Linkage disequilibrium between the beta frequency of the human EEG and a GABAA receptor gene locus (original) (raw)
Abstract
Human brain oscillations represent important features of information processing and are highly heritable. A common feature of beta oscillations (13–28 Hz) is the critical involvement of networks of inhibitory interneurons as pacemakers, gated by γ-aminobutyric acid type A (GABAA) action. Advances in molecular and statistical genetics permit examination of quantitative traits such as the beta frequency of the human electroencephalogram in conjunction with DNA markers. We report a significant linkage and linkage disequilibrium between beta frequency and a set of GABAA receptor genes. Uncovering the genes influencing brain oscillations provides a better understanding of the neural function involved in information processing.
Since the initial discovery of the human electroencephalogram (EEG) by Berger (1), it has been speculated that neural oscillations play a broad role in nervous systems and form the basis for higher cognitive functions and consciousness (2–4). Although it is not possible yet to assign a specific functional role to each frequency, the presence of a beta/gamma oscillation (18–50 Hz) is thought to represent an activated state of the underlying neuronal network. These beta (12–29 Hz) and gamma (30–50 Hz) brain rhythms involve γ-aminobutyric acid type A (GABAA) receptor action (5).
Neuronal networks can display different states of synchrony characterized by their oscillation frequencies measured by EEG (6, 7). Most EEG parameters are stable and to a large extent are determined genetically. Test-retest correlation coefficients for EEG power, after a 12–16-week interval between measurements, are high (≈0.8; refs. 8–10). The earliest extensive genetic studies of the human resting EEG were carried out by Vogel (11, 12). A number of investigators have reported higher concordance rates in the spectral characteristics of EEG from monozygotic twins when compared with dizygotic twins (8, 13–15). In the largest twin study to date (16, 17), the eyes-closed, resting EEG recorded from 14 scalp locations was measured from 213 twin pairs. The average heritability of the spectral power in four frequency bands, delta (1.5–3.5 Hz), theta (4.0–7.5 Hz), alpha (8.0–12.5 Hz), and beta (13–25 Hz), was found to be 76, 89, 89, and 86%, respectively. Although the data on the heritability of EEG frequencies are quite compelling, the genes influencing EEGs have not been identified yet. We report here a linkage and linkage-disequilibrium study concerning the beta frequency band of the human EEG.
Methods
Subjects in this study were recruited and tested in the multisite Collaborative Study on the Genetics of Alcoholism (COGA). COGA sites providing data for this study include State University of New York Downstate Medical Center (New York), University of Connecticut Health Science Center, Indiana University School of Medicine, University of Iowa School of Medicine, University of California School of Medicine (San Diego), and Washington University School of Medicine (St. Louis). Ascertainment and assessment procedures have been described (18, 19). The sample included in this EEG linkage study was drawn from 250 families and consisted of 1,553 individuals ranging from 7 to 70 years of age. The laboratory and data-collection procedures were identical at each of the sites (20). In addition to EEG activity, blood was collected for DNA extraction. Marker genotyping and Mendelian error detection have been described (21). Maximum likelihood estimates of marker allele frequencies were obtained from data on all genotyped individuals in the COGA data set by using the USERM13 program (22). CRIMAP (23) was used to calculate marker order and distances. Data in this report are based on a whole-genome screen with a total of 351 markers.
Subjects were seated comfortably in a dimly lit sound-attenuated temperature-regulated booth (Industrial Acoustics, Bronx, NY). They were instructed to keep their eyes closed and remain relaxed but to not fall asleep. Each subject wore a fitted electrode cap (Electro-Cap International, Eaton, OH) using the 19-channel montage as specified according to the 10–20 international system (FP1, FP2, F7, F3, Fz, F4, F8, T7, C3, Cz, C4, T8, P7, P3, Pz, P4, P8, O1, and O2; Fig. 1). The nose was used as a reference, and a forehead electrode served as the ground electrode. Both vertical and horizontal eye movements were monitored with electrodes that were placed supraorbitally and at the outer canthus of the left eye to perform ocular artifact correction. Electrode impedances were maintained below 5 kΩ. Electrical activity was amplified 10,000 times by Sensorium (Charlotte, VT) EPA-2 electrophysiology amplifiers with a bandpass between 0.02 and 50 Hz and digitized on a Concurrent (Atlanta, GA) 5550 computer. EEG data were collected in the awake, eyes-closed condition at a sampling rate of 256 Hz for 4.25 min.
