Genome Scan Meta-Analysis of Schizophrenia and Bipolar Disorder, Part II: Schizophrenia (original) (raw)
Abstract
Schizophrenia is a common disorder with high heritability and a 10-fold increase in risk to siblings of probands. Replication has been inconsistent for reports of significant genetic linkage. To assess evidence for linkage across studies, rank-based genome scan meta-analysis (GSMA) was applied to data from 20 schizophrenia genome scans. Each marker for each scan was assigned to 1 of 120 30-cM bins, with the bins ranked by linkage scores (1 = most significant) and the ranks averaged across studies (_R_avg) and then weighted for sample size (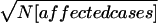 ). A permutation test was used to compute the probability of observing, by chance, each bin’s average rank (_P_AvgRnk) or of observing it for a bin with the same place (first, second, etc.) in the order of average ranks in each permutation (_P_ord). The GSMA produced significant genomewide evidence for linkage on chromosome 2q (P AvgRnk<.000417). Two aggregate criteria for linkage were also met (clusters of nominally significant P values that did not occur in 1,000 replicates of the entire data set with no linkage present): 12 consecutive bins with both _P_AvgRnk and P ord<.05, including regions of chromosomes 5q, 3p, 11q, 6p, 1q, 22q, 8p, 20q, and 14p, and 19 consecutive bins with P ord<.05, additionally including regions of chromosomes 16q, 18q, 10p, 15q, 6q, and 17q. There is greater consistency of linkage results across studies than has been previously recognized. The results suggest that some or all of these regions contain loci that increase susceptibility to schizophrenia in diverse populations.
). A permutation test was used to compute the probability of observing, by chance, each bin’s average rank (_P_AvgRnk) or of observing it for a bin with the same place (first, second, etc.) in the order of average ranks in each permutation (_P_ord). The GSMA produced significant genomewide evidence for linkage on chromosome 2q (P AvgRnk<.000417). Two aggregate criteria for linkage were also met (clusters of nominally significant P values that did not occur in 1,000 replicates of the entire data set with no linkage present): 12 consecutive bins with both _P_AvgRnk and P ord<.05, including regions of chromosomes 5q, 3p, 11q, 6p, 1q, 22q, 8p, 20q, and 14p, and 19 consecutive bins with P ord<.05, additionally including regions of chromosomes 16q, 18q, 10p, 15q, 6q, and 17q. There is greater consistency of linkage results across studies than has been previously recognized. The results suggest that some or all of these regions contain loci that increase susceptibility to schizophrenia in diverse populations.
Introduction
Schizophrenia (SCZ; locus SCZD [MIM #181500]) is a common disorder with a heritability of 0.70–0.85 and a 10-fold increase in risk to siblings of probands (Levinson and Mowry 2000). A large number of genome scan projects have been completed (see below). Genomewide significant evidence for linkage has been reported in three chromosomal regions, each with some degree of support in other data sets. In a study of 265 Irish pedigrees, Straub et al. (1995) reported a LOD score of 3.51 on chromosome 6p24-22, a region where evidence for linkage was also reported by Moises et al. (1995), Maziade et al. (2001), Lindholm et al. (2001), Schwab et al. (2000), and a multicenter analysis (Schizophrenia Linkage Collaborative Group for Chromosomes 3, 6 and 8 [hereafter referred to as “SLCG”] 1996). Straub et al. (2002_a_) recently presented evidence supporting dysbindin (DTNBP1; 6p22.3) as an SCZ candidate gene in this region. In 54 European-ancestry pedigrees, Blouin et al. (1998) reported a nonparametric linkage (NPL) score (_Z_all) of 4.18 (P<.00002) on chromosome 13q32, as well as positive evidence for linkage in 51 additional pedigrees, with a second report of significant evidence for linkage (Z _max_=4.42) in 22 Canadian pedigrees (Brzustowicz et al. 1999). In the same Canadian data set, Brzustowicz et al. (2000) reported a _Z_max of 6.50 on chromosome 1q21-22 (D1S1679), and Gurling et al. (2001) reported supportive evidence in the region in 13 U.K. and Icelandic pedigrees. In two other regions, several groups have produced highly suggestive evidence for linkage. On chromosome 8p21-22, Blouin at al. (1998) reported a _Z_all of 3.64, with support from Brzustowicz et al. (2000), Gurling et al. (2001), Kendler et al. (1996), a multicenter analysis (SLCG 1996), and Stefansson et al. (2002). The latter group also presented evidence for NRG1 (8p21-p12; neuregulin 1) as an SCZ candidate gene, although it is located 9–13 cM centromeric to other maximal linkage results. Finally, Cao et al. (1997) reported suggestive evidence for linkage on chromosome 6q21-22.3 (_P_=.00024), a finding supported by data from the National Institute of Mental Health (NIMH) Genetics Initiative data set (Martinez et al. 1999), a multicenter analysis (Levinson et al. 2000), and Lindholm et al. (2001).
None of these regions has produced strong support for linkage in the majority of genome scan projects. There are many possible explanations for this inconsistency of results, such as genotyping and diagnostic errors, differences in statistical strategies, or true differences in genetic effects of these and other loci across ethnic groups or types of pedigrees. Given the large number of projects and analyses that have been undertaken, spurious findings could also be reported several times (Lander and Kruglyak 1995). Another possibility is that each of these loci has a small populationwide effect on susceptibility and is thus difficult to detect consistently without very large samples. Suarez et al. (1994) demonstrated this problem in a simulation study of polygenic disease models, and Göring et al. (2001) recently showed that, for small genetic effect sizes, genome scans of pedigree samples with low power tend to overestimate the genetic effects at loci with the highest scores in the scan—that is, genetic parameters have effectively been maximized. This would be true whether a locus had a small effect on risk in each individual carrier (polygene model) or a larger effect on risk in a small proportion of cases (locus heterogeneity model). Thus, although optimistic observers might conclude that strong evidence has been emerging for linkage to SCZ in a number of chromosomal regions, skeptics have been more impressed by the many failures to replicate SCZ linkage findings. A major difficulty here is that SCZ genome scan sample sizes have been small—the largest scan included 294 small pedigrees (DeLisi et al. 2002_b_), and the typical sample has been 20–100, whereas samples of (for example) 600–1,000 affected sibling pairs would be required to reliably demonstrate locus-specific genetic effects causing a 27%–30% populationwide increase in risk to siblings (Hauser et al. 1996), and there are multiplicative gene effects that are even more difficult to detect (Rybicki and Elston 2000).
To address this problem, we applied the rank-based genome scan meta-analysis (GSMA) method (Wise et al. 1999) to data from the 20 complete genome scans of SCZ described in table 1. The first article in this series (Levinson et al. 2003 [in this issue]) described the GSMA method in greater detail and presented a simulation study of the power of the method to detect linkage in data sets resembling the SCZ and bipolar disorder scans that we have studied. In brief, in GSMA, the autosomes are divided into 30-cM bins, the evidence for linkage in each study is rank-ordered across bins with and without weights for sample size, and the average ranks across studies are evaluated for statistically significant evidence for linkage in several ways, as discussed in the first article in this series (Levinson et al. 2003 [in this issue]) and summarized below.
Table 1.
