzed were still selected on the basis of an affected index case patient, so that, in the presence of modifying risk factors, the estimated risks will be higher than the risks to a completely unselected mutation carrier. Nevertheless, since one affected relative would usually represent an absolute minimum criterion for genetic testing, it seems unlikely that risk estimates that lie much outside this range will be needed in any practical situation.
There are two other important advantages of this approach. First, it provides estimates for site-specific cancer risks that are largely uncorrelated, whereas the breast and ovarian cancer risk estimates derived by the maximum-LOD-score approach in multiple-case families tend to be strongly correlated. Second, relative to the maximum-LOD-score approach, the estimates at early ages, when the risks are low, are more precise. A major disadvantage of this approach is that the prevalence of mutations in unselected case series is low and, therefore, very large numbers of cases need to be tested to provide precise estimates. Thus, the studies in the present analysis included >8,000 index cases, yielding 282 BRCA1 and 218 BRCA2 mutations. Despite this, the width of many of the confidence limits still exceeds 10%.
Another important issue is the accuracy of reporting of family history. Although some of the studies did attempt to confirm cancer diagnoses in relatives, this was not always possible, and only three studies were able to identify routinely all cancer diagnoses in relatives through national records. In an attempt to minimize the effects of inaccurate reporting, we restricted our main analyses to first-degree relatives. Previous studies (e.g., Claus et al. 1998) have found that reporting of cancer in more-distant relatives is less accurate. Although data on more-distant relatives were easily incorporated in the analysis, we found that some of the penetrance estimates were higher. This suggests either inaccurate reporting of cancer diagnoses or incompleteness in the enumeration of relatives that correlated with the extent of family history.
The techniques used for mutation detection in the different studies—and, therefore, their sensitivity to detect mutations of different types—varied widely. Certain studies tested only for specific founder mutations (T300G, 185delAG, 4158delA, and 5382insC in BRCA1; 999del5 and 6174delT in BRCA2), but these mutations still represent a minority of the total set. Most of the groups used screening techniques that are most sensitive for small deletions and insertions, so these will be overrepresented in the data set; however, these are the mutations that account for the majority of mutations in families with linkage, and they represent the most important mutation type encountered in genetic testing. Since we restricted the present analysis to mutation types generally regarded as pathogenic, almost all the mutations are predicted to be protein truncating (the only exception being the T300G mutation in the ring finger of BRCA1). Thus, although these results are likely to be applicable generally to protein-truncating mutations, they will not be applicable to missense changes.
In a small number of families, carrier status of relatives was available, and we were able to incorporate this into the analysis. In most cases, however, carrier status had to be assigned probabilistically. The method relies on the assumption of Mendelian segregation of the mutation—which seems reasonable but also ignores the possibility of new mutation events. However, few new mutations in BRCA1 or BRCA2 have been reported, and the new-mutation rate is generally assumed to be low.
Overall Estimates
The overall estimates confirm most of the qualitative features, of age-specific risks in BRCA1- and _BRCA2-_mutation carriers, that have been suggested from studies based on multiple-case families or individual population studies, but with much more precise quantification. Thus, the average risks of both breast and ovarian cancer are higher in _BRCA1-_mutation carriers than in _BRCA2-_mutation carriers, but the difference is much more marked for ovarian cancer and for breast cancer at earlier ages than for breast cancer at >50 years of age. The RR of breast cancer in _BRCA1-_mutation carriers, relative to general population rates, declines with age from >30-fold at <40 years of age to 14-fold at >60 years of age; by contrast, the RR in _BRCA2-_mutation carriers is ∼11-fold in all age groups at >40 years of age and is not significantly higher at earlier ages. As a consequence of this, the incidences in _BRCA1-_mutation carriers rise to a plateau of ∼3%–4% per annum in the 40–49-years age group and are roughly constant thereafter, whereas the BRCA2 rates show a pattern similar to that in the general population, rising steeply up to age 50 years and more slowly thereafter.