Figure 1.

Diagram of aerial view of the scalp with the nose up (front) designating the positions of the electrodes in the 10–20 international System. F, frontal; C, central; P, parietal; O, occipital; T, temporal. Odd numbers indicate leads on the left side of the head, even numbers indicate leads on the right side, and Z indicates zero or midline.
The raw data were subjected to wavelet filtering and reconstruction to eliminate very high and low frequencies (24, 25). The s12 wavelet was used to perform a six-level analysis, and the output signal was reconstructed with levels d6–d3, roughly equivalent to applying a bandpass filter with a range of 2–64 Hz to the data. Subsequently, eye movements were removed by using the method developed by Gasser et al. (26, 27). This method subtracts a portion of the observed ocular activity from the observed EEG to obtain the true EEG, in which the proportionality is based on the difference between the cross-spectral values of trials with high ocular activity and those with low ocular activity. Visual inspection of corrected data showed satisfactory artifact removal. The filtered artifact-free data were transformed into 11 bipolar derivations (F7–F3, F8–F4, T7–C3, T8–C4, P7–P3, P8–P4, O1–O2, CZ–C3, CZ–C4, PZ–P3, and PZ–P4) to improve sensitivity to local electrical sources (6, 28, 29). The 19-channel montage used in this study would not be sufficient for current source density analyses. Bipolar derivations using closely adjacent electrodes provide a high-pass spatial filter (i.e., counteract some of the smearing of cortical potentials) and are more effective in capturing a greater amount of cerebral energy output than other referencing strategies. Bipolar derivations were analyzed in 254 overlapping 2-sec epochs by use of a Fourier transform. After windowing effects were minimized by application of a Hamming function (30), the resulting spectral densities, sampled at 0.5-Hz intervals, were aggregated into bands, divided by the bandwidth, and then averaged across epochs.
This study focused on the absolute power in the EEG (3–28 Hz), which was subdivided into theta (3.0–7.0 Hz), alpha 1 (7.5–9.0 Hz), alpha 2 (9.5–12.0 Hz), beta 1 (12.5–16.0 Hz), beta 2 (16.5–20.0 Hz), and beta 3 (20.5–28 Hz) bands. Band-power distributions for the population sample were examined, and values beyond 4 standard deviations from the mean were eliminated. Linear regressions were performed on the spectral values by using gender and age as covariates. After appropriate correction, the quantitative traits (theta, alpha 1 and 2, and beta 1, 2, and 3) were computed as residuals from the predicted value.
Information describing the spatial/spectral signature of an individual's EEG was compressed by employing the trilinear modeling method, a singular value decomposition procedure (31, 32). Trilinear modeling represents the EEG as superpositions of a few pairs of spatial and spectral components. The spatial components are obtained by pooling the individual's data together by electrodes and performing a singular value decomposition. The spectral components are obtained by pooling the individual's data together by frequency bins and performing a singular value decomposition. The resultant components then are rotated by using the orthogonal matrices from a singular value decomposition on the average of the fitting coefficient matrices; hence the average of the fitting coefficients to the individual's EEG is a diagonal matrix. The representation of the data is unique given the spatial and spectral components. We extracted the first spatial and spectral component pair of the five EEG bands (theta, alpha 1 and 2, and beta 1, 2, and 3) and used the fitting coefficients for each subject as the phenotypic data in subsequent analysis, greatly reducing the number of EEG variables examined.
The quantitative EEG phenotypes were used in a variance component linkage analysis using the sequential oligogenic linkage analysis routines (SOLAR; ref. 33). SOLAR uses all relative pairs by constructing likelihood functions for whole pedigrees. The appropriate variance/covariance matrix for a pedigree depends on the predicted proportion of genes shared identical by descent at a hypothesized quantitative trait locus (QTL); this proportion in turn depends on the proportion shared identical by descent at genotyped markers and on the type of relative pair. Maximum likelihood estimates for the variance component parameters are obtained, and a lod score is computed as log 10 of the likelihood ratio comparing two models: a model for which the additive genetic variance σ2a for the QTL is estimated versus a model for which σ2a is constrained to be 0 (no linkage). Variance component analyses were carried out at 1-cM intervals across all chromosomes using SOLAR (34). Although the variance component method generally assumes multivariate normality within pedigrees, the method is quite robust to distributional violations (35–38). Because of slight kurtosis in the distributions of the EEG traits (kurtosis values of 0.9–1.2), analyses were performed by using the multivariate t distribution rather than the multivariate normal distribution option of SOLAR (34).
