A Novel Functional Human Eukaryotic Translation Initiation Factor 4G (original) (raw)
Abstract
Mammalian eukaryotic translation initiation factor 4F (eIF4F) is a cap-binding protein complex consisting of three subunits: eIF4E, eIF4A, and eIF4G. In yeast and plants, two related eIF4G species are encoded by two different genes. To date, however, only one functional eIF4G polypeptide, referred to here as eIF4GI, has been identified in mammals. Here we describe the discovery and functional characterization of a closely related homolog, referred to as eIF4GII. eIF4GI and eIF4GII share 46% identity at the amino acid level and possess an overall similarity of 56%. The homology is particularly high in certain regions of the central and carboxy portions, while the amino-terminal regions are more divergent. Far-Western analysis and coimmunoprecipitation experiments were used to demonstrate that eIF4GII directly interacts with eIF4E, eIF4A, and eIF3. eIF4GII, like eIF4GI, is also cleaved upon picornavirus infection. eIF4GII restores cap-dependent translation in a reticulocyte lysate which had been pretreated with rhinovirus 2A to cleave endogenous eIF4G. Finally, eIF4GII exists as a complex with eIF4E in HeLa cells, because eIF4GII and eIF4E can be purified together by cap affinity chromatography. Taken together, our findings indicate that eIF4GII is a functional homolog of eIF4GI. These results may have important implications for the understanding of the mechanism of shutoff of host protein synthesis following picornavirus infection.
In eukaryotes, mRNA translation is a complex process that involves the concerted interactions of numerous components (29). The cap structure m7GpppX, where X is any nucleotide, is present at the 5′ end of all cellular mRNAs (except organellar). The cap is bound by the cap-binding protein complex, eukaryotic initiation factor 4F (eIF4F). This complex is believed to promote, in conjunction with eIF4B, the unwinding of mRNA 5′ secondary structure to facilitate ribosome binding (reviewed in reference 41). Mammalian eIF4F consists of three subunits: eIF4E, eIF4A, and eIF4G. eIF4E is the subunit which specifically interacts with the cap. eIF4A is a bidirectional RNA helicase (35, 36), and eIF4G (formerly called eIF4γ or p220) is a scaffolding polypeptide which interacts with eIF4E, eIF4A, eIF3, and 40S ribosomes (reviewed in references 14 and 37). An eIF4E binding site exists in the N-terminal one-third region of eIF4G (23), whereas eIF4A interacts with the C-terminal one-third, and eIF3 binds to the central region (18). It is thought that through these interactions, eIF4G is responsible for recruiting the 43S preinitiation complex to the mRNA (18, 34). Yeast eIF4G contains an RNA recognition motif-like sequence (12), and mammalian eIF4G has been shown to interact with the encephalomyocarditis virus (EMCV) internal ribosome entry site (IRES) in vitro (34). eIF4F exhibits sequence-independent RNA-binding activity (17). This activity could partially explain the finding that eIF4F is approximately 20 times more potent as an RNA helicase than the free form of its catalytic subunit, eIF4A (36).
Many lines of evidence support the idea that eIF4G participates in both cap-dependent and cap-independent translation (26–28, 31, 33, 34). Several members of the picornavirus family, including poliovirus, human rhinovirus 2 (HRV2), and foot-and-mouth disease virus, inhibit cellular mRNA translation by cleaving eIF4G into two fragments (10, 19, 22, 39, 40), which effectively separates the N-terminal eIF4E binding site from the C-terminal binding sites for eIF4A and eIF3. This separation leads to the shutoff of host cell mRNA translation. Picornavirus RNAs, which are uncapped and which are translated in a cap-independent fashion, utilize the C-terminal fragment of eIF4G for translation (26–28, 33, 34).
eIF4G is encoded by two distinct genes in wheat germ (3, 6) and in yeast (12), while in mammals, only one cDNA has been reported (45). Recently, the existence of a truncated homolog of eIF4G that is functionally different from eIF4G was described (15, 21, 44). This homolog, p97/NAT1/DAP-5, does not interact with eIF4E and appears to be a negative regulator of translation (15, 44). Here we describe the isolation and functional characterization of a novel, closely related functional homolog of eIF4G.
MATERIALS AND METHODS
Materials.
Materials were obtained from the following sources: restriction enzymes, New England Biolabs; T7 RNA polymerase, RNasin, and rabbit reticulocyte lysate, Promega; T7 DNA polymerase sequencing kit, m7G(5′)ppp(5′)G, agarose adipic acid hydrazide, glutathione-Sepharose, and Ready-to-go labeling kit, Pharmacia Biotech, Inc.; protein A-Sepharose, Repligen; QIAexpress type IV vector kit, QIAGEN; Hybond-N+ nylon membrane, chemiluminescence system, and 125I-labeled protein A (30 mCi/mg), Amersham; nitrocellulose membrane, Schleicher & Schuell; Lipofectin and 5′ rapid amplification of cDNA ends (RACE) kit, GIBCO-BRL; 5′/3′ RACE kit, Boehringer Mannheim; elastatinal and bovine heart muscle kinase (HMK), Sigma; and [γ-32P]ATP, [α-32P]dCTP (3,000 Ci/mmol), [35S]methionine (1,000 Ci/mmol), and En3Hance, DuPont, NEN. Oligonucleotides were prepared at the Sheldon Biotechnology Center, McGill University, or at Dalton Chemical Laboratories, North York, Ontario, Canada.
Isolation of eIF4GII cDNA clones and DNA sequence analysis.
EST06315/GenBank accession no. T08424 cDNA from a human fetal brain cDNA library was obtained from the American Type Culture Collection. Human placenta (in λgt11), human ovarian follicle cell, and fetal brain cDNA libraries (in λ Uni-ZAP-XR), kindly provided by M. Park, K. H. Scheit, and G. Rouleau, were screened with several 5′ end PCR probes.
Construction of plasmids.
pcDNA3-HA (hemagglutinin)-eIF4GI and pcDNA3-HA-La were described previously (15). pcDNA3-HA-luciferase was generated by insertion in frame into pcDNA3-HA of the luciferase cistron, which had been excised from the pGEM-luc plasmid DNA (Promega). The _Eco_RI restriction site of eIFGII cDNA (see Fig. 1B) was used to construct pcDNA3-HA-eIF4GII. For the preparation of baculovirus-expressed recombinant protein, pBlueBacHis2 (version A; Invitrogen) was employed as a polyhedrin promoter transfer vector. _Eco_RI fragments (Fig. 1B) of eIFGII cDNA and of human eIF4GI cDNA (45) were used to construct pBlueBacHis2-eIF4GII and pBlueBacHis2-eIF4GI. Note that because the _Eco_RI site was utilized to generate pcDNA3 and pBlueBacHis2-eIF4GII, the expressed protein lacks the first 158 terminal amino acids (aa). Constructs for truncated glutathione _S_-transferase (GST)-eIF4GII proteins (see Fig. 5B) were generated by PCR with primers in which an _Eco_RI site had been engineered. Cleavage of the PCR products with _Eco_RI and ligation in the pGEX expression vector (4) creates GST-FLAG-HMK fusion proteins. pGEX2T(128/129) (4) was digested with _Eco_RI and ligated in frame with the PCR products to create GST-FLAG-HMK-eIF4GII 445–604 and GST-FLAG-HMK-eIF4GII 445–718. For both constructs, the forward primer was 5′-GTG GAA TTC GCC ATC ACA GTC CAG AGAG-3′, whereas the reverse primers were 5′-CAC GAA TTC CCC ATC ACC AGA ACT ATC TG-3′ and 5′-TTG CAG AAT TCT ACC AGA ACT GTG ATG ATC TTTC-3′, respectively. To confirm that no mutations had been introduced by the PCR, the cDNAs were subjected to DNA sequencing on both strands. pGEMCAT/EMC/LUC, containing the coding regions of chloramphenicol acetyltransferase (CAT) and luciferase separated by the IRES of EMCV, has been described previously (30). FLAG-HMK-eIF4E cDNA has been described previously (30). 5′-RACE was performed with 2 μg of HeLa poly(A)+ RNA as the template for avian myeloblastosis virus reverse transcriptase (RT) and sequence-specific primers. The RT primers were 5′-GCC ATT AGC TTG AGG CTC CAG ACC GCCT-3′ for eIF4GI and 5′-ATA CAC CGG CTG ACT TGGG-3′ for eIF4GII. The PCR primers were 5′-CAT GAT CTC CTC TGT GAT ATC CT-3′ for eIF4GI and 5′-GTT GGG GGG GCC CAA CAT AAG GAG GGC-3′ for eIF4GII.