Ovarian cancer risks in _BRCA1-_mutation carriers were low (in absolute terms) at <40 years of age (no cases at all were observed at <30 years of age). Thereafter, the incidences were ∼1% at 40–59 years of age and 2% at >60 years of age (the latter estimate is, however, particularly imprecise, since there are relatively few unaffected carriers in this age group). Ovarian cancer risks in _BRCA2-_mutation carriers are, in contrast, very low at <50 years of age but then increase sharply in the 50–59-years age group, perhaps declining somewhat thereafter. These differences in age-specific risks are mirrored by other important differences in the pathological characteristics of tumors in carriers (e.g., the estrogen-receptor–negative status of most breast tumors in _BRCA1-_mutation carriers but not in _BRCA2-_mutation carriers) and must reflect some important functional differences between the two proteins.
Absolute versus Relative Risk Models—and Cohort and Center Effects
We chose to model the BRCA1 and BRCA2 penetrances primarily in terms of RRs compared to a single set of background rates (those for England and Wales), thus estimating a single set of incidences for carriers from all populations. We also performed an alternative analysis in which the penetrance was expressed in terms of RRs relative to the population-specific incidences (so that the absolute risks would be higher in populations with higher background incidence rates). Such a model may be more appropriate if risks in carriers were modified by important lifestyle risk factors to a similar (relative) extent as in noncarriers. In fact, we found little evidence to favor one model over the other. Although we did find some evidence of variation in penetrance among populations, this did not correlate directly with population rates—for example, breast cancer risks were lower in families from the Polish center than in those from the U.K. centers but were similarly lower in families from the Canadian centers.
We found that year of birth had a marked effect on breast cancer risk in BRCA1, with a slightly weaker (and nonsignificant) effect in _BRCA2-_mutation carriers and for ovarian cancer risk in _BRCA1-_mutation carriers. The breast cancer effect was slightly weaker when analyses were performed relative to cohort-specific background rates but was still highly significant. Most of this effect was due to a markedly lower risk in women born before 1920. A possible explanation for this effect is the incomplete reporting of cancers among women born in this generation. In practice, the before-1920 birth cohort is not relevant to current genetic counselling, and exclusion of this birth cohort from the present analysis made little difference to the overall penetrance estimates (data not shown). There was also some evidence of a higher breast cancer risk in the 1960-onward birth cohort. This result seems less likely to be due to the underreporting of cancers in relatives. It could conceivably reflect changing patterns of reproductive risk factors, such as age at first pregnancy, breast feeding, or oral contraceptive use. Changes in screening practices may also account for some of the cohort effect. This will require more-detailed investigation.
Type of Index Case
We found some evidence of variation in penetrance estimates according to the type of index case. The breast cancer risk estimates for both BRCA1- and _BRCA2-_mutation carriers were higher when the index case was a breast cancer case, rather than an ovarian cancer case, and were markedly higher when the index case was a breast cancer case diagnosed at <35 years of age. A similar effect has been reported previously (Eccles et al. 1994). The ovarian cancer risks in _BRCA1-_mutation carriers were also highest in families selected on the basis of a breast cancer index case diagnosed at <35 years of age, but, for _BRCA2-_mutation carriers, the risks were higher when based on ovarian cancer index cases (albeit with wide confidence limits). Such differences in penetrance estimates are generally consistent with the hypothesis that other genes modify risks in carriers. Alleles conferring a higher risk of breast cancer will be more frequent among index cases diagnosed at earlier ages, leading to higher breast cancer risks in carriers' relatives. The more complicated pattern of ovarian cancer risk in _BRCA1-_mutation carriers may be explicable if some modifiers of breast cancer risk also modified ovarian cancer risk. No genetic modifiers have been definitively implicated yet, although several have been suggested; these include, for breast cancer, the lengths of triplet repeats in the androgen-receptor (Rebbeck et al. 1999) and AIB1 (Rebbeck et al. 2001) genes and polymorphisms in the progesterone-receptor (Runnebaum et al. 2001) and (for BRCA2) RAD51 (Levy-Lahad et al. 2001) genes. Rare alleles at the HRAS1 minisatellite locus have been suggested to be associated with ovarian cancer risk in _BRCA1-_mutation carriers (Phelan et al. 1996).