Characteristics of SCZ Genome Scans
| No. ofd | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Study Sitea | First Authorb | Diagnostic Model(s) (Criteria)c | Peds | Aff | Aff/Ped | Markers | Genetic Model(s)e | Type ofAnalysis | Program | Output Usedf | Reported Ethnicitiesg |
| U.S./International | DeLisi (1)1 | SCZ,SA (D3R) | 294 | 669 | 2.28 | 392 | Nonpar | Multipoint | MM/Sibs | MLS | Eur, Chile |
| Finland-gen pop | Paunio (1)2 | SCZ; +SA (D4) | 185 | 390 | 2.11 | 398 | Dom, Rec | Single-point | LINKAGE | LOD | Finland |
| Bonn | Schwab3 | SCZ,SA-S (RDC) | 71 | 171 | 2.41 | 463 | Nonpar | Multipoint | ASPEX | MLS | Ger, Seph, Hung |
| Cardiff | Williams4 | SCZ,SA (D4) | 81 | 170 | 2.10 | 229 | Nonpar | Multipoint | MM/Sibs | MLS | UK, Eire |
| MCV/Ireland (a) | Straub5 | SCZ,SA-PO (D3R) | 77 | 179 | 2.32 | 383 | Par (4) | Single-point | MENDEL | HLOD | Ireland |
| MCV/Ireland (b) | Straub5 | SCZ,SA-PO (D3R) | 67 | 164 | 2.45 | 374 | Par (4) | Single-point | MENDEL | HLOD | Ireland |
| MCV/Ireland (c) | Straub5 | SCZ,SA-PO (D3R) | 74 | 169 | 2.28 | 385 | Par (4) | Single-point | MENDEL | HLOD | Ireland |
| Johns Hopkins | Blouin6 | SCZ,SA (D4) | 54 | 146 | 2.70 | 456 | Nonpar | Multipoint | GH 1.1 | NPL | Eur-Am, Afr-Am |
| Costa Rica | DeLisi (2)7 | SCZ,SA (D4) | 60 | 132 | 2.20 | 397 | Nonpar | Multipoint | GH+ | LOD | Costa Rica |
| U.S./Australia | Levinson8 | SCZ,SA,PNOS (D3R) | 43 | 126 | 2.93 | 310 | Nonpar | Multipoint | GH 1.1 | NPL | Eur-Am, Afr-Am |
| Finland-isolate | Paunio (2)2 | SCZ; +SA (D4) | 53 | 113 | 2.13 | 398 | Dom, Rec | Single-point | LINKAGE | LOD | Finland (isolate) |
| NIMH, Eur-Am | Faraone9 | SCZ,SA-D (D3R) | 43 | 96 | 2.23 | 442 | Nonpar | Multipoint | GH 1.1 | NPL | Eur-Am, Afr-Am |
| NIMH, Afr-Am | Kaufmann10 | SCZ,SA-D (D3R) | 30 | 79 | 2.63 | 442 | Nonpar | Multipoint | GH 1.1 | NPL | Afr-Am |
| Canada | Brzustowicz11 | SCZ,SA (D3R) | 22 | 79 | 3.59 | 381 | Dom, Rec | Single-point | FASTLINK | HLOD | Canada (Celtic) |
| Texas | Garver12 | SCZ (D4) | 21 | 58 | 2.76 | 644 | Nonpar | Multipoint | GH+ | NPL | Afr-Am, Eur-Am |
| UC London | Gurling13 | SCZ,SA,UFP (RDC) | 13 | 56 | 4.31 | 365 | Model-free | Single-point | MFLINK | P values | Iceland,UK |
| Sweden | Lindholm14 | SCZ; +SA-D; +PNOS; +SA-B (D4) | 1 | 43 | 43.00 | 352 | Dom | Single-point | LINKAGE | LOD | Sweden |
| Kiel | Moises15 | SCZ,SA (RDC or D3R) | 5 | 37 | 7.40 | 413 | Nonpar | Single-point | WRPC | P values | Iceland |
| Utah | Coon16 | SCZ,SA-C (RDC) | 9 | 35 | 3.89 | 329 | Dom, Rec | Single-point | LINKAGE | LOD | Eur-Am |
| Palau | Byerley17 | SCZ,SA-S (RDC) | 5 | 33 | 6.60 | 496 | Dom, Rec | Single-point | LINKAGE | LOD | Palau |
| Total | 1,208 | 2,945 |
Badner and Gershon (2002_a_) reported a meta-analysis of published SCZ genome scans using their multiple scan probability (MSP) method (Badner and Gershon 2002_b_), which combines P values across scans in regions with clusters of positive scores after adjusting for the size of the region. Differences between these methods have been discussed in the first article in this series (Levinson et al. 2003 [in this issue]). The GSMA presented here included studies that were not available to Badner and Gershon (2002_b_) and excluded some of the studies they analyzed (see the “Discussion” section for details); our analysis also included data provided by the investigators for every marker in each scan, whereas the MSP used only published data (from scans with data in all nominally significant regions) and substituted P values of .5 or 1.0 for missing data points. By including only complete whole-genome data from each scan, the present analysis may be expected to avoid two problems inherent in publication bias: (1) investigators tend to present their most positive results, as well as weakly positive results that seem to confirm others’ findings, which could inflate meta-analysis results in those regions; and (2) conversely, GSMA can detect significant cross-study results for regions that are weakly positive in many studies but not sufficiently positive to have been presented as “one of the best results” in any study.
The GSMA for SCZ has been performed under a single narrow diagnostic model (with a few exceptions, as described below), which includes SCZ (psychotic symptoms such as delusions and hallucinations and deficits in emotional and social functioning, usually with a chronic course) and schizoaffective disorder (mixed schizophrenic and manic or depressive disorders with persistence of psychosis when mood symptoms remit), which coaggregates with SCZ in families (Levinson and Mowry 2000). Although family study data suggest that a broader spectrum of disorders coaggregate with SCZ in families (Levinson and Mowry 2000), the “core” diagnoses involving overt psychosis and chronic course are made most reliably across research groups (Faraone et al. 1996) and have been the focus of most linkage studies.
If at least weak linkage is present in many or most samples, larger samples would be expected to contribute more information. Therefore, we consider our primary analysis to be one in which the ranks of bins for each scan are weighted to account for sample size, as described below. Unweighted analyses are also reported for comparison. This method of meta-analysis should be most useful in detecting loci that increase disease susceptibility in a substantial proportion of the samples under study, although the observed effect in each sample may be both small and variable because of stochastic or other effects. One would not expect the method to be useful in cases where a locus is having an effect only in a few unique samples, such as those with specific pedigree structures or ethnic or geographical backgrounds. However, we have included samples collected in more isolated populations, because the similar clinical features and prevalence of SCZ in different cultures suggest that at least some of the susceptibility loci are common to many populations (Risch 1990). We followed the same rationale in including studies of one or a few extended pedigrees and of many smaller pedigrees.
Material and Methods
Selection of Genome Scans and Genotyping Data
Genome scans of SCZ were identified through publications and oral presentations. Scans were selected if DNA markers at 30 cM or finer density were typed throughout the genome in families selected through a proband with SCZ defined by Diagnostic and Statistical Manual (DSM)-IIIR or DSM-IV criteria or by Research Diagnostic Criteria (RDC) (and not through a particular subtype), in nonoverlapping samples with at least 30 affected genotyped cases with SCZ or schizoaffective disorder. Partial scans and candidate region studies were excluded. These criteria establish consistency across studies in several important respects, including reasonably even marker coverage of the genome, well-established linkage statistical methods, and the use of a diagnostic model with good reliability across research teams, as well as the predominantly European ethnicity of most of the samples; however, the studies did vary in sample size, the degree of ethnic diversity, the number of linkage analyses that were applied, and the density and evenness of marker spacing. For all selected scans (table 1), either the required data were already published or posted on the Web, or the investigators provided the requested data used in analyses that were published or in press, except for the Palau study (Coon et al. 1998), for which unpublished complete genome scan data were provided by the investigator (W.B.).
The 20 scans were performed by 14 different research groups in 16 projects. Straub et al. (2002_b_) typed three different interdigitated 30-cM marker maps (with follow-up of positive scores) in each of three pedigree subsets, which were treated here as three independent scans; the Finnish group separately analyzed families from the general Finnish population and from a genetically isolated subpopulation (Paunio et al. 2001); and the NIMH group analyzed European American and African American families as separate samples (Kaufman et al. 1998; Faraone et al. 1998). The GSMA method assumes a uniform map in each scan, so we did not consider the second stages of genome scans where candidate regions were more densely mapped or samples were modified. For example, we used only the first-phase uniform genome scan for the Cardiff study (Williams et al. 1999). For most scans, however, we could not achieve uniform density, because markers had been added at various times in candidate regions and any post hoc decision to remove certain markers would have been biased. The SD of the number of markers per bin ranged from close to 1, in some studies, to 4.19. Further study will be required to elucidate the degree of bias introduced into GSMA by variable marker densities.
Diagnostic Models
The most common phenotypic definition included both SCZ and schizoaffective disorders by any of the diagnostic criteria noted above, which are historically related and similar but not identical. For 16 of the 20 scans, we ignored any broader definitions and used only scores from analyses that considered these two diagnoses as affected; if the strategy had included sequential analyses considering only SCZ and then adding schizoaffective cases, we maximized scores at each marker over these two models. For one study (Garver et al. 2001), we used scores based only on cases of SCZ, because the alternative was a very broad definition. For three studies (Levinson et al. 1998; Gurling et al. 2001; Lindholm et al. 2001), the narrowest or only definition included SCZ, schizoaffective disorder, and other “nonaffective” psychoses, which have been shown to coaggregate with SCZ in families (Levinson and Mowry 2000). Note that, unlike traditional meta-analysis methods, which typically require an identical statistical test for each sample, GSMA can accommodate methodological differences, as long as each study uses an apparently valid approach to arrive at a single score, for each marker, that can be rank ordered across the genome. Since there were too few studies that used each alternative definition of the phenotype to permit separate GSMAs (as was done for bipolar disorder in the following article in this series [Segurado et al. 2003 {in this issue}]), we used the analysis from each study that came closest to the predominant diagnostic model of SCZ plus schizoaffective disorder. If the study’s strategy involved maximizing LOD scores over two or more diagnostic models within this range, then we followed that approach (see table 1). One limitation of the present analysis is that, in our simulation studies presented in the first article in this series (Levinson et al. 2003 [in this issue]), we did not consider the effect on GSMA of maximizing scores over multiple diagnostic and/or transmission models.