FIG. 1.

(A) Protein sequence alignment of human eIF4GI and eIF4GII. The pattern-induced multisequence alignment program (38) was used to align eIF4GI and eIF4GII amino acid sequences. Identical and conserved amino acid residues are in solid and shaded boxes, respectively. The eIF4E binding site (23) is indicated by dashed lines above and below the sequences. The rhinovirus 2Apro cleavage site (19) is indicated by arrows. The published ATG initiation codon of eIF4GI is marked with an asterisk. (The sequence of eIF4GI is revised from the original entry of reference 45 [accession no. AF012088], but it does not contain the newly discovered extension at the N terminus [see panel C].) (B) Alignment of nucleotide and deduced amino acid sequences of eIF4GI and eIF4GII flanking the eIF4GI assigned initiator AUG (45). The published ATG initiation codon and the first methionine of eIF4GI are marked as +1. _Eco_RI is a conserved restriction site in both eIF4GI and eIF4GII cDNA. A putative splice acceptor site (SA) for an intron in eIF4GI is indicated by an arrow. The putative intron sequence is in lowercase letters. (C) Protein alignment of human eIF4GI and eIF4GII N-terminal regions (accession no. AF012088 and AF012072). Identical and conserved amino acid residues are in solid and shaded boxes, respectively. The published ATG initiation codon of eIF4GI is marked with an asterisk.
FIG. 5.

eIF4GII interacts with eIF4E. (A) Alignment of eIF4E-binding regions of eIF4G proteins from several species and human 4E-binding proteins (3, 12, 30, 45). (B) Schematic representation of GST-HMK-eIF4GII fusion constructs. (C) eIF4E interaction with eIF4GII. GST-eIF4GII fusion fragments (positions 445 to 604 and 445 to 718) were expressed in E. coli and resolved by SDS-PAGE (10% polyacrylamide). Proteins were blotted onto nitrocellulose membranes as described in Materials and Methods. Immunoblot analysis (left panel) was performed with an anti-GST polyclonal antibody (1:1,000), and far-Western analysis of an identical membrane (right panel) was conducted with 32P-labeled FLAG-HMK-eIF4E. _E. coli_-purified FLAG-HMK-eIF4E fusion protein (30) (3 μg) was 32P-labeled with the catalytic subunit of bovine HMK (Sigma) (4). Processing of the nitrocellulose filters through a denaturation-renaturation cycle and hybridization (3 to 4 h, 4°C) with the probe (105 cpm/ml) were performed as described previously (4). Membranes were washed twice with the hybridization buffer and processed for autoradiography. (D) Coimmunoprecipitation (IP) of eIF4E with eIF4GII. HA-tagged protein expression plasmids pcDNA3-HA-luciferase (lane 1), pcDNA3-HA-eIF4GI (lane 2), pcDNA3-HA-eIF4GII (lane 3), and pcDNA3-HA (lane 4) were transfected into HeLa cells after infection with vTF7-3 as described in Materials and Methods. Proteins were immunoprecipitated with the 16B12 anti-HA monoclonal antibody (Babco), and immunoprecipitates were resolved by SDS-PAGE (12% polyacrylamide). Western blotting was performed with anti-eIF4E (upper panel) or anti-HA (lower panel) antibody (α) as described in Materials and Methods.
Cell culture, virus infections, and transient transfections.
The Mahoney strain of poliovirus type 1 was used to infect HeLa R19 cells grown in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum. Cells at ∼80% confluency were infected in serum-free medium at a multiplicity of infection of 100 PFU per cell. After adsorption of the virus at room temperature for 30 min, cells were further incubated at 37°C for 4.5 h. Cells were scraped into cold phosphate-buffered saline, pelleted by slow centrifugation, and lysed in cold lysis buffer (20 mM Tris-HCl [pH 7.5], 150 mM KCl, 1 mM dithiothreitol [DTT], 1 mM EDTA) by three freeze-thaw cycles. Cell debris was pelleted by centrifugation, and the concentration of protein in the supernatant was measured by the Bio-Rad assay.
HeLa cells were infected with recombinant vaccinia virus vTF7-3 (11) and transfected with plasmid DNA (5 μg) with Lipofectin (GIBCO-BRL) according to the manufacturer’s recommendations.
Spodoptera frugiperda (Sf9) insect cells were kindly provided by Paul Lasko (McGill University). Recombinant baculoviruses expressing His-tagged eIF4GI or eIF4GII were generated with a Bac-N-Blue transfection kit (Invitrogen). The His-tagged recombinant proteins were purified with Ni2+-nitrilotriacetic acid-agarose (QIAGEN). Recombinant mouse FLAG-HMK-eIF4E was purified as previously described (30).
Antibodies and Western blotting.
Cell extracts were prepared as described above. Proteins were denatured with an equal volume of 2× Laemmli sample buffer and boiled for 2 min. Proteins were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and blotted onto 0.45-μm-pore-diameter nitrocellulose membranes. The membranes were blocked for 2 h at 4°C in 5% skim milk and probed with various antibodies against initiation factors overnight at 4°C in 10 mM Tris-HCl (pH 8.0)–150 mM NaCl–0.5% Tween 20–5% skim milk. The blot was washed and subsequently incubated with either 125I-labeled protein A at a 1:1,000 dilution or with donkey anti-rabbit immunoglobulin conjugated with horseradish peroxidase (Amersham) at a 1:5,000 dilution in the same buffer for 1 h at room temperature. After extensive washing, the blot was either exposed to Kodak X-OMAT AR film or developed with the Renaissance enhanced chemiluminescence system (ECL; Amersham).
A GST-eIF4GII fusion protein of a region of eIF4GII which is not conserved between the two eIF4G polypeptides (aa 445 to 604 [Fig. 1A]) was purified on glutathione Sepharose (Pharmacia) as described previously (25). Two rabbits were immunized once with 0.5 mg of _Escherichia coli_-purified protein followed by additional injections of 250 μg at 4-week intervals. The crude serum was then utilized at 1:500. Monoclonal anti-eIF4A, polyclonal anti-eIF3, and polyclonal anti-eIF4GI antibodies were kind gifts from H. Trachsel, J. Hershey, and L. Carrasco, respectively. Polyclonal anti-eIF4E antibody and polyclonal anti-p97 antibody have been described previously (15, 20). The 12CA5 and 16B12 monoclonal antibodies directed against the HA epitope were obtained from M. Tremblay (McGill University) and Babco (Richmond, Calif.), respectively.
Coimmunoprecipitation.
Following transfection, HeLa cells were cultured for 16 h in 6-cm-diameter petri dishes. Cells were lysed in 1 ml of buffer B (100 mM KCl, 0.5 mM EDTA, 20 mM HEPES-KOH [pH 7.6], 0.4% Nonidet P-40, 20% glycerol, 1 mM DTT, 1 mM phenylmethylsulfonyl fluoride, 5 μg of pepstatin per ml, 5 μg of leupeptin per ml). Following centrifugation, an aliquot (0.5 ml) was mixed with 2 μg of anti-HA antibody for 6 h at 4°C. Protein G-Sepharose (30 μl of a 50% slurry) was added, and the mixture was incubated for an additional 2 h. After being washed with buffer B (1 ml, three times), immunoprecipitates were collected by centrifugation, and proteins were eluted with Laemmli buffer and subjected to SDS-PAGE.
Northern analysis.
Northern blots were prehybridized at 68°C for 2 h with ExpressHyb hybridization solution according to the manufacturer’s instructions (Clontech). As probes, portions of the 3′ untranslated region (UTR) of eIF4GI and eIF4GII cDNA spanning nucleotides (nt) 4598 to 5018 and 4973 to 5560, respectively, were utilized. Hybridization was carried out at 68°C for 3 h in the same ExpressHyb solution containing boiled randomly labeled cDNA probe (2 × 109 to 4 × 109 cpm/μg [2 × 106 cpm/ml]). Blots were washed for 40 min at room temperature in 2× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate)–0.05% SDS and for 15 to 30 min at 50°C in 0.1× SSC–0.1% SDS, and then they were exposed for 48 h to a Kodak BioMax film with intensifying screens.
In vitro transcription and translation.
pGEMCAT/EMC/LUC (30) was linearized with _Xho_I. In vitro transcription was performed with T7 RNA polymerase (Promega) for 2 h at 37°C. Capped RNA was synthesized in the presence of the cap analog m7GpppG at a 10-fold molar excess over GTP. RNA integrity was examined by gel electrophoresis. For translation, rabbit reticulocyte lysate (Promega) was programmed with mRNA in the presence of [35S]methionine (20 μCi) according to the manufacturer’s recommendations. Translation reaction mixtures were incubated at 30°C for 90 min and analyzed by SDS-PAGE. Gels were fixed with 40% methanol–7% acetic acid, treated with En3Hance (Dupont, NEN) and processed for autoradiography. The intensity of the bands was determined with a BAS-2000 PhosphorImager (Fuji).