Mutation-Specific Risks
Mutation-specific differences in risk have been suggested for both BRCA1 and BRCA2 from differential cancer risks in multiple-case families, and we were able to test these hypotheses in this data set. In the case of BRCA2, Thompson and Easton (2001) have found a higher risk of ovarian cancer but a lower risk of breast cancer in carriers of mutations in the ovarian cancer cluster region. The RR estimates from the current data set were of a similar magnitude (0.57 for breast cancer and 2.1 for ovarian cancer), but neither was significant. Furthermore, the ovarian cancer effect disappeared once the 999del5 and 6174delT mutations were removed from the analysis. In the case of BRCA1, Thompson and Easton (2002) have found a lower risk of breast cancer associated with mutations in a central region of the gene (nucleotides 2401–4184), together with a lower risk of ovarian cancer for mutations 3′ of nucleotide 4184. The estimate RRs from our data set were in the same direction (and, in the case of the breast cancer risk, of a similar magnitude) as those reported by Thompson and Easton (2002), but neither effect was statistically significant. Although the consistency of these results is reassuring, they emphasize that detailed analyses of mutation-specific risks can be achieved only through studies of multiple-case families.
Conclusions
The present analysis has provided breast and ovarian cancer risks in BRCA1- and _BRCA2-_mutation carriers that are based on the majority of available data from studies of mutation screening in series of patients with breast and ovarian cancer who were unselected for family history. It has confirmed that the lifetime risks based on this design are lower than those based on high-risk families, suggestive of some modification of risk by other factors, but the differences are smaller than has been suggested by some previous studies. The variation in risks by type of index case and by age at diagnosis of index case is also suggestive of risk modifiers. Risk estimates for counselling should take into account both mutation status and family history, as well as other risk factors once their effects become reliably known.
Acknowledgments
These analyses were supported by Cancer Research U.K. (formerly Cancer Research Campaign) and National Institutes of Health (NIH) grant 1R01 CA81203. D.F.E. is a Principal Research Fellow of Cancer Research U.K., and P.D.P.P. is a Senior Clinical Research Fellow of Cancer Research U.K. Individual studies were supported by NIH grant 1R01 CA63682, the Italian Association and Foundation for Cancer Research, the Swedish Cancer Society, European Community grant QLRI-CT-1999-00063, Hungarian Research Grants from the Ministry of Education Szechenyi Project NKFP1/48/2001 and OTKA T030039, the Icelandic Cancer Society and the University of Iceland Research Fund, and the Wessex Cancer Trust. We also thank the Department of Pathology, University Hospital Iceland; the Nordic Cancer Union; and the Cancer Society and Cancer Registry of Finland.
Electronic-Database Information
URLs for data presented herein are as follows:
- Breast Cancer Information Core, http://research.nhgri.nih.gov/bic/
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for BRCA1 and BRCA2)
References
- Anglian Breast Cancer Study Group (2000) Prevalence and penetrance of BRCA1 and BRCA2 in a population based series of breast cancer cases. Br J Cancer 83:1301–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anton-Culver H, Cohen PF, Gildea ME, Ziogas A (2000) Characteristics of BRCA1 mutations in a population-based case series of breast and ovarian cancer. Eur J Cancer 36:1200–1208 [DOI] [PubMed] [Google Scholar]