Linkage Scores Used in the Rank-Ordering Procedure
For each scan, we obtained linkage scores at each marker, the numbers of pedigrees and of affected genotyped cases for the selected diagnostic models, and the phenotypic criteria and the nature of the statistical tests (table 1). The rank-ordering procedure (below) requires one score for each marker. We used scores from a single nonparametric test (e.g., maximum LOD score [MLS] or NPL) of one diagnostic model, except as follows: for Utah (Coon et al. 1994), Palau (W.B., unpublished data), and Canada (Brzustowicz et al. 2000), we used the maximum of the dominant or recessive parametric LOD scores for any recombination fraction and proportion of linked families at each marker (one diagnostic model). We use the maximum LOD score over four parametric models (one diagnostic model) at each marker for Medical College of Virginia (MCV)/Ireland (Straub et al. 2002_b_), over four diagnostic models (one genetic model) for Sweden (Lindholm et al. 2001), over two “model-free” linkage tests (minimum P values for MFLOD and MALOD scores) for University College London (Gurling et al. 2001), and, for Finland (Paunio et al. 2001), over eight tests, including all combinations of two LOD scores (dominant and recessive), two diagnostic models, and two definitions of family structure (nuclear families, or with connections retained between more distantly related pedigrees, which was most relevant to the isolated population).
GSMA
GSMA (Wise et al. 1999) was performed as described in the first article of this series (Levinson et al. 2003 [in this issue]). Key terms are summarized in appendix A. In brief, the autosomes were divided into 120 30-cM bins defined by Généthon markers (CEPH-Généthon Integrated Map Web site). On the Marshfield map (Center for Medical Genetics Web site), the average bin width was 29.1 cM, as shown in figure 1. Each marker was placed within one of these bins, on the basis of its location on the Généthon, Marshfield, or Southampton map (Genetic Location Database Web site) or on the human genome map (National Center for Biotechnology Web site). For each study, each bin was assigned a within-study rank (_R_study) based on the maximum linkage score or minimum P value within the bin. Bins were ranked in ascending order (1 = most significant result). Negative or zero scores were considered ties, and their _R_study values were averaged (e.g., 20 such scores would all be assigned an _R_study of 110.5). The average rank across studies was then computed for each bin (_R_Avg). For the weighted analysis, each _R_study value was multiplied by its study’s weight (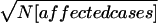 ), divided by the mean of this value over all studies), as discussed in the first article in this series (Levinson et al. 2003 [in this issue]). Two pointwise P values were determined, _P_AvgRnk and _P_ord, as defined in appendix A. These were computed by permutation test, as described in the first article of this series (Levinson et al. 2003 [in this issue]) (here, 5,000 permutations of the unweighted analysis and 30,000 of the weighted analysis).
), divided by the mean of this value over all studies), as discussed in the first article in this series (Levinson et al. 2003 [in this issue]). Two pointwise P values were determined, _P_AvgRnk and _P_ord, as defined in appendix A. These were computed by permutation test, as described in the first article of this series (Levinson et al. 2003 [in this issue]) (here, 5,000 permutations of the unweighted analysis and 30,000 of the weighted analysis).
Figure 1.

Ranks by study and average ranks. The following are shown: within-study ranks (_R_study) grouped as shown in the legend; the average rank (_R_avg) for each bin across studies (low values are best), weighted proportional to 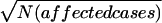 for each study; and the overall place of each bin in ascending order of average ranks (the lowest/best average rank is first place). Average ranks with significant _P_AvgRnk values are highlighted above the columns (black for P AvgRnk<.01, gray for P AvgRnk<.05). Exact _P_AvgRnk values are shown in table 2. Tied ranks sometimes resulted in uneven numbers of bins in some groupings, particularly for lower ranks when there were many zero or negative scores. Marshfield (“Mfd”) locations are shown for the marker at the distal boundary of each bin. Bin boundaries were selected at ∼30 cM spacing on the Généthon map; mean bin width is 29.1 cM on the Marshfield map. Peds = number of pedigrees; Aff = number of genotyped affected cases. See table 1 for the references associated with each study.
for each study; and the overall place of each bin in ascending order of average ranks (the lowest/best average rank is first place). Average ranks with significant _P_AvgRnk values are highlighted above the columns (black for P AvgRnk<.01, gray for P AvgRnk<.05). Exact _P_AvgRnk values are shown in table 2. Tied ranks sometimes resulted in uneven numbers of bins in some groupings, particularly for lower ranks when there were many zero or negative scores. Marshfield (“Mfd”) locations are shown for the marker at the distal boundary of each bin. Bin boundaries were selected at ∼30 cM spacing on the Généthon map; mean bin width is 29.1 cM on the Marshfield map. Peds = number of pedigrees; Aff = number of genotyped affected cases. See table 1 for the references associated with each study.
One might think of these two values by analogy in terms of a race. Consider runner X, who finished in fourth place. If the _R_Avg values in the randomly permuted analyses might be compared to the times of many runners in many previous races, then _P_AvgRnk is analogous to the probability of any previous runner having run a race at least as fast as X, and _P_ord is analogous to the probability of any previous fourth-place finisher having run a race at least as fast as X. _P_ord compares _R_Avg values to those with the same place in the order (was this _R_Avg particularly low, compared with other bins in this place in the order?), whereas _P_AvgRnk is a typical pointwise P value that ignores order.
The simulation studies presented in the first article in this series (Levinson et al. 2003 [in this issue]) suggested several criteria for determining the significance of GSMA results, corrected for multiple testing (i.e., genomewide significance). These are shown in appendix B. P AvgRnk<.000417 (.05 corrected for 120 tests) suggests that a bin is likely to contain a linked locus or loci. A number of aggregate criteria were also determined to suggest that linkage is likely to present in some or all of a set of bins. When aggregate criteria for linkage are met, bins that have achieved both P AvgRnk<.05 and P ord<.05 are the most likely to contain linked loci or to be adjacent to linked bins.
_P_ord can be computed by permuting ranks bin by bin or by permuting chromosomal segments so that average ranks for bins at the edges of chromosomes, where linkage information is reduced, will be compared with the distribution of other telomeric bins, as described in the first article in this series through use of simulated data (Levinson et al. 2003 [in this issue]). Here the by-chromosome permutation procedure was not more conservative, and we report _P_ord values that were computed by permuting by bin within each scan.
Results
Figure 1 shows the markers at the boundaries of each bin and their location on the Marshfield map, the average rank for each bin across all scans weighted by the square root of the number of genotyped affected cases, and a condensed representation of each bin’s within-study ranks (“1” denotes R _study_=1–5, etc., as shown in the legend). A complete list of weighted and unweighted _R_study and _R_Avg values is available at the URL listed in the “Electronic-Database Information” section. In the row labeled “bin” for each half of the figure, the bins with P AvgRnk<.01 are shaded in black, and those with P AvgRnk<.05 are shaded in gray. This figure illustrates which studies contributed the lowest ranks to the average rank for each bin. For example, bin 2.5, which achieved the best _R_Avg, had _R_study values between 1 and 10 in 8 of 20 scans, whereas bin 3.2, with the third-best _R_Avg, was ranked between 1 and 20 in only 4 scans, and its low _R_Avg was due to lower _R_study values than the average in most scans. The scans are sorted in descending size so that the relative contributions of larger and smaller data sets can be appreciated.
Figure 2 is a scatterplot of the weighted and unweighted average ranks for each bin, with solid diamonds representing the primary, weighted analysis, and open diamonds representing the unweighted analysis for comparison. The 99%, 95%, and 90% thresholds of significance are shown; the _Y_-axes for the two analyses have been adjusted so that the lines marking the thresholds are the same.
Figure 2.

Average ranks (_R_avg) for 20 SCZ genome scans. Shown are the average ranks for each bin, comparing the results for the unweighted (◊) and weighted (⧫) analyses. _P_AvgRnk thresholds are shown. See table 2 for exact _P_AvgRnk and _P_ord values for the most significant bins.
Table 2 lists _P_AvgRnk and _P_ord values for weighted and unweighted analyses for the 19 bins with either value <.05 by either analysis. Results for the primary weighted analysis will be discussed here and are illustrated in figure 3. Only bin 2.5 (2p11.1-q21.1) met the criterion for genomewide significance of P AvgRnk<.000417. The nominally significant _P_AvgRnk of the adjacent bin, 2.6, provides additional evidence for linkage here, because, in simulated data, bins adjacent to those containing disease loci often also achieve nominal significance. This bin had one of the five lowest ranks in six scans (U.S./International, MCV/Ireland [c], U.S./Australia, NIMH-Eur-Am, Kiel, and Palau), and ranks between 6 and 10 in two others (Cardiff and Utah).