Nucleotide sequence accession number.
The GenBank nucleotide and protein sequence accession numbers for eIF4GII and eIF4GI are AF012072 and AF012088, respectively.
RESULTS
Isolation and characterization of eIF4GII.
A partial human cDNA clone (EST06315/GenBank accession no. T08424) (1) was found to encode a predicted protein with homology to the human and rabbit eIF4G proteins (45). This cDNA was used to screen a human placenta cDNA library. In the screening of 3 × 106 plaques, 10 positive clones were isolated and sequenced. With the 5′ sequence of the longest clone, we sequentially screened human ovarian follicle cell and fetal brain cDNA libraries. Three overlapping partial cDNA fragments were ligated to form a contiguous cDNA of 5.6 kb. An oligonucleotide probe from the 5′ end of this cDNA was used in a 5′-RACE to obtain an extended sequence of 280 nt. The first ATG in this sequence, which is located 257 nt from the 5′ end of the cDNA, begins an open reading frame of 4,755 nt. It is very likely that this ATG is the authentic initiation codon, because there exists an in-frame upstream stop codon. The 3′ UTR is 836 nt in length. We termed the novel putative protein eIF4GII to distinguish it from the existing eIF4G protein (45), which hereafter is referred to as eIF4GI. The eIF4GII cDNA encodes a predicted polypeptide of 1,585 aa with a predicted molecular mass of 176.4 kDa. The two eIF4G proteins share 46% identity at the amino acid level, with an overall similarity of 56% (Fig. 1A). The homology is distributed throughout the entire protein, but less so for the amino terminus, and is particularly high in some areas of the central and carboxy-terminal portions. For example, a stretch of 70 aa in the middle portion of the proteins (aa 701 to 770 in eIF4GI and aa 890 to 959 in eIF4GII [Fig. 1A]) is completely conserved. The predicted amino terminus of eIF4GII is 158 aa longer than the published eIF4GI sequence (but see below). We noted, however, that eIF4GI and eIF4GII show a striking complete conservation at the amino acid level in sequences that lie 5′ to the assigned initiator AUG of eIF4GI (93% identity at the nucleotide level and 100% identity at the corresponding amino acid level [Fig. 1B]). The high similarity between the two cDNAs ends abruptly at a position (indicated by an arrow) that includes a canonical splice acceptor site (a stretch of pyrimidines followed by AG) in the eIF4GI DNA. Thus, the published eIF4GI cDNA sequence (45) might contain an intron at its 5′ end. To address this possibility a 5′-RACE was performed with a 5′ end probe of eIF4GI cDNA on HeLa poly(A)+ RNA. Several cDNA clones were obtained, and all contained an overlapping sequence upstream of the predicted 3′ splice acceptor site, which differed from the published eIF4GI cDNA. Strikingly this sequence was highly homologous to the eIF4GII sequence (Fig. 1C). Thus, it is highly likely that we have obtained the authentic amino terminus of eIF4GI. However, unlike the case for eIF4GII, it is not clear whether we have identified the initiator ATG of eIF4GI, because there is no upstream in-frame termination codon for the first ATG. This ATG corresponds to an internal ATG in eIF4GII. Similar results from the extension of the published eIF4GI sequence were obtained by Poncet et al. (34a). (Because we are unsure about the assignment of the initiator ATG codon in eIF4GI, we will use the numbering published in the original eIF4GI cloning publication [45] throughout the paper.)
eIF4GII mRNA expression was examined by Northern blotting (Fig. 2). Specific hybridization probes derived from the divergent eIF4GII and eIF4GI 3′ UTRs detected two major mRNA species of ∼6.0 and ∼5.2 kb, respectively (Fig. 2A). The apparent size of the eIF4GII mRNA suggests that the cloned cDNA is nearly full length. eIF4GII transcripts are ubiquitously expressed (Fig. 2B, upper panel). However, there do appear to be some differences in levels of expression in different tissues between eIF4GI and eIF4GII (e.g., brain [Fig. 2B, middle and upper panels]).
FIG. 2.

Northern analysis. (A) A Northern blot of human fetal brain poly(A)+ RNA (2 μg [Clontech]) was probed with a labeled PCR product derived from the 3′ UTR of eIF4GI cDNA (lane 1), as described in Materials and Methods. The membrane was then stripped and reprobed with a random-labeled PCR probe derived from the 3′ UTR of the eIF4GII cDNA (lane 2). Autoradiograms of lanes 1 and 2 were superimposed (lane 3). RNA size markers (in kilobases) are indicated to the left. (B) Tissue distribution. Membranes containing poly(A)+ RNA from human adult or fetal tissues or human cancer cell lines (2 μg [Clontech]) were probed with PCR probes derived from the 3′ UTRs of eIF4GII (upper panel), eIF4GI (middle panel), or a human β-actin cDNA (lower panel). RNA samples were used as indicated in the figure, and the cell lines were as follows: lane 20, peripheral blood leukocytes (PBL); lane 21, promyelocytic leukemia HL-60; lane 22, HeLa S3; lane 23, chronic myelogenous leukemia K-562; lane 24, lymphoblastic leukemia MOLT-4; lane 25, Burkitt’s lymphoma Raji; lane 26, colorectal adenocarcinoma SW480; lane 27, lung carcinoma A549; lane 28, melanoma G361. RNA size markers (in kilobases) are indicated to the left of the figure.
eIF4GII protein expression.
To examine eIF4GII protein expression, an antibody directed against an N-terminal fragment (aa 445 to 604) of the predicted eIF4GII protein sequence, which does not share amino acid similarity with eIF4GI (Fig. 1A), was produced. To determine the specificity of the anti-eIF4GII serum, Western analysis of baculovirus recombinant eIF4GI and eIF4GII proteins was performed. The proteins were first resolved by SDS-PAGE and visualized by Coomassie staining. Both proteins migrated at ∼195 kDa, although eIF4GII migrated slightly faster than eIF4GI (Fig. 3A). Both recombinant eIF4GI and eIF4GII migrated faster than the native polypeptides, which migrate at about 220 kDa, largely because both proteins were expressed from the AUG corresponding to that published for eIF4GI as the initiator AUG (45) (Fig. 1B), and thus missing 158 aa for eIF4GII and at least 109 aa for eIF4GI (Fig. 1C). Anti-eIF4GI serum, which was generated against short synthetic peptides (which are divergent from eIF4GII) from the amino and carboxy termini of the eIF4GI protein (2), recognized recombinant eIF4GI but not eIF4GII (Fig. 3B, compare lane 1 to lane 2). Similarly, the anti-eIF4GII serum recognized the recombinant eIF4GII, but not eIF4GI (compare lane 4 to lane 3). The presence of recombinant eIF4GII and eIF4GI in lanes 2 and 3 was confirmed by incubation of the membranes with anti-eIF4GII and anti-eIF4GI antibodies, respectively (data not shown).
FIG. 3.

Expression of eIF4GII protein. (A) Coomassie blue staining of recombinant proteins. His-eIF4GII and His-eIF4GI were produced in Sf9 cells, and equal amounts of each protein were resolved by SDS-PAGE (8% polyacrylamide) as described in Materials and Methods. Molecular masses (in kilodaltons) of protein standards (Bio-Rad) are indicated to the left. (B) Immunological identification of eIF4GI and eIF4GII proteins. Recombinant eIF4GI (0.5 μg [lanes 1 and 3]) and eIF4GII (0.5 μg [lanes 2 and 4]) were resolved on an SDS–5 to 10% gradient polyacrylamide gel, and proteins were transferred onto a nitrocellulose membrane, which was probed with anti-eIF4GI (lanes 1 and 2) or anti-eIF4GII (lanes 3 and 4) antibodies (α). Positions of molecular mass standards (in kilodaltons) are indicated to the left. (C) Cytoplasmic extracts were prepared from mock-infected (lanes 1 and 2) or poliovirus-infected (lanes 3 and 4) HeLa cells as described in Materials and Methods. Extracts (75 μg) were resolved on an SDS–5 to 10% gradient polyacrylamide gel, and proteins were transferred onto a nitrocellulose membrane, which was probed with anti-eIF4GI (lanes 1 and 3) or anti-eIF4GII (lanes 2 and 4) antibodies. N-terminal fragments are bracketed, and the C-terminal fragment of eIF4GI is indicated by a dot. Protein size markers (Bio-Rad) are indicated (in kilodaltons) to the left.