- Antoniou AC, Gayther SA, Stratton JF, Ponder BAJ, Easton DF (2000) Risk models for familial breast and ovarian cancer. Genet Epidemiol 18:173–190 [DOI] [PubMed] [Google Scholar]
- Antoniou AC, Pharoah PDP, McMullen G, Day NE, Ponder BAJ, Easton DF (2001) Evidence for further breast cancer susceptibility genes in addition to BRCA1 and BRCA2 in a population based study. Genet Epidemiol 21:1–18 [DOI] [PubMed] [Google Scholar]
- Basham VM, Lipscombe JP, Ward JM, Easton DF, Gayther SA, Ponder BAJ, Pharoah PDP (2002) BRCA1 and BRCA2 mutations in a population based study of male breast cancer. Breast Cancer Res 4:R2.1–R2.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begg CB (2002) On the use of familial aggregation in population-based case probands for calculating penetrance. J Natl Cancer Inst 94:1221–1226 [DOI] [PubMed] [Google Scholar]
- Claus EB, Schildkraut J, Iversen ES Jr, Berry D, Parmigiani G (1998) Effect of BRCA1 and BRCA2 on the association between breast cancer risk and family history. J Natl Cancer Inst 90:1824–1829 [DOI] [PubMed] [Google Scholar]
- Clerget-Darpoux, F (2001) Extension of the lod score: the mod score. Adv Genet 42:115–124 [DOI] [PubMed] [Google Scholar]
- De Benedetti VM, Radice P, Pasini B, Stagi L, Pensotti V, Mondini P, Manoukian S, Conti A, Spatti G, Rilke F, Pierotti MA (1998) Characterization of ten novel and 13 recurring BRCA1 and BRCA2 germline mutations in Italian breast and/or ovarian carcinoma patients: mutation in brief no. 178. Online. Hum Mutat 12:215 [PubMed] [Google Scholar]
- Easton DF, Bishop DT, Ford D, Crockford GP, Breast Cancer Linkage Consortium (1993) Genetic linkage analysis in familial breast and ovarian cancer: results from 214 families. Am J Hum Genet 52:678–701 [PMC free article] [PubMed] [Google Scholar]
- Easton DF, Ford D, Bishop DT, Breast Cancer Linkage Consortium (1995) Breast and ovarian cancer incidence in _BRCA1-_mutation carriers. Am J Hum Genet 56:265–271 [PMC free article] [PubMed] [Google Scholar]
- Easton DF, Peto J, Babiker AG (1991) Floating absolute risk: an alternative to relative risk in survival and case-control analysis avoiding an arbitrary reference group. Stat Med 10:1025–1035 [DOI] [PubMed] [Google Scholar]
- Eccles DM, Englefield P, Soulby MA, Campbell IG (1998) BRCA1 mutations in southern England. Br J Cancer 77:2199–2203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eccles D, Marlow A, Royle G, Collins A, Morton NE (1994) Genetic epidemiology of early onset breast cancer. J Med Genet 31:944–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford D, Easton DF, Bishop DT, Narod SA, Goldgar DE, Breast Cancer Linkage Consortium (1994) Risks of cancer in BRCA1 mutation carriers. Lancet 343:692–695 [DOI] [PubMed] [Google Scholar]
- Ford D, Easton DF, Stratton M, Narod S, Goldgar D, Devilee P, Bishop DT, et al (1998) Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. Am J Hum Genet 62:676–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman LS, Gayther SA, Kurosaki T, Gordon D, Noble B, Casey G, Ponder BA, Anton-Culver H (1997) Mutation analysis of BRCA1 and BRCA2 in a male breast cancer population. Am J Hum Genet 60:313–319 [PMC free article] [PubMed] [Google Scholar]
- Gayther SA, Mangion J, Russell P, Seal S, Barfoot R, Ponder BA, Stratton MR, Easton D (1997) Variation of risks of breast and ovarian cancer associated with different germline mutations of the BRCA2 gene. Nat Genet 15:103–105 [DOI] [PubMed] [Google Scholar]
- Gayther SA, Warren W, Mazoyer S, Russell PA, Harrington PA, Chiano M, Seal S, Hamoudi R, van Rensburg EJ, Dunning AM, Love R, Evans G, Easton D, Clayton C, Stratton MR, Ponder BAJ (1995) Germline mutations of the BRCA1 gene in breast and ovarian cancer families provide evidence for a genotype-phenotype correlation. Nat Genet 11:428–433 [DOI] [PubMed] [Google Scholar]