Results
The strongest evidence for linkage with EEG power was observed on the short arm of chromosome 4 for the beta traits; there were no other statistically significant lod scores for other EEG frequency bands or chromosomes. Therefore, this study focused on the findings from the three beta bands (beta 1, 2, and 3) using the first spatial and spectral component pairs.
The first component for beta 1 accounted for 54% of the spatial variance and 73% of the spectral variance. The first component for beta 2 accounted for 52% of the spatial variance and 78% of the spectral variance. The first component for beta 3 accounted for 46% of the spatial variance and 70% of the spectral variance. The analysis of beta 1 provided a significant linkage on chromosome 4 at GABRB1 between the markers D4S1627 and D4S1645 (lod = 3.39). Beta 2 demonstrated significant linkage with a lod = 5.01 at the same locus. Beta 3 yielded a nonsignificant but suggestive linkage at the same locus (lod = 2.17). The maximum lod score of the EEG linkages for all three beta frequency bands on chromosome 4 were at GABRB1, a microsatellite marker (Fig. 2). The 1-lod support interval for the location of the QTL encompassed ≈16 cM flanking the locus. Multivariate analyses were performed between the beta 1, 2, and 3 phenotypes (39). The overall genetic correlations between these traits were very high; ρg was 0.88 between beta 1 and 2 and 0.91 between beta 2 and 3, indicating substantial overlap in the genes influencing these traits. In linkage analyses at the chromosome 4 peak, the QTL-specific correlations between the traits were estimated at 1 and were not significantly different from 1, providing evidence for pleiotropy and supporting the hypothesis that the same chromosome 4 QTL influences all three beta EEG phenotypes.
Figure 2.

Linkage of beta frequencies: beta 1 (12.5–16 Hz), beta 2 (16.5–20 Hz), and beta 3 (20.5–28 Hz) on chromosome 4. For all three beta bands, the maximum lod scores were at GABRB1, a microsatellite marker situated within a cluster of GABAA receptor genes (beta 2, lod = 5.01; beta 1, lod = 3.39; beta 3, lod = 2.17).
Combined linkage/linkage disequilibrium analysis was used to test for association between the beta 2 EEG phenotype and the GABRB1 microsatellite marker (40). This approach uses identity by state sharing between unrelated individuals to augment the linkage information obtained from identity by descent sharing among relatives. In the present analyses, the identity by state matrix was constructed such that only individuals in the same ethnic group were allowed to share alleles identity by state, effectively constraining the disequilibrium analyses to within-group comparisons. Additionally, using the contrast between the locus-specific variance predicted by identity by descent and that predicted by identity by state provides an estimate of the strength of the linkage disequilibrium between the genotyped marker and the true QTL. Using this method, the lod score in this region of chromosome 4 increased to 6.53, and strong evidence for association between the beta 2 EEG phenotype and the GABRB1 microsatellite marker was observed (P = 0.004). The disequilibrium parameter (ρd = 0.57) indicated linkage disequilibrium between the GABRB1 microsatellite and the functional QTL. ρd is a function of the difference between the actual haplotype frequencies for the marker and the QTL and those predicted by the constituent allele frequencies, standardized by the allele frequencies such that it is a proportion of the maximum possible disequilibrium between the marker and the QTL. For example, for a two allele system
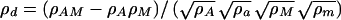 |
1 |
|---|
where ρAM is the frequency of the AM haplotype and ρA, ρa, ρM, and ρm are the allele frequencies at marker M and QTL A. In theory ρd varies between −1 and +1, with the sign depending on how the alleles at the marker and QTL are designated (i.e., which allele is assigned to A and which to a). In the combined linkage/linkage disequilibrium analysis described above, the square of ρd is estimated from the covariance among unrelated individuals. Taking the square root of this estimate produces a ρd figure varying from 0 to 1 with 0 indicating no disequilibrium and 1 indicating the maximum possible disequilibrium.