Table 2.
_P_AvgRnk and _P_ord for Bins with the Lowest Average Ranks[Note]
| Unweighted Analysis | Weighted analysis | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Bin | Marshfield Location(Begin–End)(cM) | CytogeneticLocation | Rank | _P_AvgRnk | _P_ord | Rank | _P_AvgRnk | _P_ord | Both_P_ Values<.05 |
| 2.5 | 101.6–128.4 | 2p12-q22.1 | 1 | .0001 | .0111 | 1 | .0004 | .0327 | * |
| 5.5 | 131.5–164.2 | 5q23.2-q34 | 4 | .0139 | .0765 | 2 | .0032 | .0491 | * |
| 3.2 | 32.4–63.1 | 3p25.3-p22.1 | 2 | .0039 | .0739 | 3 | .0060 | .0311 | * |
| 11.5 | 99.0–123.0 | 11q22.3-q24.1 | 3 | .0075 | .0543 | 4 | .0060 | .0040 | * |
| 6.1 | 0–32.6 | 6pter-p22.3 | 7 | .0220 | .0101 | 5 | .0159 | .0328 | * |
| 2.6 | 128.4–154.5 | 2q22.1-q23.3 | 8 | .0240 | .0042 | 6 | .0230 | .0448 | * |
| 1.6 | 142.2–170.8 | 1p13.3-q23.3 | 10 | .0598 | .1372 | 7 | .0235 | .0136 | * |
| 22.1 | 0–33.8 | 22pter-q12.3 | 9 | .0420 | .0411 | 8 | .0310 | .0216 | * |
| 8.2 | 27.4–55.0 | 8p22-p21.1 | 6 | .0184 | .0169 | 9 | .0310 | .0068 | * |
| 6.2 | 32.6–65.1 | 6p22.3-p21.1 | 13 | .0817 | .1288 | 10 | .0330 | .0024 | * |
| 20.2 | 21.2–47.5 | 20p12.3-p11 | 5 | .0173 | .0479 | 11 | .0460 | .0098 | * |
| 14.1 | 0–40.1 | 14pter-q13.1 | 21 | .1559 | .2514 | 12 | .0470 | .0043 | * |
| 16.2 | 32.1–67.6 | 16p13-q12.2 | 18 | .1297 | .2262 | 13 | .0560 | .0069 | |
| 18.4 | 96.5–126.0 | 18q22.1-qter | 15 | .1020 | .1841 | 14 | .0650 | .0103 | |
| 10.1 | 0–29.2 | 10pter-p14 | 24 | .1818 | .2706 | 15 | .0680 | .0046 | |
| 1.7 | 170.8–201.6 | 1q23.3-q31.1 | 11 | .0662 | .1283 | 16 | .0820 | .0142 | |
| 15.3 | 52.3–85.6 | 15q21.3-q26.1 | 12 | .0679 | .0697 | 17 | .0950 | .0293 | |
| 6.4 | 99.0–131.1 | 6q15-q23.2 | 27 | .2048 | .2592 | 18 | .0980 | .0177 | |
| 17.3 | 63.6–94.0 | 17q21.33-q24.3 | 23 | .1621 | .1458 | 19 | .1120 | .0349 |
Figure 3.

SCZ average ranks versus random distribution. The blackened diamonds (⧫) denote the weighted average ranks (_R_avg) for the bins with the best (i.e., lowest) 19 ranks in the SCZ GSMA, sorted in order of _R_avg. Bin numbers are labeled in boldface type for bins with _P_AvgRnk and P ord<.05 and labeled in normal type for bins with only P ord<.05. The gray data points indicate the mean _R_avg for each ordered bin (first place, second place, etc.) in 5,000 randomly permuted replicates, and the vertical gray bars represent 2 SDs above and below these means. The figure illustrates how, for this cluster of 19 bins, average ranks are significantly lower than expected from the random distribution. See table 2 for exact P values and cytogenetic locations of the best 19 bins.
Three aggregate criteria for genomewide significant linkage were met:
- 1.
Twelve bins had P AvgRnk<.05 (shown in boldface italic type in table 2). This exceeds the threshold of ⩾11 such bins observed in <5% of simulated whole-genome replicates of similar data sets with no linkage present. - 2.
Each of these 12 bins also had P ord<.05, indicated by asterisks in table 2 and illustrated in figure 3. This exceeds the 5% empirical threshold of ⩾4 bins with _P_AvgRnk and _P_ord values <.05 for GSMA replicates of 14 or 20 studies (the threshold was 5 with only 9 studies); 12 such bins were never observed in 1,000 unlinked replicates of 20 studies resembling those in the SCZ GSMA data set. - 3.
The 19 bins with the lowest average ranks had _P_ord values <.05 (see fig. 3). The 5% threshold in unlinked simulated data was ⩾4 such values among the 10 best bins, and a cluster of 19 was never observed in 1,000 unlinked replicates of 20 studies.
In simulated data with linkage present, all three of these thresholds were exceeded with increasing frequency as the number of linked bins and their genetic effect increased.
Finally, to determine whether the placement of bin boundaries was having a critical effect on results, adjacent bins were combined into 60-cM bins, for two possible combinations (e.g., for chromosome 1, bins 1+2, 3+4, etc., and then 2+3, 4+5, etc.), for the weighted analysis. Results were unchanged: the same regions had nominally significant _P_AvgRnk P values, as are shown in table 2 for 30-cM bins, with similar levels of significance, and no new regions of interest were identified. Thus, it appears that the placement of bin boundaries did not, in this case, bias the results against any region.
Discussion
Genomewide evidence for linkage was observed in this meta-analysis of 20 SCZ genome scans. The average rank of bin 2.5 (chromosome 2p12-q22.1) was associated with a genomewide significant P value after correction for multiple testing. In addition, all three empirical aggregate criteria for linkage were met: more bins than expected by chance achieved nominally significant values of _P_AvgRnk (pointwise P value of the average rank), of _P_ord (pointwise P value of the average rank taking into account its place in the order of average ranks), and of both _P_AvgRnk and _P_ord. The number of bins meeting each of the latter two criteria was highly significant—that is, it was greater than was observed in any of 1,000 GSMAs of simulated whole-genome replicates of 20 similar studies (genomewide P<.001).
The aggregate criteria suggest that these 20 scans support linkage of multiple loci to SCZ. However, there is no straightforward way to determine which of the implicated bins represent true positive results. In simulations of genomes containing 5–10 linked loci, each of small effect, bins with nominal significance for _P_AvgRnk and _P_ord are most likely to contain a linked locus or to be immediately adjacent to a bin that does; when clusters of bins meet this criterion, few, if any, are false positives. Therefore, the 10 chromosomal regions represented by the first 12 bins in table 2 (marked with asterisks) should be considered strong SCZ candidate regions. In simulated data, both the linked bin and an adjacent bin often achieve nominally significant _P_AvgRnk and _P_ord, and it is possible that this phenomenon is being observed in the pairs of significant bins on chromosomes 2q (2.5 and 2.6) and 6p (6.1 and 6.2) and perhaps also on 1q (1.6 and 1.7), where bin 1.7 achieved a nominally significant _P_ord.
Six additional chromosomal regions were included in the cluster of 19 consecutive bins with nominally significant values of _P_ord (table 2 ). In simulated data (see table 7 in the first article in this series [Levinson et al. 2003 {in this issue}]), when there are 5–10 weakly linked loci in the genome, there are still more unlinked than linked bins with P ord<.05, but these tend to be distributed throughout the overall order of average ranks rather than clustered among the best average ranks. Thus, it is possible that some or all of these regions are also linked to SCZ, but the support for these regions must be considered weaker than for the cluster with nominally significant _P_AvgRnk and _P_ord.
The simulation studies reported in the previous article (Levinson et al. 2003 [in this issue]) did not consider a sufficiently broad range of situations to interpret these data more definitively. For example, we did not study the effects on GSMA of placing two or more very weakly linked loci in the same bin or in adjacent bins or of varying the genetic effect of multiple linked loci. However, the single-bin and aggregate criteria for genomewide significance discussed above were validated by data from simulations of genomes with no linkage present, so that these conclusions do not depend on the comprehensiveness of the simulations that assumed linkage.
Data for the unweighted analysis, with each study weighted equally, are shown in table 2 for comparison. Genomewide significance was still observed for bin 2.5. Only 9 bins had nominally significant _P_AvgRnk values, fewer than the aggregate significance threshold of 11, but 6 of the best 10 bins had both _P_AvgRnk and _P_ord values <.05. In discussions among the authors and with colleagues, it has been debated whether small studies with larger numbers of cases per pedigree should be given relatively more weight, because of the possible advantages of such pedigrees for linkage analysis. The unweighted analysis can be considered the most extreme possible alternative, in that the smallest study is weighted equally with the largest study, but it produced less evidence for linkage in most regions, including those that have been supported by studies of larger pedigrees.