- Hopper JL, Southey MC, Dite GS, Jolley DJ, Giles GG, McCredie MR, Easton DF, Venter DJ, Australian Breast Cancer Family Study (1999) Population-based estimate of the average age-specific cumulative risk of breast cancer for a defined set of protein-truncating mutations in BRCA1 and BRCA2. Cancer Epidemiol Biomarkers Prev 8:741–747 [PubMed] [Google Scholar]
- Lange K, Weeks D, Boehnke M (1988) Programs for pedigree analysis: MENDEL, FISHER, and dGENE. Genet Epidemiol 5:471–472 [DOI] [PubMed] [Google Scholar]
- Levy-Lahad E, Lahad A, Eisenberg S, Dagan E, Paperna T, Kasinetz L, Catane R, Kaufman B, Beller U, Renbaum P, Gershoni-Baruch R (2001) A single nucleotide polymorphism in the RAD51 gene modifies cancer risk in BRCA2 but not BRCA1 carriers. Proc Natl Acad Sci USA 98:3232–3236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loman N, Johannsson O, Kristoffersson U, Olsson H, Borg Å (2001) Family history of breast and ovarian cancers and BRCA1 and BRCA2 mutations in a population-based series of early-onset breast cancer. J Natl Cancer Inst 93:1215–1223 [DOI] [PubMed] [Google Scholar]
- Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harsham K, Tavtigian S, Liu Q, et al (1994) A strong candidate for the 17 linked breast and ovarian cancer susceptibility gene BRCA1. Science 266:66–71 [DOI] [PubMed] [Google Scholar]
- Moslehi R, Chu W, Karlan B, Fishman D, Risch H, Fields A, Smotkin D, Ben-David Y, Rosenblatt J, Russo D, Schwartz P, Tung N, Warner E, Rosen B, Friedman J, Brunet JS, Narod SA (2000) BRCA1 and BRCA2 mutation analysis of 208 Ashkenazi Jewish women with ovarian cancer. Am J Hum Genet 66:1259–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir C, Waterhouse JAH, Mack T, Powell J, Whelan S (1987) Cancer incidence in five continents, vol V. IARC, Lyon, France [Google Scholar]
- Parkin DM, Whelan SL, Ferlay J, Raymond L, Young J (1992) Cancer incidence in five continents, vol VI. IARC, Lyon, France [Google Scholar]
- ——— (1997) Cancer incidence in five continents, vol VII. IARC, Lyon, France [Google Scholar]
- Peto J, Collins N, Barfoot R, Seal S, Warren W, Rahman N, Easton DF, Evans C, Deacon J, Stratton MR (1999) Prevalence of BRCA1 and BRCA2 gene mutations in patients with early-onset breast cancer. J Natl Cancer Inst 91:943–949 [DOI] [PubMed] [Google Scholar]
- Phelan CM, Rebbeck TR, Weber BL, Devilee P, Ruttledge MH, Lynch HT, Lenoir GM, Stratton MR, Easton DF, Ponder BA, Cannon-Albright L, Larsson C, Goldgar DE, Narod SA (1996) Ovarian cancer risk in BRCA1 carriers is modified by the HRAS1 variable number of tandem repeat (VNTR) locus. Nat Genet 12:309–311 [DOI] [PubMed] [Google Scholar]
- Rebbeck TR, Kantoff PW, Krithivas K, Neuhausen S, Blackwood MA, Godwin AK, Daly MB, Narod SA, Garber JE, Lynch HT, Weber BL, Brown M (1999) Modification of _BRCA1-_associated breast cancer risk by the polymorphic androgen-receptor CAG repeat. Am J Hum Genet 64:1371–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebbeck TR, Wang Y, Kantoff PW, Krithivas K, Neuhausen SL, Godwin AK, Daly MB, Narod SA, Brunet JS, Vesprini D, Garber JE, Lynch HT, Weber BL, Brown M (2001) Modification of BRCA1- and BRCA2-associated breast cancer risk by AIB1 genotype and reproductive history. Cancer Res 61:5420–5424 [PubMed] [Google Scholar]
- Risch HA, McLaughlin JR, Cole DE, Rosen B, Bradley L, Kwan E, Jack E, Vesprini DJ, Kuperstein G, Abrahamson JL, Fan I, Wong B, Narod SA (2001) Prevalence and penetrance of germline BRCA1 and BRCA2 mutations in a population series of 649 women with ovarian cancer. Am J Hum Genet 68:700–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runnebaum IB, Wang-Gohrke S, Vesprini D, Kreienberg R, Lynch H, Moslehi R, Ghadirian P, Weber B, Godwin AK, Risch H, Garber J, Lerman C, Olopade OI, Foulkes WD, Karlan B, Warner E, Rosen B, Rebbeck T, Tonin P, Dube MP, Kieback DG, Narod SA (2001) Progesterone receptor variant increases ovarian cancer risk in BRCA1 and BRCA2 mutation carriers who were never exposed to oral contraceptives. Pharmacogenetics 11:635–638 [DOI] [PubMed] [Google Scholar]