When tested on a HeLa cell extract, anti-eIF4GII interacted with a set of polypeptides (approximately three) that migrated on an SDS-polyacrylamide gel at similar, but not identical, positions to eIF4GI (Fig. 3C, compare lane 2 to lane 1). Infection of HeLa cells with poliovirus caused the disappearance of eIF4GI and the appearance of cleavage products (lane 3 [the N-terminal fragments are bracketed, and the C-terminal fragment is indicated by a dot]). A similar but not identical pattern of cleavage products was detected with anti-eIF4GII antiserum (lane 4 [the antibody recognizes only the bracketed N-terminal fragments]). These results indicate that eIF4GII is expressed in HeLa cells and that, as documented for eIF4GI, it is probably a substrate for the poliovirus 2Apro. Indeed, the sequence for the 2A cleavage site (R485-G487 in human eIF4GI) (19) is conserved in eIF4GII (R692-G693 [Fig. 1A]).
To directly demonstrate the ability of picornavirus 2Apro to cleave recombinant eIF4GII, in vitro cleavage was performed with purified rhinovirus 2Apro. Recombinant eIF4GI and eIF4GII were digested with recombinant HRV2 2Apro, subjected to electrophoresis, and transferred to a nitrocellulose membrane. The membrane was probed with a monoclonal antibody (anti-Xpress; Invitrogen) that recognizes the sequence D-L-Y-D-D-D-D-Y encoded in the baculovirus vector. Cleavage of eIF4GI by 2Apro is significantly increased in the presence of eIF4E (13, 26), and incubation of recombinant eIF4GI with 2Apro in the presence of eIF4E resulted in efficient cleavage of eIF4GI (Fig. 4, compare lane 2 to lane 1). Similarly, incubation of recombinant eIF4GII with 2Apro in the presence of eIF4E also resulted in the efficient cleavage of the protein (compare lane 5 to lane 3). In the absence of eIF4E, only partial proteolysis of eIF4GII by 2Apro was observed (lane 4). Cleavage of eIF4GII also resulted in the appearance of a faster-migrating protein of ∼30 kDa which may indicate the presence of a second cleavage site for the viral protease.
FIG. 4.
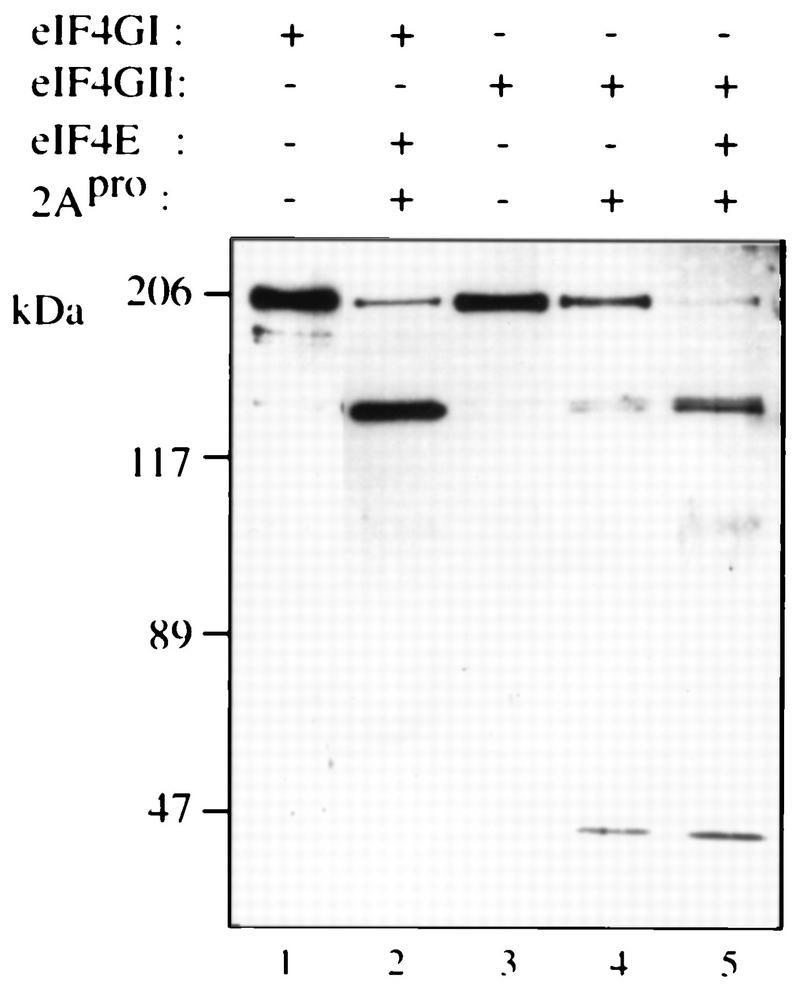
eIF4GII is a substrate for rhinovirus 2Apro. Recombinant eIF4GI (0.5 μg) and eIF4GII (0.5 μg) were incubated in the absence (lanes 1 and 3) or presence (0.1 μg [lanes 2, 4, and 5]) of rhinovirus 2Apro in buffer containing 100 mM KOAc–20 mM Tris-HCl (pH 7.6)–2.5 mM MgOAc–10% glycerol for 30 min at 30°C in a final volume of 10 μl. In lanes 2 and 5, samples were supplemented with 0.1 μg of eIF4E. Laemmli buffer was added to stop the reaction. Samples were resolved by SDS-PAGE (8% polyacrylamide), and proteins were transferred onto a nitrocellulose membrane which was blotted with anti-Xpress antibody (Invitrogen), a monoclonal antibody which recognizes an epitope located in the baculovirus vector. Protein size markers (Bio-Rad) are indicated (in kilodaltons) to the left.
eIF4GII interacts with eIF4E.
The site on human eIF4GI interacting with eIF4E was previously shown to encompass aa 414 to 426 (23). This region contains several evolutionarily conserved hydrophobic amino acids (Fig. 5A). Point mutations of Y416 and two L residues (L421 and L422) prevent the interaction of eIF4GI with eIF4E (23). Because this 13-aa region is highly conserved in eIF4GII (Fig. 1A [aa 622 to 634] and 5A), it was predicted that eIF4GII would also interact with eIF4E. To examine this, we used far-Western analysis (overlay assay) with two GST-eIF4GII fragments, one (aa 445 to 718) containing and another (aa 445 to 604) not containing the putative eIF4E binding domain (Fig. 5B). Crude extracts of E. coli expressing the two different fragments of eIF4GII were resolved on SDS-polyacrylamide gels, transferred to nitrocellulose membranes, and probed with a 32P-labeled FLAG-HMK-eIF4E protein. The two GST-eIF4GII fragments were expressed at similar levels, as determined by Western blotting with a polyclonal antibody directed against GST (Fig. 5C, left panel). FLAG-HMK-eIF4E was capable of interacting with the GST-eIF4GII 445–718 fragment containing the conserved eIF4E-binding motif (right panel), but failed to interact with either the GST-eIF4GII 445–604 fragment, which lacks the eIF4E binding domain, or with GST alone (right panel). These data indicate that the eIF4GII region between aa 605 and 718 (which includes the conserved 13-aa motif) is necessary for eIF4E binding.
The interaction between eIF4E and eIF4GII was further substantiated by coimmunoprecipitation of influenza virus HA epitope-tagged proteins expressed in HeLa cells by the vaccinia virus expression system (11). HA-tagged eIF4G proteins were precipitated with a monoclonal anti-HA antibody, and the immunoprecipitates were probed by Western blotting with an anti-eIF4E antibody. As negative controls, infections with vector alone or vector expressing luciferase were used, and for a positive control, HA-eIF4GI was used. Anti-HA antibody immunoprecipitated the HA-luciferase protein and the HA-eIF4G proteins (Fig. 5D, lower panel [some degradation products of the HA-tagged eIF4G proteins are evident and are comigrating with an ∼75-kDa band present in all lanes]). eIF4E did not coprecipitate with HA-luciferase (lane 1, upper panel), nor was it detected when vector alone was used for infection (lane 4). However, eIF4E coprecipitated with both eIF4GI and eIF4GII (lanes 2 and 3).
eIF4GII interacts with eIF4A and eIF3.