These data do not exclude the possibility that other chromosomal regions harbor SCZ susceptibility loci that could not be detected by the present methods or that substantially influence risk in only one or a few populations. GSMA does not currently consider X or Y chromosome data, so no conclusions can be reached concerning possible linkage on those chromosomes (DeLisi et al. 2000). There are many other limitations to our analysis—for example, we made decisions about multiple tests (we maximized over them), subdivided data sets (we considered them separate scans), used only the first stage of two-stage scans, accepted variable map density both within and across studies, as well as variable numbers of incomplete families (missing parents), and, in some cases, used data with denser maps of certain candidate regions. It is possible that other decisions regarding these or other issues would have produced different and perhaps more accurate results.
Regarding variable marker density, the mean number of markers genotyped in the 12 bins with P AvgRnk<.05 was 5.21, versus 3.1 for all other bins (_t_=3.39; _P_=.006). Wise (2000) simulated an unlinked 30-cM region in 120 sib pairs (10 markers, 500 replicates). Multipoint sib pair analysis using 3, 5, 7, or 10 markers yielded mean maximum MLS values (within the bin) of 0.272, 0.333, 0.373, and 0.385, respectively, because of increasing SDs. A GSMA could thus be biased by extreme variation in density, especially if there was biased selection of regions (e.g., increased density in candidate regions only where positive scores had been observed by chance). In the present data, the densest typing was in a few studies that first reported evidence for linkage in a region; however, because the bin had one of the best ranks before the extra markers were typed, the biasing effect should be small. For example, in bin 6.1, mean coverage was 8.65 markers, but without the Bonn and three MCV/Ireland scans (18–27 markers) the mean was 4.85. For bin 22.1, the mean was 6.85, but without Johns Hopkins, U.S./International, and Bonn (13–16 markers) it was 4.82. However, this type of bias was not systematically studied here, and an effect cannot be ruled out. Future investigators should be aware that it is advantageous to perform an initial analysis of a screening map with uniform spacing prior to adding additional markers in candidate regions.
One previous meta-analysis of SCZ genome scan data (Badner and Gershon 2002_a_) applied a method for combining P values from many of these studies and found significant results on chromosomes 8p and 13q. They analyzed published results, whereas we obtained linkage data for all markers from each investigator, to permit detection of evidence for linkage in regions that had not come to attention because scores were not strikingly positive. The differences in results could also have been due primarily to the use of different sets of scan analyses. Badner and Gershon (2002_a_) included nine data sets that were used in this analysis (Bonn, Canada, Johns Hopkins, Kiel, the two NIMH scans, U.K./Iceland, U.S./Australia, and Utah), a different data set for one scan (the second-stage analysis of candidate regions in the Cardiff scan, using an enlarged sample), incomplete data for four scans (U.S./International, the two Finnish scans, and Sweden), no data for six scans (the three MCV/Ireland subsets, and the Costa Rica, Texas, and Palau scans), and data from four scans that we excluded because of small sample size (Bailer et al. 2002), incomplete genome coverage (Barr et al. 1994), selection of probands for periodic catatonia rather than SCZ (Stober et al. 2000), or possible overlap with another sample (Rees et al. 1999) leading us to include the larger sample (Williams et al. 1999). The same region of chromosome 8p was significant in both analyses, and it seems understandable that we identified additional significant regions with a larger data set and more complete data. It is more difficult to explain why the 13q region was highly significant in the Badner and Gershon (2002_a_) analysis but did not approach significance here. Evidence for a susceptibility gene in this region has recently been published (Chumakov et al. 2002) and awaits confirmation.
In the present analysis, bin 2.5 (2p11.1-q21.1) had the best average rank (31.8), with a genomewide significant P _AvgRnk_=.0004. This bin had ranks between 1 and 5 in six scans (U.S./International, MCV/Ireland [c], U.S./Australia, NIMH-Eur-Am, Kiel, and Palau) and had ranks between 6 and 10 in two scans (Cardiff and Utah). DeLisi et al. (2002_b_) observed slightly more positive results in bin 2.4, with the peak MLS in the initial genome scan at the boundary marker for bins 2.4 and 2.5 (D2S139), whereas the other scans with high ranks in this region maximized within bin 2.5 or the more distal bin 2.6.
Among the other bins listed in table 2, previous findings on chromosomes 6p and 8p have been discussed in the Introduction. For 6p, maximal evidence for linkage was in bin 6.1 (Lindholm et al. 2001; Straub et al. 2002_b_) or 6.2 (Schwab et al. 2000), whereas all of the highly positive linkage reports on 8p have been in bin 8.2 (Kendler et al. 1996; SLCG 1996; Blouin et al. 1998; Brzustowicz et al. 1999), except for the linkage peak and candidate gene locus (NRG1) reported recently by Stefansson et al. (2002), which are in bin 8.3. Positive findings in bin 5.5 have previously been presented by Straub et al. (1997) and by Schwab et al. (1997). Pulver et al. (1995) reported a modestly positive LOD score on chromosome 3p in bin 3.1, whereas the current finding is in bin 3.2; a multicenter study that found no evidence for linkage on 3p (SLCG 1996) did not include all of bin 3.2. Gurling et al. (2001) reported evidence for linkage in bin 11.5. On chromosome 1q, the maximum evidence for linkage reported by Brzustowicz et al. (2000) was in bin 1.6, and the nearby peak reported by Gurling et al. (2001) was in bin 1.7. Bin 22.1 on chromosome 22q contains the deletion region for velo-cardio-facial syndrome, which is associated with schizophrenic symptoms (Pulver et al. 1994_b_; Karayiorgou et al. 1995; Murphy et al. 1998) and which came to the attention of the field after Pulver et al. (1994_a_) reported modestly positive evidence for linkage in the region. Chiu et al. (2002) reported an interaction between linkage at markers in bins 8.2 and 14.1 in the Johns Hopkins data set.
Among the bins in the cluster of nominally significant _P_ord values, bin 10.1 had one of the highest ranks in four scans (U.S./International, MCV/Ireland [a], Bonn, and NIMH-Eur-Am) and had been proposed as a candidate region by Faraone et al. (1998), Schwab et al. (1998), and Straub et al. (1998), with a multicenter study providing only weak support (Levinson et al. 2000). Bin 6.4 contains the region of chromosome 6q discussed above. No previous reports have called attention to chromosomes 16p-q, 15q, or 17q as SCZ candidate regions. Bin 15.3 is considerably distal to the location of the α-7-nicotinic receptor in bin 15.1 (Freedman et al. 1997), where positive evidence for linkage to SCZ has been reported (Freedman et al. 2001_a,_ 2001_b_; Gejman et al. 2001; Tsuang et al. 2001).
It is of interest that, in several cases, the GSMA provided support for linkages that had been observed previously in small samples of extended pedigrees from more homogeneous or isolated populations (e.g., regions of chromosomes 1q, 2q, 6q, and 11q, as discussed above), despite the fact that the weighting procedure greatly reduced the contributions of these smaller samples to the results. This supports the hypothesis that there are at least some loci that contribute to SCZ susceptibility in many or most populations and that loci whose effects are detected particularly strongly in a unique population may also prove relevant in many other populations.
In conclusion, we can suggest with considerable confidence that SCZ susceptibility loci are likely to exist in some or all of the chromosomal regions noted above. A meta-analysis is one step toward defining genomic regions that harbor SCZ susceptibility loci. Investigators might wish to consider performing collaborative linkage analyses using raw genotypes and integrated marker maps or regenotyping available pedigrees with a common, dense map. Investigators of other complex disorders might be further stimulated to collaborate in various ways, to study larger data sets. Additional details of the present analysis, including ranks for each study, are available at the University of Pennsylvania Web site listed in the Electronic-Database Information section. These results are cause for considerable optimism about the prospects for identifying SCZ susceptibility genes.
We plan to update this analysis when several new SCZ genome scans are completed, and we invite investigators with new genome scan data to contact the present authors.