- Satagopan JM, Offit K, Foulkes W, Robson ME, Wacholder S, Eng CM, Karp SE, Begg CB (2001) The lifetime risks of breast cancer in Ashkenazi Jewish carriers of BRCA1 and BRCA2 mutations. Cancer Epidemiol Biomarkers Prev 10:467–473 [PubMed] [Google Scholar]
- Southey MC, Tesoriero AA, Andersen CR, Jennings KM, Brown SM, Dite GS, Jenkins MA, Osborne RH, Maskiell JA, Porter L, Giles GG, McCredie MR, Hopper JL, Venter DJ (1999) BRCA1 mutations and other sequence variants in a population-based sample of Australian women with breast cancer. Br J Cancer 79:34–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struewing JP, Hartge P, Wacholder S, Baker SM, Berlin M, McAdams M, Timmerman MM, Brody LC, Tucker MA (1997) The risk of cancer associated with specific mutations of BRCA1 and BRCA2 among Ashkenazi Jews. N Engl J Med 336:1401–1408 [DOI] [PubMed] [Google Scholar]
- Syrjäkoski K, Vahteristo P, Eerola H, Tamminen A, Kivinummi K, Sarantaus L, Holli K, Blomqvist C, Kallioniemi OP, Kainu T, Nevanlinna H (2000) Population-based study of BRCA1 and BRCA2 mutations in 1035 unselected Finnish breast cancer patients. J Natl Cancer Inst 92:1529–1531 [DOI] [PubMed] [Google Scholar]
- Tang NL, Pang CP, Yeo W, Choy KW, Lam PK, Suen M, Law LK, King WW, Johnson P, Hjelm M (1999) Prevalence of mutations in the BRCA1 gene among Chinese patients with breast cancer. J Natl Cancer Inst 91:882–885 [DOI] [PubMed] [Google Scholar]
- Tavtigian SV, Simard J, Rommens J, Couch F, Shattuck Eidens D, Neuhausen S, Merajver S, et al (1996) The complete BRCA2 gene and mutations in chromosome 13q-linked kindreds. Nat Genet 12:333–337 [DOI] [PubMed] [Google Scholar]
- Thompson D, Easton D (2001) Variation in cancer risks, by mutation position, in BRCA2 mutation carriers. Am J Hum Genet 68:410–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— (2002) Variation in BRCA1 cancer risks by mutation position. Cancer Epidemiol Biomarkers Prev 11:329–336 [PubMed] [Google Scholar]
- Thorlacius S, Struewing JP, Hartge P, Olafsdottir GH, Sigvaldason H, Tryggvadottir L, Wacholder S, Tulinius H, Eyfjord JE (1998) Population-based study of risk of breast cancer in carriers of BRCA2 mutation. Lancet 352:1337–1339 [DOI] [PubMed] [Google Scholar]
- Van Der Looij M, Szabo C, Besznyak I, Liszka G, Csokay B, Pulay T, Toth J, Devilee P, King MC, Olah E (2000) Prevalence of founder BRCA1 and BRCA2 mutations among breast and ovarian cancer patients in Hungary. Int J Cancer 86:737–740 [DOI] [PubMed] [Google Scholar]
- Warner E, Foulkes W, Goodwin P, Meschino W, Blondal J, Paterson C, Ozcelik H, Goss P, Allingham-Hawkins D, Hamel N, Di Prospero L, Contiga V, Serruya C, Klein M, Moslehi R, Honeyford J, Liede A, Glendon G, Brunet JS, Narod S (1999) Prevalence and penetrance of BRCA1 and BRCA2 gene mutations in unselected Ashkenazi Jewish women with breast cancer. J Natl Cancer Inst 91:1241–1247 [DOI] [PubMed] [Google Scholar]
- Waterhouse JAH, Muir C, Correa P, Powell J (1976) Cancer incidence in five continents, vol III. IARC, Lyon, France [Google Scholar]
- Waterhouse JAH, Muir C, Shanmurgaratnam K, Powell J (1982) Cancer incidence in five continents, vol IV. IARC, Lyon, France [Google Scholar]
- Wooster R, Bignell G, Lancaster J, Swift S, Seal S, Mangion J, Collins N, et al (1995) Identification of the breast cancer susceptibility gene BRCA2. Nature 378:789–792 [DOI] [PubMed] [Google Scholar]