Alignment of the eIF4GII and eIF4GI amino acid sequences reveals a very high degree of identity in some regions of the central and carboxy portions (Fig. 1A). The C-terminal two-thirds region of eIF4GI binds to eIF3 and eIF4A (18). Thus, it was considered probable that eIF4GII would also interact with eIF4A and eIF3. To examine this, HA-tagged eIF4GII was expressed in HeLa cells by the vaccinia virus expression system (11), and cell extracts were immunoprecipitated with a monoclonal anti-HA antibody. As controls, HA-eIF4GI, HA-La, and an empty vector were utilized. Immunoprecipitates were assayed by Western blotting for eIF3 (Fig. 6, upper panel), eIF4A (middle panel), and HA-tagged protein (lower panel) expression. Both eIF4A and eIF3 were coprecipitated with eIF4GI and eIF4GII (Fig. 6, lanes 2 and 3; the p115 subunit of eIF3 is shown because it exhibits the strongest reactivity among all subunits towards the anti-eIF3 antibody), while an unrelated RNA-binding protein, the La autoantigen (7), failed to coprecipitate either factor (lane 1; note that HA-La comigrates with a nonspecific band at ∼50 kDa also observed in lane 4, which might represent the immunoglobulin heavy chain). Also, no immunoprecipitate was detected when vector alone was used (lane 4). Thus, eIF4GII, like eIF4GI and p97 (15), specifically interacts with eIF4A and eIF3. The interaction of eIF4A with eIF4GII was further substantiated by in vitro synthesis of eIF4GII in a reticulocyte lysate and the demonstration that it could interact with a FLAG-eIF4A column (data not shown).
FIG. 6.
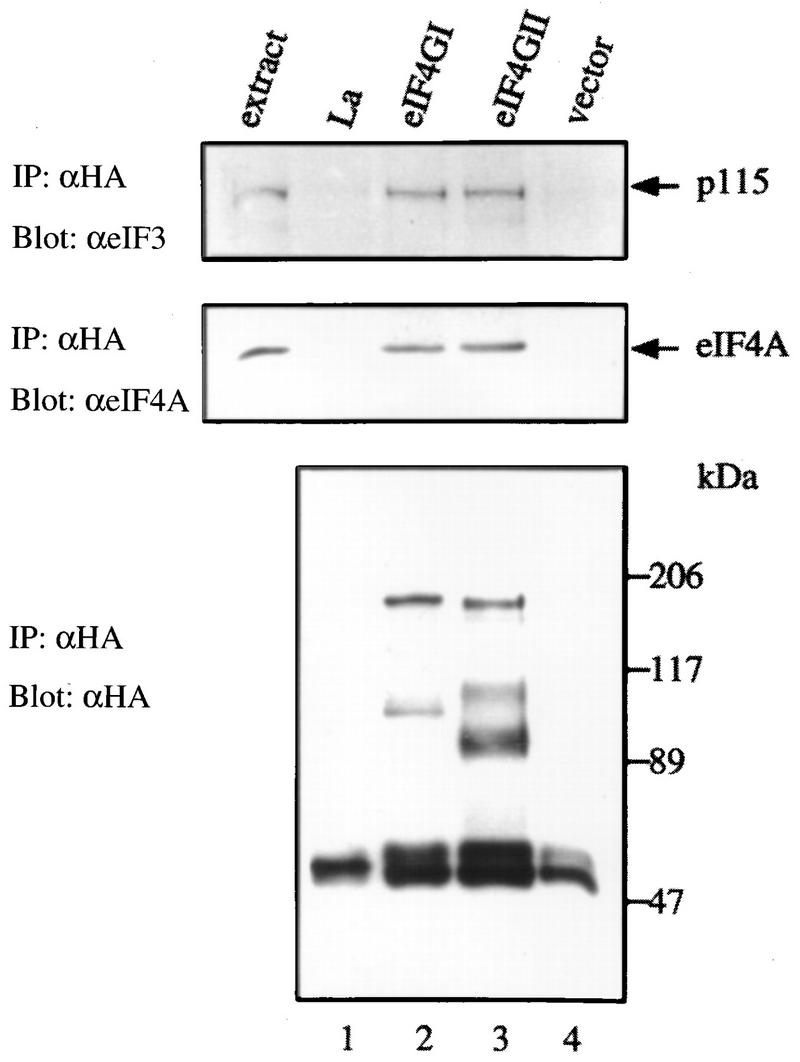
Interaction of eIF4GII with eIF4A and eIF3. HA-tagged protein expression plasmids pcDNA3-HA-La (lane 1), pcDNA3-HA-eIF4GI (lane 2), pcDNA3-HA-eIF4GII (lane 3), and pcDNA3-HA (lane 4) were transfected into HeLa cells after infection with vTF7-3 as described in Materials and Methods. Proteins were immunoprecipitated (IP) with the 12CA5 anti-HA monoclonal antibody, and immunoprecipitates were resolved by SDS-PAGE (10% polyacrylamide). Western blotting was performed with anti-eIF3 (upper panel), anti-eIF4A (middle panel), or anti-HA (lower panel) antibody (α) as described in Materials and Methods. Mock-infected cell extracts (60 μg of protein) are shown to the left of lane 1. Positions of molecular mass standards (in kilodaltons) are indicated to the right.
eIF4GII is retained on a cap affinity resin.
Since eIF4GII interacts with all of the initiation factors previously demonstrated to interact with eIF4GI, and in particular the subunits of eIF4F (eIF4E and eIF4A), it is highly likely that eIF4GII forms an eIF4F-like complex in the cell. To address this possibility, affinity chromatography of an m7GDP resin (9) was performed. A HeLa high-salt ribosomal wash (RSW) was applied to m7GDP-Sepharose, and after being washed with buffer, proteins were eluted first with GDP and then with m7GDP. The load and eluted fractions were probed by Western analysis with antibodies against eIF4GI, eIF4GII, and p97. p97 is not expected to bind to the column because it does not interact with eIF4E (15, 21, 44). As expected, eIF4GI bound to the m7GDP matrix and was eluted specifically with m7GDP (Fig. 7, compare lane 3 to lane 2). eIF4E and eIF4A also bound to the column and eluted with m7GDP (compare lane 3 to lane 2). (These proteins are also present in the GDP eluate in smaller amounts; the combined signals in the eluate lanes exceed the signal in the load lanes, because a smaller fraction of the load was applied to the gel.) The elution profile of eIF4GII (lanes 4 to 6) was very similar to that of eIF4GI, whereas p97 did not bind to the m7GDP matrix. These results demonstrate that eIF4GII forms a complex with eIF4E that can interact with the cap structure. It is very likely that eIF4A is also present in this complex, because as shown by the coimmunoprecipitation assay (Fig. 6), it interacts with eIF4GII.
FIG. 7.

eIF4GII co-purifies with eIF4E by m7GDP affinity chromatography. RSW was prepared from HeLa R19 cells according to the method of Merrick (24). An m7GDP-coupled Sepharose resin (9) was incubated with 1 mg of RSW for 60 min at 4°C, washed three times with 1 ml of buffer A containing 20 mM Tris-HCl (pH 7.5)–100 mM KCl–2 mM DTT–2 mM EDTA–0.5% Triton X-100, and further incubated for 30 min at 4°C with 100 μl of GDP (200 μM). After being washed once with 1 ml of buffer A, the resin was further incubated for 20 min at 4°C with 100 μl of m7GDP (200 μM). Aliquots of the eluted fractions (25 μl) were resolved by SDS-PAGE (12% polyacrylamide) (with 1/10 of the input RSW [10 μg] loaded in lanes 1, 4, and 7). Proteins were transferred to a nitrocellulose membrane, which was cut and probed with anti-eIF4GI, anti-eIF4GII, anti-p97, anti-eIF4A, or anti-eIF4E antibodies (α) as indicated to the left of the blots.
eIF4GII is a functional homolog of eIF4GI.