Acknowledgments
This work was funded by NIMH grant R01-MH61602 (to D.F.L.). The genome scan projects were supported, in part, by NIMH grant R01-MH44245, Axys Pharmaceuticals and Schizophrenia A National Emergency (SANE) (support to L.E.D.); NIMH grants R01-MH45390 (to R.E.S.), R01-MH41953 (to K.S.K.), and R01-MH52537 (to C. J. MacLean); the Academy of Finland and the MacDonald Foundation, USA (support to L.P.); the Medical Research Council of the U.K. (support to M.J.O.); Deutsche Forschungsgemeinschaft grant SFB 400 and German-Israeli Foundation for Scientific Research (support to D.B.W.); NIMH grant R01-MH4558 and Novartis Pharmaceuticals (1995–1998) (support to A.E.P.); NIMH grants R01-MH45097, K02-01207, K24-MH64197 (to D.F.L.); National Health and Medical Research Council of Australia (NHMRC) grants 910234, 941087, and 971095, the Rebecca L. Cooper Medical Research Foundation, the Queensland Department of Health, and the NHMRC Network for Brain Research into Mental Disorders (support to B.J.M.); NIMH grants 1 R01-MH41874-01, 5 U01-MH46318, and 1 R37-MH43518 (to M.T.T.), U01-MH46289 and R01-MH44292 (to C.A.K.), and U01-MH46276 (to C.R.C.); the Distinguished Investigator Award from the National Alliance for Research on Schizophrenia and Depression (to C.A.K.); Millennium Pharmaceuticals, sponsored by Wyeth-Ayerst Pharmaceuticals (NIMH project); Canadian Institutes of Health Research grant MT-1225, the Ontario Mental Health Foundation, the Bill Jeffries Schizophrenia Endowment Fund, the Ian Douglas Bebensee Foundation (support to A.B.); NIMH grants K08-MH01392 and R01-MH62440, Independent Investigator Award from the National Alliance for Research on Schizophrenia and Depression, and the EJLB Foundation Scholar Research Programme (support to L.M.B.); the Center for Inherited Disease Research (Canada project); the Veterans Administration Merit Review award (to D.L.G.); MRC project grant G880473N, The European Science Foundation, SANE, the Iceland Department of Health, the General Hospital Reykjavik, the Joseph Levy Charitable Foundation, Wellcome Trust grant 055379, The Priory Hospital, the Neuroscience Research Charitable Trust, the University of Iceland and the Icelandic Science Council, and Généthon (Paris) (support to H.M.D.G.); Deutsche Forschungsgemeinschaft (support to H.W.M.); and NIMH grants R01-MH42643, K02-MH01089, and R01-MH56098 (to W.B.). The authors gratefully acknowledge the many additional collaborators who contributed to these projects. Finally we acknowledge the families without whose patience and assistance, in the face of the many burdens of coping with familial schizophrenia, progress in this field would be impossible.
Appendix A: Summary of Terminology
Bin:
One of 120 30-cM autosomal segments used as units of analysis in GSMA; bin 2.1 is the first 30 cM of chromosome 2.
_R_study (within-study rank):
The rank of each bin within a single study, based on the maximum linkage score (or lowest P value) within it. The bin containing the best score has a rank of 1. All negative and 0 scores are considered to be tied. For weighted analyses, each raw rank is multiplied by the study’s weighting factor.
_R_Avg (average rank):
The average of a bin’s within-study ranks or weighted ranks across all studies.
_P_AvgRnk (probability of _R_Avg):
The pointwise probability of observing a given _R_Avg for a bin in a GSMA of N studies, determined by theoretical distribution (unweighted analysis only) or by permutation test.
_P_ord (probability of _R_Avg given the order):
The pointwise probability that, for example, a first-place, second-place, third-place, etc., bin would achieve _R_Avg at least this extreme in a GSMA of N studies.
Genomewide significance:
For α=0.05, correction for 120 bins yields a threshold for genomewide significance of .000417 for _P_AvgRnk or _P_ord. For suggestive linkage (a result observed once per scan by chance), α=1/120=0.0083.
Appendix B: Criteria for Genomewide Significance
For individual bins, the criterion for genomewide significance is P AvgRnk<.000417. When linkage is likely to be present in one or more bins, the aggregate criteria are as follows: ⩾11 bins with P AvgRnk<.05, ⩾4 bins with P ord<.05 among the 10 best values of _R_Avg, or ⩾5 bins with P AvgRnk<.05 and P ord<.05. Bins with P AvgRnk<.05 and P ord<.05 are most likely to contain linked loci. No valid combined significance criterion was identified.
Electronic-Database Information
The URLs for data presented herein are as follows:
- Center for Medical Genetics, http://research.marshfieldclinic.org/genetics/ (for the Marshfield map)
- CEPH-Généthon Integrated Map, http://www.cephb.fr/ceph-genethon-map.html
- D. F. Levinson Research, http://depressiongenetics.med.upenn.edu/meta-analysis.html (for further details, including weighted and unweighted ranks for each bin for each study)
- Genetic Location Database, http://cedar.genetics.soton.ac.uk/public_html/ldb.html (for the Cedar genetic map)
- National Center for Biotechnology Information Home Page, http://www.ncbi.nlm.nih.gov/ (for cytogenetic locations obtained from the April 2002 freeze of the Human Genome Project)
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for SCZD) [PubMed]
References
- Badner JA, Gershon ES (2002_a_) Meta-analysis of whole-genome linkage scans of bipolar disorder and schizophrenia. Mol Psychiatry 7:405–411 [DOI] [PubMed] [Google Scholar]
- ——— (2002_b_) Regional meta-analysis of published data supports linkage of autism with markers on chromosome 7. Mol Psychiatry 7:56–66 [DOI] [PubMed] [Google Scholar]
- Bailer U, Leisch F, Meszaros K, Lenzinger E, Willinger U, Strobl R, Heiden A, Gebhardt C, Doge E, Fuchs K, Sieghart W, Kasper S, Hornik K, Aschauer HN (2002) Genome scan for susceptibility loci for schizophrenia and bipolar disorder. Biol Psychiatry 52:40–52 [DOI] [PubMed] [Google Scholar]
- Barr CL, Kennedy JL, Pakstis AJ, Wetterberg L, Sjogren B, Bierut L, Wadelius C, Wahlstrom J, Martinsson T, Giuffra L (1994) Progress in a genome scan for linkage in schizophrenia in a large Swedish kindred. Am J Med Genet 54:51–58 [DOI] [PubMed] [Google Scholar]
- Blouin JL, Dombroski BA, Nath SK, Lasseter VK, Wolyniec PS, Nestadt G, Thornquist M, et al (1998) Schizophrenia susceptibility loci on chromosomes 13q32 and 8p21. Nat Genet 20:70–73 [DOI] [PubMed] [Google Scholar]
- Brzustowicz LM, Hodgkinson KA, Chow EW, Honer WG, Bassett AS (2000) Location of a major susceptibility locus for familial schizophrenia on chromosome 1q21-q22. Science 288:678–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzustowicz LM, Honer WG, Chow EW, Little D, Hogan J, Hodgkinson K, Bassett AS (1999) Linkage of familial schizophrenia to chromosome 13q32. Am J Hum Genet 65:1096–1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Q, Martinez M, Zhang J, Sanders AR, Badner JA, Cravchik A, Markey CJ, Beshah E, Guroff JJ, Maxwell ME, Kazuba DM, Whiten R, Goldin LR, Gershon ES, Gejman PV (1997) Suggestive evidence for a schizophrenia susceptibility locus on chromosome 6q and a confirmation in an independent series of pedigrees. Genomics 43:1–8 [DOI] [PubMed] [Google Scholar]
- Chiu YF, McGrath JA, Thornquist MH, Wolyniec PS, Nestadt G, Swartz KL, Lasseter VK, Liang KY, Pulver AE (2002) Genetic heterogeneity in schizophrenia. II. Conditional analyses of affected schizophrenia sibling pairs provide evidence for an interaction between markers on chromosome 8p and 14q. Mol Psychiatry 7:658–664 [DOI] [PubMed] [Google Scholar]