The ability of rhinovirus 2Apro to cleave eIF4GII (Fig. 4) was used as a basis for an assay to examine the activity of recombinant eIF4GII. Addition of recombinant picornavirus 2A or L proteases to a Krebs cell extract or to a reticulocyte lysate results in the abrogation of cap-dependent translation due to the cleavage of eIF4G, but does not inhibit cap-independent translation (13, 28). Cap-dependent translation in Krebs cell extracts can be restored by the addition of eIF4F or recombinant eIF4GI in combination with eIF4E (13). Since recombinant eIF4GII is cleaved by 2Apro (Fig. 4), endogenous eIF4GII is also expected to be cleaved in a 2Apro-treated extract. To determine whether recombinant eIF4GII could restore translation in a 2Apro-treated rabbit retyculocyte lysate, the lysate was programmed with a bicistronic mRNA in which translation of the first cistron, encoding the CAT protein, is cap dependent. Translation of the second cistron, which is preceded by the IRES of EMCV (and produces the luciferase enzyme), is cap independent. Treatment of the lysate with 2Apro drastically reduced (7-fold) CAT translation, but slightly stimulated (1.7-fold), cap-independent (luciferase) translation (Fig. 8, compare lane 2 to lane 1). Addition of recombinant eIF4E alone increased CAT translation to a small extent (∼1.5-fold, compare lane 3 to lane 2), while addition of eIF4GI alone stimulated CAT translation 2.5-fold (compare lane 4 to lane 2). Addition of eIF4E and eIF4GI in combination enhanced translation (fivefold) to 73% of that of the control extract (compare lane 5 to lane 1), in agreement with earlier reports (e.g., reference 13). eIF4GII behaved similarly to eIF4GI, because its addition alone stimulated translation moderately (lane 6), and when combined with eIF4E, translation was enhanced (4.5-fold) to 62% of the control level (lane 7). eIF4F completely restored translation (lane 8) and only slightly stimulated (1.8-fold) cap-independent translation. It is noteworthy that neither of the eIF4G forms restored translation as efficiently as eIF4F (in two different experiments). Possible explanations are addressed in the Discussion. Consistent with earlier reports (13), the effect of the addition of the eIF4Gs on luciferase translation was modest. Similar results were obtained when a lower concentration of mRNA was employed (data not shown). Taken together, these results demonstrate that after 2Apro treatment, eIF4GII in conjunction with eIF4E is capable of restoring cap-dependent translation. Thus, eIF4GII is a functional homolog of eIF4GI.
FIG. 8.
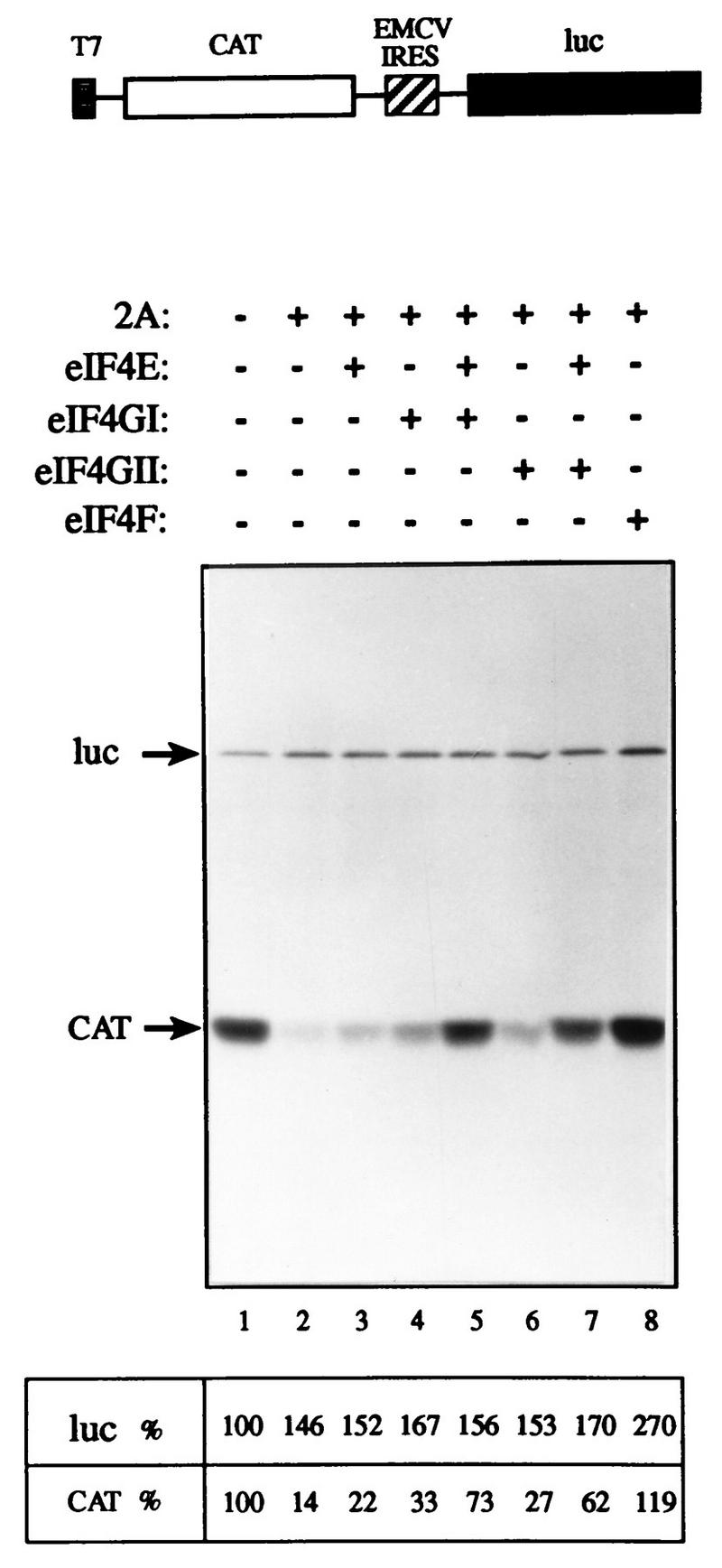
eIF4GII restores cap-dependent translation to an eIF4G-deficient extract. Rabbit reticulocyte lysate (90 μl) was mock treated (lane 1) or treated with 3.6 μg of rhinovirus 2Apro (13) (lanes 2 to 8) for 5 min at 30°C, followed by a 10-min incubation on ice in the presence of 0.8 mM elastatinal (Sigma). Aliquots (12.5 μl) were programmed for translation with 0.1 μg of capped bicistronic pGEMCAT/EMC/LUC mRNA (shown schematically at the top of the figure) in the presence of [35S]methionine and supplemented with the indicated initiation factors as follows: lanes 1 and 2, buffer alone; lane 3, 0.05 μg of eIF4E; lane 4, 0.45 μg of eIF4GI; lane 5, 0.05 μg of eIF4E and 0.45 μg of eIF4GI; lane 6, 0.5 μg of eIF4GII; lane 7, 0.05 μg of eIF4E and 0.5 μg of eIF4GII; lane 8, 0.5 μg of rabbit reticulocyte eIF4F. Translation and processing for electrophoresis were conducted as described in Materials and Methods. Following autoradiography, the bands corresponding to luciferase (luc) and CAT were quantified densitometrically. The efficiency of translation of the luciferase and CAT products is given as a percentage of that of the control (lane 1). The positions of the luciferase and CAT proteins are indicated to the left by arrows.
DISCUSSION
We have isolated and characterized a novel homolog of eIF4G referred to as eIF4GII. eIF4GII is the second homolog of eIF4GI to be reported, the first being p97/NAT1/DAP-5. p97 differs from both eIF4GI and eIF4GII in that it does not interact with eIF4E, which is consistent with the lack of an eIF4E binding site (15, 21, 44). As expected from the sequence homology, eIF4GII interacts with eIF4E, eIF4A, and eIF3. We have demonstrated that eIF4GII is a bona fide functional homolog of eIF4G by showing that eIF4GII in combination with eIF4E, but not alone, is capable of restoring cap-dependent translation in reticulocyte lysate treated with rhinovirus 2Apro. In addition, eIF4GII is present in an eIF4F complex from HeLa cells, because it could be purified by m7GDP affinity chromatography. We were not able to determine whether rabbit eIF4F preparations contain both eIF4GI and eIF4GII, because anti-eIF4GII antibody did not react with the rabbit eIF4F high-molecular-weight polypeptides (12a).
Why have two different eIF4G proteins? One interesting possibility is that the two eIF4G proteins could be required for the translation of different classes of mRNAs. Although we found that eIF4GII can stimulate cap-dependent translation of CAT almost as efficiently as eIF4GI in a translation extract devoid of intact eIF4G, it is possible that eIF4GI and eIF4GII activities exhibit preferential mRNA binding. Alternatively, it is possible that both eIF4G forms are required for maximal translation of a given mRNA. Consistent with this, neither of the eIF4G forms could restore the translation of CAT mRNA in a 2Apro-treated extract as efficiently as eIF4F (Fig. 8). (We could not further address this issue because of our inability to achieve higher concentrations of the proteins for the limited volume of the in vitro translation system.) However, it is also possible that neither of the eIF4G isoforms was as active as endogenous eIF4G, because the recombinant eIF4GII and eIF4GI lack N-terminal sequences. It is of interest that there are also two forms of eIF4A, and both of these forms were shown to be associated with eIF4F, but in different amounts (8). Since eIF4F might contain a mixture of eIF4GI and eIF4GII, it would be interesting to determine if each of the eIF4G polypeptides recognizes a different form of eIF4A.
Different functions for the two forms of eIF4G in yeast and plants have not been described. Recently, it was reported that the two eIF4G forms of yeast (TIF4631 and TIF4632 [12]) could synergize with the poly(A) binding protein (Pabp) in stimulating cap-dependent translation (42, 43). Since eIF4G in yeast interacts directly with Pabp, it was of interest to determine if the two human eIF4G species could also interact with mammalian Pabp. We could not demonstrate binding of either of the human eIF4G species to Pabp (8a). Perhaps there are other forms of eIF4G-like proteins that mediate the action of mammalian Pabp in translation initiation, or perhaps an extended N-terminal form of eIF4GI that is not yet available can bind to mammalian Pabp.
The finding of a new homolog of eIF4G may have important implications for the understanding of the shutoff of host protein synthesis following infection with several picornaviruses. The cleavage of eIF4G precedes the shutoff of host protein synthesis and is thought to be the major cause of the inhibition of translation (10). However, under certain conditions, for example, when replication of poliovirus is inhibited with drugs, cleavage of eIF4GI occurs without complete shutoff of host protein synthesis (5). These results led Bonneau and Sonenberg to conclude that cleavage of eIF4G (p220) is not sufficient for complete inhibition of host cell protein synthesis and that an additional event is required (5). Pérez and Carrasco concluded, based on similar results, that eIF4G cleavage is not responsible for the shutoff of host protein synthesis (16, 32). However, the presence of a functional homolog of eIF4G raises the intriguing possibility that eIF4GII is more resistant to cleavage by 2Apro than eIF4GI. Although we showed here that eIF4GII is degraded in infected cells, preliminary evidence suggests that it is less sensitive to cleavage, in that infection with poliovirus in the presence of drugs results in cleavage of eIF4GI, but not eIF4GII (12a).
eIF4G is thought to play a major role as an adapter molecule in the assembly of the 43S ribosomal preinitiation complex (14, 18, 37, 41). The discovery of a functional homolog of eIF4G in this study, as well as a truncated eIF4G homolog, p97, earlier (15), establishes the existence of an eIF4G family and raises the possibility that other eIF4G-like molecules could exist. It is possible that the various eIF4G adapters play important and specific roles in regulating translation in response to different stimuli or during different stages of development and differentiation.
ACKNOWLEDGMENTS
We thank W. Merrick, T. Skern, L. Carrasco, H. Trachsel, J. Hershey, M. Tremblay, and A.-C. Gingras for the rabbit eIF4F, HRV2 2Apro, anti-eIF4GI, anti-eIF4A, anti-eIF3, and anti-HA antibodies. We are indebted to Colin Lister for excellent technical assistance; J. Gerlach, M. Miron, and F. Poulin for advice on sequence alignments; and M. Park, K. H. Scheit, H. von der Kammer, and G. Rouleau for cDNA libraries. We thank J. Pelletier, N. Méthot, and members of our laboratory for fruitful discussions and for critical reading of the manuscript.
A.G. was supported by Istituto Pasteur Fondazione Cenci-Bolognetti. This work was supported by a grant from the Medical Research Council of Canada to N.S. N.S. is a Distinguished Scientist of the Medical Research Council of Canada and a Howard Hughes Institute International Scholar.
REFERENCES
- 1.Adams M D, Soares M B, Kerlavage A R, Fields C F, Venter J C. Rapid cDNA sequencing (expressed sequence tags) from a directionally cloned human infant brain cDNA library. Nat Genet. 1993;4:373–380. doi: 10.1038/ng0893-373. [DOI] [PubMed] [Google Scholar]
- 2.Aldabe R, Feduchi E, Novoa I, Carrasco L. Efficient cleavage of p220 by poliovirus 2A protease expression in mammalian cells: effects on vaccinia virus. Biochem Biophys Res Commun. 1995;215:928–936. doi: 10.1006/bbrc.1995.2553. [DOI] [PubMed] [Google Scholar]
- 3.Allen M L, Metz A M, Timmer R T, Rhoads R E, Browning K S. Isolation and sequence of the cDNAs encoding the subunits of the isozyme form of wheat protein synthesis initiation factor 4F. J Biol Chem. 1992;267:23232–23236. [PubMed] [Google Scholar]
- 4.Blanar M, Rutter W. Interaction cloning: identification of a helix-loop-helix zipper protein that interacts with c-fos. Science. 1992;256:1014–1018. doi: 10.1126/science.1589769. [DOI] [PubMed] [Google Scholar]
- 5.Bonneau A-M, Sonenberg N. Proteolysis of the p220 component of the cap-binding protein complex is not sufficient for complete inhibition of host cell protein synthesis after poliovirus infection. J Virol. 1987;61:986–991. doi: 10.1128/jvi.61.4.986-991.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Browning K S, Webster C, Roberts J K, Ravel J M. Identification of an isozyme form of protein synthesis initiation factor 4F in plants. J Biol Chem. 1992;267:10096–10100. [PubMed] [Google Scholar]
- 7.Chambers J C, Kenan D, Martin B J, Keene J D. Genomic structure and amino acid sequence domains of the human La autoantigen. J Biol Chem. 1988;263:18043–18051. [PubMed] [Google Scholar]
- 8.Conroy S C, Dever T E, Owens C L, Merrick W C. Characterization of the 46,000-dalton subunit of eIF-4F. Arch Biochem Biophys. 1990;282:363–371. doi: 10.1016/0003-9861(90)90130-q. [DOI] [PubMed] [Google Scholar]
- 8a.Craig, A., and N. Sonenberg. Unpublished observations.
- 9.Edery I, Altmann M, Sonenberg N. High-level synthesis in Escherichia coli of functional cap-binding eukaryotic initiation factor eIF-4E and affinity purification using a simplified cap-analog resin. Gene. 1988;74:517–525. doi: 10.1016/0378-1119(88)90184-9. [DOI] [PubMed] [Google Scholar]
- 10.Etchison D, Milburn S C, Edery I, Sonenberg N, Hershey J W. Inhibition of HeLa cell protein synthesis following poliovirus infection correlates with the proteolysis of a 220,000-dalton polypeptide associated with eucaryotic initiation factor 3 and a cap binding protein complex. J Biol Chem. 1982;257:14806–14810. [PubMed] [Google Scholar]
- 11.Fuerst T R, Niles E G, Studier F W, Moss B. Eukaryotic transient expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc Natl Acad Sci USA. 1986;83:8122–8126. doi: 10.1073/pnas.83.21.8122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goyer C, Altmann M, Lee H S, Blanc A, Deshmukh M, Woolford J, Jr, Trachsel H, Sonenberg N. TIF4631 and TIF4632: two yeast genes encoding the high-molecular-weight subunits of the cap-binding protein complex (eukaryotic initiation factor 4F) contain an RNA recognition motif-like sequence and carry out an essential function. Mol Cell Biol. 1993;13:4860–4874. doi: 10.1128/mcb.13.8.4860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12a.Gradi, A. Unpublished observations.