- Chumakov I, Blumenfeld M, Guerassimenko O, Cavarec L, Palicio M, Abderrahim H, Bougueleret L, et al (2002) Genetic and physiological data implicating the new human gene G72 and the gene for D-amino acid oxidase in schizophrenia. Proc Natl Acad Sci USA 99:13675–13680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coon H, Jensen S, Holik J, Hoff M, Myles-Worsley M, Reimherr F, Wender P, Waldo M, Freedman R, et al (1994) Genomic scan for genes predisposing to schizophrenia. Am J Med Genet 54:59–71 [DOI] [PubMed] [Google Scholar]
- Coon H, Myles-Worsley M, Tiobech J, Hoff M, Rosenthal J, Bennett P, Reimherr F, Wender P, Dale P, Polloi A, Byerley W (1998) Evidence for a chromosome 2p13–14 schizophrenia susceptibility locus in families from Palau, Micronesia. Mol Psychiatry 3:521–527 [DOI] [PubMed] [Google Scholar]
- DeLisi LE, Mesen A, Rodriguez C, Bertheau A, LaPrade B, Llach M, Riondet S, Razi K, Relja M, Byerley W, Sherrington R (2002_a_) Genome-wide scan for linkage to schizophrenia in a Spanish-origin cohort from Costa Rica. Am J Med Genet 114:497–508 [DOI] [PubMed] [Google Scholar]
- DeLisi LE, Shaw SH, Crow TJ, Shields G, Smith AB, Larach VW, Wellman N, Loftus J, Nanthakumar B, Razi K, Stewart J, Comazzi M, Vita A, Heffner T, Sherrington R (2002_b_) A genome-wide scan for linkage to chromosomal regions in 382 sibling pairs with schizophrenia or schizoaffective disorder. Am J Psychiatry 159:803–812 [DOI] [PubMed] [Google Scholar]
- DeLisi LE, Shaw S, Sherrington R, Nanthakumar B, Shields G, Smith AB, Wellman N, Larach VW, Loftus J, Razi K, Stewart J, Comazzi M, Vita A, De Hert M, Crow TJ (2000) Failure to establish linkage on the X chromosome in 301 families with schizophrenia or schizoaffective disorder. Am J Med Genet 96:335–341 [DOI] [PubMed] [Google Scholar]
- Faraone SV, Blehar M, Pepple J, Moldin SO, Norton J, Nurnberger JI, Malaspina D, Kaufmann CA, Reich T, Cloninger CR, DePaulo JR, Berg K, Gershon ES, Kirch DG, Tsuang MT (1996) Diagnostic accuracy and confusability analyses: an application to the Diagnostic Interview for Genetic Studies. Psychol Med 26:401–410 [DOI] [PubMed] [Google Scholar]
- Faraone SV, Matise T, Svrakic D, Pepple J, Malaspina D, Suarez B, Hampe C, Zambuto CT, Schmitt K, Meyer J, Markel P, Lee H, Harkavy Friedman J, Kaufmann C, Cloninger CR, Tsuang MT (1998) Genome scan of European-American schizophrenia pedigrees: results of the NIMH Genetics Initiative and Millennium Consortium. Am J Med Genet 81:290–295 [PubMed] [Google Scholar]
- Freedman R, Coon H, Myles-Worsley M, Orr-Urtreger A, Olincy A, Davis A, Polymeropoulos M, Holik J, Hopkins J, Hoff M, Rosenthal J, Waldo MC, Reimherr F, Wender P, Yaw J, Young DA, Breese CR, Adams C, Patterson D, Adler LE, Kruglyak L, Leonard S, Byerley W (1997) Linkage of a neurophysiological deficit in schizophrenia to a chromosome 15 locus. Proc Natl Acad Sci USA 94:587–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman R, Leonard S, Gault JM, Hopkins J, Cloninger CR, Kaufmann CA, Tsuang MT, Farone SV, Malaspina D, Svrakic DM, Sanders A, Gejman P (2001_a_) Linkage disequilibrium for schizophrenia at the chromosome 15q13-14 locus of the alpha7-nicotinic acetylcholine receptor subunit gene (CHRNA7). Am J Med Genet 105:20–22 [PubMed] [Google Scholar]
- Freedman R, Leonard S, Olincy A, Kaufmann CA, Malaspina D, Cloninger CR, Svrakic D, Faraone SV, Tsuang MT (2001_b_) Evidence for the multigenic inheritance of schizophrenia. Am J Med Genet 105:794–800 [DOI] [PubMed] [Google Scholar]
- Garver DL, Holcomb J, Mapua FM, Wilson R, Barnes B (2001) Schizophrenia spectrum disorders: an autosomal-wide scan in multiplex pedigrees. Schizophr Res 52:145–160 [DOI] [PubMed] [Google Scholar]
- Gejman PV, Sanders AR, Badner JA, Cao Q, Zhang J (2001) Linkage analysis of schizophrenia to chromosome 15. Am J Med Genet 105:789–793 [DOI] [PubMed] [Google Scholar]
- Göring HHH, Terwilliger JD, Blangero J (2001) Large upward bias in estimation of locus-specific effects from genome-wide scans. Am J Hum Genet 69:1357–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurling HM, Kalsi G, Brynjolfson J, Sigmundsson T, Sherrington R, Mankoo BS, Read T, Murphy P, Blaveri E, McQuillin A, Petursson H, Curtis D (2001) Genomewide genetic linkage analysis confirms the presence of susceptibility loci for schizophrenia, on chromosomes 1q32.2, 5q33.2, and 8p21-22 and provides support for linkage to schizophrenia, on chromosomes 11q23.3-24 and 20q12.1-11.23. Am J Hum Genet 68:661–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser ER, Boehnke M, Guo SW, Risch N (1996) Affected sib-pair interval mapping and exclusion for complex genetic traits: sampling considerations. Genet Epidemiol 13:117–138 [DOI] [PubMed] [Google Scholar]
- Karayiorgou M, Morris MA, Morrow B, Shprintzen RJ, Goldberg R. Borrow J, Gos A, Nestadt G, Wolyniec PS, Lasseter VK (1995): Schizophrenia susceptibility associated with interstitial deletions of chromosome 22q11. Proc Natl Acad Sci USA 92:7612–7616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann CA, Suarez B, Malaspina D, Pepple J, Svrakic D, Markel PD, Meyer J, Zambuto CT, Schmitt K, Matise TC, Harkavy Friedman JM, Hampe C, Lee H, Shore D, Wynne D, Faraone SV, Tsuang MT, Cloninger CR (1998) NIMH Genetics Initiative Millennium Schizophrenia Consortium: linkage analysis of African-American pedigrees. Am J Med Genet 81:282–289 [PubMed] [Google Scholar]
- Kendler KS, MacLean CJ, O’Neill FA, Burke J, Murphy B, Duke F, Shinkwin R, Easter SM, Webb BT, Zhang J, Walsh D, Straub RE (1996) Evidence for a schizophrenia vulnerability locus on chromosome 8p in the Irish Study of High-Density Schizophrenia Families. Am J Psychiatry 153:1534–1540 [DOI] [PubMed] [Google Scholar]
- Lander E, Kruglyak L (1995) Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet 11:241–247 [DOI] [PubMed] [Google Scholar]
- Levinson DF, Holmans P, Straub RE, Owen MJ, Wildenauer DB, Gejman PV, Pulver AE, Laurent C, Kendler KS, Walsh D, Norton N, Williams NM, Schwab SG, Lerer B, Mowry BJ, Sanders AR, Antonarakis SE, Blouin JL, DeLeuze JF, Mallet J (2000) Multicenter linkage study of schizophrenia candidate regions on chromosomes 5q, 6q, 10p, and 13q: schizophrenia linkage collaborative group III. Am J Hum Genet 67:652–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levinson DF, Levinson MD, Segurado R, Lewis CM (2003) Genome scan meta-analysis of schizophrenia and bipolar disorder, part I: methods and power analysis. Am J Hum Genet 73:17–33 (in this issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levinson DF, Mahtani MM, Nancarrow DJ, Brown DM, Kruglyak L, Kirby A, Hayward NK, Crowe RR, Andreasen NC, Black DW, Silverman JM, Endicott J, Sharpe L, Mohs RC, Siever LJ, Walters MK, Lennon DP, Jones HL, Nertney DA, Daly MJ, Gladis M, Mowry BJ (1998) Genome scan of schizophrenia. Am J Psychiatry 155:741–750 [DOI] [PubMed] [Google Scholar]
- Levinson DF, Mowry BJ (2000) Genetics of schizophrenia. In: Pfaff DW, Berrettini WH, Maxson SC, Joh TH (eds) Genetic influences on neural and behavioral functions. CRC Press, New York, pp 47–82 [Google Scholar]
- Lindholm E, Ekholm B, Shaw S, Jalonen P, Johansson G, Pettersson U, Sherrington R, Adolfsson R, Jazin E (2001) A schizophrenia-susceptibility locus at 6q25, in one of the world’s largest reported pedigrees. Am J Hum Genet 69:96–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez M, Goldin LR, Cao Q, Zhang J, Sanders AR, Nancarrow DJ, Taylor JM, Levinson DF, Kirby A, Crowe RR, Andreasen NC, Black DW, Silverman JM, Lennon DP, Nertney DA, Brown DM, Mowry BJ, Gershon ES, Gejman PV (1999) Follow-up study on a susceptibility locus for schizophrenia on chromosome 6q. Am J Med Genet 88:337–343 [PubMed] [Google Scholar]