- 13.Haghighat A, Svitkin Y, Novoa I, Kuechler E, Skern T, Sonenberg N. The eIF4G-eIF4E complex is the target for direct cleavage by the rhinovirus 2A proteinase. J Virol. 1996;70:8444–8450. doi: 10.1128/jvi.70.12.8444-8450.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hentze M W. eIF4G: a multipurpose ribosome adapter? Science. 1997;275:500–501. doi: 10.1126/science.275.5299.500. [DOI] [PubMed] [Google Scholar]
- 15.Imataka H, Olsen H S, Sonenberg N. A new translational regulator with homology to eukaryotic translation initiation factor 4G. EMBO J. 1997;16:817–825. doi: 10.1093/emboj/16.4.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Irurzun A, Sánchez-Palomino S, Novoa I, Carrasco L. Monensin and nigericin prevent the inhibition of host translation by poliovirus, without affecting p220 cleavage. J Virol. 1995;69:7453–7460. doi: 10.1128/jvi.69.12.7453-7460.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jaramillo M, Dever T E, Merrick W C, Sonenberg N. RNA unwinding in translation: assembly of helicase complex intermediates comprising eukaryotic initiation factors eIF-4F and eIF-4B. Mol Cell Biol. 1991;11:5992–5997. doi: 10.1128/mcb.11.12.5992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lamphear B J, Kirchweger R, Skern T, Rhoads R E. Mapping of functional domains in eukaryotic protein synthesis initiation factor 4G (eIF4G) with picornaviral proteases. Implications for cap-dependent and cap-independent translational initiation. J Biol Chem. 1995;270:21975–21983. doi: 10.1074/jbc.270.37.21975. [DOI] [PubMed] [Google Scholar]
- 19.Lamphear B J, Yan R, Yang F, Waters D, Liebig H D, Klump H, Kuechler E, Skern T, Rhoads R E. Mapping the cleavage site in protein synthesis initiation factor eIF-4 γ of the 2A proteases from human coxsackievirus and rhinovirus. J Biol Chem. 1993;268:19200–19203. [PubMed] [Google Scholar]
- 20.Lazaris-Karatzas A, Montine K S, Sonenberg N. Malignant transformation by a eukaryotic initiation factor subunit that binds to mRNA 5′ cap. Nature. 1990;345:544–547. doi: 10.1038/345544a0. [DOI] [PubMed] [Google Scholar]
- 21.Levy-Strumpf N, Deiss L P, Berissi H, Kimchi A. DAP-5, a novel homolog of eukaryotic translation initiation factor 4G isolated as a putative modulator of gamma interferon-induced programmed cell death. Mol Cell Biol. 1997;17:1615–1625. doi: 10.1128/mcb.17.3.1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liebig H D, Ziegler E, Yan R, Hartmuth K, Klump H, Kowalski H, Blaas D, Sommergruber W, Frasel L, Lamphear B, Rhoads R E, Kuechler E, Skern T. Purification of two picornaviral 2A proteinases: interaction with eIF-4 γ and influence on in vitro translation. Biochemistry. 1993;32:7581–7588. doi: 10.1021/bi00080a033. [DOI] [PubMed] [Google Scholar]
- 23.Mader S, Lee H, Pause A, Sonenberg N. The translation initiation factor eIF-4E binds to a common motif shared by the translation factor eIF-4γ and the translational repressors 4E-binding proteins. Mol Cell Biol. 1995;15:4990–4997. doi: 10.1128/mcb.15.9.4990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Merrick W C. Assays for eukaryotic protein synthesis. Methods Enzymol. 1979;60:108–123. doi: 10.1016/s0076-6879(79)60011-3. [DOI] [PubMed] [Google Scholar]
- 25.Méthot N, Pause A, Hershey J W B, Sonenberg N. The translation initiation factor eIF-4B contains an RNA-binding region that is distinct and independent from its ribonucleoprotein consensus sequence. Mol Cell Biol. 1994;14:2307–2316. doi: 10.1128/mcb.14.4.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ohlmann T, Pain V M, Wood W, Rau M, Morley S J. The proteolytic cleavage of eukaryotic initiation factor (eIF) 4G is prevented by eIF4E-binding protein (PHAS-I; 4E-BP1) in the reticulocyte lysate. EMBO J. 1997;16:844–855. doi: 10.1093/emboj/16.4.844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ohlmann T, Rau M, Morley S J, Pain V M. Proteolytic cleavage of initiation factor eIF-4G in the reticulocyte lysate inhibits translation of capped mRNAs but enhances that of uncapped mRNAs. Nucleic Acids Res. 1995;23:334–340. doi: 10.1093/nar/23.3.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ohlmann T, Rau M, Pain V M, Morley S J. The C-terminal domain of eukaryotic protein synthesis initiation factor (eIF) 4G is sufficient to support cap-independent translation in the absence of eIF4E. EMBO J. 1996;15:1371–1382. [PMC free article] [PubMed] [Google Scholar]
- 29.Pain V M. Initiation of protein synthesis in eukaryotic cells. Eur J Biochem. 1996;236:747–771. doi: 10.1111/j.1432-1033.1996.00747.x. [DOI] [PubMed] [Google Scholar]
- 30.Pause A, Belsham G J, Gingras A C, Donzé O, Lin T A, Lawrence J C, Sonenberg N. Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5′-cap function. Nature. 1994;371:762–767. doi: 10.1038/371762a0. [DOI] [PubMed] [Google Scholar]
- 31.Pause A, Méthot N, Svitkin Y V, Merrick W C, Sonenberg N. Dominant negative mutants of mammalian translation initiation factor eIF-4A define a critical role for eIF-4F in cap-dependent and cap-independent initiation of translation. EMBO J. 1994;13:1205–1215. doi: 10.1002/j.1460-2075.1994.tb06370.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pérez L, Carrasco L. Lack of direct correlation between p220 cleavage and the shut-off of host translation after poliovirus infection. Virology. 1992;189:178–186. doi: 10.1016/0042-6822(92)90693-j. [DOI] [PubMed] [Google Scholar]
- 33.Pestova T V, Hellen C U T, Shatsky I N. Canonical eukaryotic initiation factors determine initiation of translation by internal ribosomal entry. Mol Cell Biol. 1996;16:6859–6869. doi: 10.1128/mcb.16.12.6859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pestova T V, Shatsky I N, Hellen C U T. Functional dissection of eukaryotic initiation factor 4F: the 4A subunit and the central domain of the 4G subunit are sufficient to mediate internal entry of 43S preinitiation complexes. Mol Cell Biol. 1996;16:6870–6878. doi: 10.1128/mcb.16.12.6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34a.Poncet, D., et al. Personal communication.
- 35.Ray B K, Lawson T G, Kramer J C, Cladaras M H, Grifo J A, Abramson R D, Merrick W C, Thach R E. ATP-dependent unwinding of messenger RNA structure by eukaryotic initiation factors. J Biol Chem. 1985;260:7651–7658. [PubMed] [Google Scholar]
- 36.Rozen F, Edery I, Meerovitch K, Dever T E, Merrick W C, Sonenberg N. Bidirectional RNA helicase activity of eucaryotic translation initiation factors 4A and 4F. Mol Cell Biol. 1990;10:1134–1144. doi: 10.1128/mcb.10.3.1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sachs A B, Sarnow P, Hentze M W. Starting at the beginning, middle, and end: translation initiation in eukaryotes. Cell. 1997;89:831–838. doi: 10.1016/s0092-8674(00)80268-8. [DOI] [PubMed] [Google Scholar]
- 38.Smith R F, Smith T F. Pattern-induced multi-sequence alignment (PIMA) algorithm employing secondary structure-dependent gap penalties for comparative protein modelling. Protein Eng. 1992;5:35–41. doi: 10.1093/protein/5.1.35. [DOI] [PubMed] [Google Scholar]
- 39.Sommergruber W, Ahorn H, Klump H, Seipelt J, Zoephel A, Fessl F, Krystek E, Blaas D, Kuechler E, Liebig H D, Skern T. 2A proteinases of coxsackie- and rhinovirus cleave peptides derived from eIF-4 γ via a common recognition motif. Virology. 1994;198:741–745. doi: 10.1006/viro.1994.1089. [DOI] [PubMed] [Google Scholar]
- 40.Sommergruber W, Ahorn H, Zophel A, Maurer-Fogy I, Fessl F, Schnorrenberg G, Liebig H D, Blaas D, Kuechler E, Skern T. Cleavage specificity on synthetic peptide substrates of human rhinovirus 2 proteinase 2A. J Biol Chem. 1992;267:22639–22644. [PubMed] [Google Scholar]
- 41.Sonenberg N. mRNA 5′ cap-binding protein eIF4E and control of cell growth. In: Hershey J W B, Mathews M B, Sonenberg N, editors. Translational control. Plainview, N.Y: Cold Spring Harbor Laboratory Press; 1996. pp. 245–269. [Google Scholar]
- 42.Tarun S Z, Sachs A B. Association of the yeast poly(A) tail binding protein with translation initiation factor eIF4G. EMBO J. 1996;15:7168–7177. [PMC free article] [PubMed] [Google Scholar]
- 43.Tarun S Z, Wells S, Deardorff J, Sachs A B. Translation initiation factor eIF4G mediates in vitro poly(A) tail-dependent translation. Proc Natl Acad Sci USA. 1997;94:9046–9051. doi: 10.1073/pnas.94.17.9046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yamanaka S, Poksay K S, Arnold K S, Innerarity T L. A novel translational repressor mRNA is edited extensively in livers containing tumors caused by the transgene expression of the apoB mRNA-editing enzyme. Genes Dev. 1997;11:321–333. doi: 10.1101/gad.11.3.321. [DOI] [PubMed] [Google Scholar]
- 45.Yan R, Rychlik W, Etchison D, Rhoads R E. Amino acid sequence of the human protein synthesis initiation factor eIF-4γ. J Biol Chem. 1992;267:23226–23231. [PubMed] [Google Scholar]