- Maziade M, Roy MA, Rouillard E, Bissonnette L, Fournier JP, Roy A, Garneau Y, Montgrain N, Potvin A, Cliche D, Dion C, Wallot H, Fournier A, Nicole L, Lavallee JC, Merette C (2001) A search for specific and common susceptibility loci for schizophrenia and bipolar disorder: a linkage study in 13 target chromosomes. Mol Psychiatry 6:684–693 [DOI] [PubMed] [Google Scholar]
- Moises HW, Yang L, Kristbjarnarson H, Wiese C, Byerley W, Macciardi F, Arolt V, Blackwood D, Liu X, Sjogren B, et al (1995) An international two-stage genome-wide search for schizophrenia susceptibility genes. Nat Genet 11:321–324 [DOI] [PubMed] [Google Scholar]
- Murphy KC, Jones RG, Griffiths E, Thompson PW, Owen MJ (1998) Chromosome 22qII deletions: an under-recognised cause of idiopathic learning disability. Br J Psychiatry 172:180–183 [DOI] [PubMed] [Google Scholar]
- Paunio T, Ekelund J, Varilo T, Parker A, Hovatta I, Turunen JA, Rinard K, Foti A, Terwilliger JD, Juvonen H, Suvisaari J, Arajarvi R, Suokas J, Partonen T, Lonnqvist J, Meyer J, Peltonen L (2001) Genome-wide scan in a nationwide study sample of schizophrenia families in Finland reveals susceptibility loci on chromosomes 2q and 5q. Hum Mol Genet 10:3037–3048 [DOI] [PubMed] [Google Scholar]
- Pulver AE, Karayiorgou M, Wolyniec PS, Lasseter VK, Kasch L, Nestadt G, Antonarakis S, Housman D, Kazazian HH, Meyers D (1994_a_) Sequential strategy to identify a susceptibility gene for schizophrenia: report of potential linkage on chromosome 22q12-q13.1: part 1. Am J Med Genet 54:36–43 [DOI] [PubMed] [Google Scholar]
- Pulver AE, Lasseter VK, Kasch L, Wolyniec P, Nestadt G, Blouin JL, Kimberland M, Babb R, Vourlis S, Chen H, Lalioti M, Morris MA, Karayiorgou M, Ott J, Meyers D, Antonarakis SE, Housman D, Kazazian HH (1995) Schizophrenia: a genome scan targets chromosomes 3p and 8p as potential sites of susceptibility genes. Am J Med Genet 60:252–260 [DOI] [PubMed] [Google Scholar]
- Pulver AE, Nestadt G, Goldberg R, Shprintzen RJ, Lamacz M, Wolyniec PS, Morrow B, Karayiorgou M, Antonarakis SE, Housman D (1994_b_) Psychotic illness in patients diagnosed with velo-cardio-facial syndrome and their relatives. J Nerv Ment Dis 182:476–478 [DOI] [PubMed] [Google Scholar]
- Rees MI, Fenton I, Williams NM, Holmans P, Norton N, Cardno A, Asherson P, Spurlock G, Roberts E, Parfitt E, Mant R, Vallada H, Dawson E, Li MW, Collier DA, Powell JF, Nanko S, Gill M, McGuffin P, Owen MJ (1999) Autosome search for schizophrenia susceptibility genes in multiply affected families. Mol Psychiatry 4:353–359 [DOI] [PubMed] [Google Scholar]
- Risch N (1990) Genetic linkage and complex diseases, with special reference to psychiatric disorders. Genet Epidemiol 7:3–16 [DOI] [PubMed] [Google Scholar]
- Rybicki BA, Elston RC (2000) The relationship between the sibling recurrence risk and genotype relative risk. Am J Hum Genet 66:593–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schizophrenia Linkage Collaborative Group for Chromosomes 3, 6 and 8 (1996) Additional support for schizophrenia linkage on chromosomes 6 and 8: a multicenter study. Am J Med Genet 67:580–594 [DOI] [PubMed] [Google Scholar]
- Schwab SG, Eckstein GN, Hallmayer J, Lerer B, Albus M, Borrmann M, Lichtermann D, Ertl MA, Maier W, Wildenauer DB (1997) Evidence suggestive of a locus on chromosome 5q31 contributing to susceptibility for schizophrenia in German and Israeli families by multipoint affected sib-pair linkage analysis. Mol Psychiatry 2:156–160 [DOI] [PubMed] [Google Scholar]
- Schwab SG, Hallmayer J, Albus M, Lerer B, Eckstein GN, Borrmann M, Segman RH, Hanses C, Freymann J, Yakir A, Trixler M, Falkai P, Rietschel M, Maier W, Wildenauer DB (2000) A genome-wide autosomal screen for schizophrenia susceptibility loci in 71 families with affected siblings: support for loci on chromosome 10p and 6. Mol Psychiatry 5:638–649 [DOI] [PubMed] [Google Scholar]
- Schwab SG, Hallmayer J, Albus M, Lerer B, Hanses C, Kanyas K, Segman R, Borrman M, Dreikorn B, Lichtermann D, Rietschel M, Trixler M, Maier W, Wildenauer DB (1998) Further evidence for a susceptibility locus on chromosome 10p14-p11 in 72 families with schizophrenia by nonparametric linkage analysis. Am J Med Genet 81:302–307 [PubMed] [Google Scholar]
- Segurado R, Detera-Wadleigh SD, Levinson DF, Lewis CM, Gill M, Nurnberger JI Jr, Craddock N (2003) Genome scan meta-analysis of schizophrenia and bipolar disorder, part III: bipolar disorder. Am J Hum Genet 73:49–62 (in this issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson H, Sigurdsson E, Steinthorsdottir V, Bjornsdottir S, Sigmundsson T, Ghosh S, Brynjolfsson J, et al (2002) Neuregulin 1 and susceptibility to schizophrenia. Am J Hum Genet 71:877–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stober G, Saar K, Ruschendorf F, Meyer J, Nurnberg G, Jatzke S, Franzek E, Reis A, Lesch KP, Wienker TF, Beckmann H (2000) Splitting schizophrenia: periodic catatonia-susceptibility locus on chromosome 15q15. Am J Hum Genet 67:1201–1207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub RE, Jiang Y, MacLean CJ, Ma Y, Webb BT, Myakishev MV, Harris-Kerr C, Wormley B, Sadek H, Kadambi B, Cesare AJ, Gibberman A, Wang X, O’Neill FA, Walsh D, Kendler KS (2002_a_) Genetic variation in the 6p22.3 gene DTNBP1, the human ortholog of the mouse dysbindin gene, is associated with schizophrenia. Am J Hum Genet 71:337–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub RE, MacLean CJ, Ma Y, Webb BT, Myakishev MV, Harris-Kerr C, Wormley B, Sadek H, Kadambi B, O’Neill FA, Walsh D, Kendler KS (2002_b_) Genome-wide scans of three independent sets of 90 Irish multiplex schizophrenia families and follow-up of selected regions in all families provides evidence for multiple susceptibility genes. Mol Psychiatry 7:542–559 [DOI] [PubMed] [Google Scholar]
- Straub RE, MacLean CJ, Martin RB, Myakishev MV, Harris-Kerr C, O’Neill FA, Walsh D, Kendler KS (1998) A schizophrenia locus may be located in region 10p15.1. Am J Med Genet 81:296–301 [DOI] [PubMed] [Google Scholar]
- Straub RE, MacLean CJ, O’Neill FA, Burke J, Murphy B, Duke F, Shinkwin R, Webb BT, Zhang J, Walsh D, Kendler KK (1995) A potential vulnerability locus for schizophrenia on chromosome 6p-24-22 and evidence for genetic heterogeneity. Nature Genet 11:287–293 [DOI] [PubMed] [Google Scholar]
- Straub RE, MacLean CJ, O’Neill FA, Walsh D, Kendler KS (1997). Support for a possible schizophrenia vulnerability locus in region 5q22-31 in Irish families. Mol Psychiatry 2:148–155 [DOI] [PubMed] [Google Scholar]
- Suarez BK, Hampe CL, VanEerdewegh PV (1994) Problems of replicating linkage claims in psychiatry. In: Gershon ES, Cloninger CR (eds) Genetic approaches to mental disorders. American Psychiatric Press, Washington, DC, pp 23–46 [Google Scholar]
- Tsuang DW, Skol AD, Faraone SV, Bingham S, Young KA, Prabhudesai S, Haverstock SL, Mena F, Menon AS, Bisset D, Pepple J, Sauter F, Baldwin C, Weiss D, Collins J, Boehnke M, Schellenberg GD, Tsuang MT (2001). Examination of genetic linkage of chromosome 15 to schizophrenia in a large Veterans Affairs Cooperative Study sample. Am J Med Genet 105:662–668 [PubMed] [Google Scholar]
- Williams NM, Rees MI, Holmans P, Norton N, Cardno AG, Jones LA, Murphy KC, Sanders RD, McCarthy G, Gray MY, Fenton I, McGuffin P, Owen MJ (1999) A two-stage genome scan for schizophrenia susceptibility genes in 196 affected sibling pairs. Hum Mol Genet 8:1729–1739 [DOI] [PubMed] [Google Scholar]
- Wise LH (2000) Meta-analysis of genetic studies for complex diseases. PhD thesis, University of London, London [Google Scholar]
- Wise LH, Lanchbury JS, Lewis CM (1999) Meta-analysis of genome searches. Ann Hum Genet 63:263–272 [DOI] [PubMed] [Google Scholar]