These results provide strong evidence for an association between the GABRB1 locus and the beta 2 EEG phenotype and suggest that the chromosome 4 QTL is in or near GABRB1. This significant linkage/linkage disequilibrium for the beta frequencies is situated within a cluster of GABAA receptor genes located on chromosome 4. This cluster includes GABRA2, GABRA4, and GABRB1 within 1 cM (41).
Discussion
Fast synaptic inhibition in the mammalian central nervous system is mediated largely by activation of GABAA receptors (42). Most GABAA receptors consist of α, β, and γ subunits. It has been hypothesized that GABAA actions are a fundamental requirement for both gamma (30–80 Hz) and beta (12–30 Hz) oscillations to occur, and blockade of these receptors results in the loss of synchronization (5); beta rhythms can synchronize over long temporal delays between more spatially distant brain loci than gamma rhythms (43). Although the recording of electrical oscillations from a neural population reflects the firing of multiple excitatory pyramidal cells, the mechanism underlying beta and gamma oscillations depends on the firing patterns of a network of inhibitory interneurons (43, 44) gated by their mutually induced GABAA action (45). Two kinds of beta have been described. In the first, both excitatory pyramidal cells and inhibitory interneurons fire at beta frequencies and approximately in phase; the mechanism seems to be a similar but slower version of gamma oscillations in which both pyramidal cells and interneurons participate. In the second kind of beta, pyramidal cells fire at beta frequencies, but interneurons fire at gamma frequencies; the pyramidal cells generate synchronous action potentials that fire at subharmonics of the inhibitory gamma oscillation (“missed beats”; ref. 44).
Classical benzodiazepines such as diazepam bind to GABAA receptors containing the α subunits α1, α2, α3 or α5, whereas the receptors containing the α4 or α6 subunits are insensitive to diazepam. It has been reported recently that the sleep and waking EEG spectral changes observed with diazepam are mediated by GABAA α subunits other than α1, namely by α2, α3, or α5 (42); anxiolytic activity is mediated by α2- but not α3-containing receptors (46). These pharmacological findings suggest that the GABAA α2 subunit may be the most likely candidate for our linkage/linkage disequilibrium findings with the EEG beta spectral band. Benzodiazepines produce a strong increase in EEG beta power that is more marked in frontal regions. Benzodiazepines disrupt the beta (excitatory pyramidal cell)/gamma (inhibitory interneuron) oscillations at the cellular level and produce a “beta buzz.” This beta buzz can be produced experimentally by pressure injection of glutamate or specific metabotropic glutamate agonists into a hippocampal slice (47, 48). In this case, the observed beta rhythm occurs as a slower pyramidal-interneuron network oscillation determined by benzodiazepine GABAergic synaptic potentials in pyramidal cells and interneurons; this occurs without an underlying inhibitory gamma rhythm (there are no missed beats; ref. 49). These drug-induced beta oscillations can be considered as “slow gamma,” because they are caused by the same fundamental mechanisms; they differ from the beta (excitatory pyramidal cell)/gamma (inhibitory interneuron) oscillations, which involve both an underlying inhibitory gamma component as well as recurrent excitatory synaptic activity.
Beta activity is indicative of background excitation involving a frequency potentiation mechanism at the synaptic level of the recurrent loops (45, 50). In a spontaneously active network of interneurons, inhibitory GABA and glycine receptors generate periodic oscillatory burst patterns with remarkable regularity in burst period and duration observed at many brain sites (45, 51). This finding suggests that beta frequencies typically observed in the human EEG reflect a state of central nervous system activation, with GABAA receptor action as pacemakers (52, 53). Our findings of a strong linkage/linkage disequilibrium between the beta frequencies and the region on chromosome 4 containing a cluster of GABAA receptor genes represent the identification of a genetic locus associated with these fundamental human brain oscillations.
Acknowledgments
The superlative technical assistance of Arthur Stimus, Aquanette Sass, Marty Krakowsky, Ed Babington, Sandi Watson, Vladimir Kotlyarevsky, Elizabeth Iskander, Marc Ostrega, and Sergio Valentini on this project is gratefully acknowledged. The Collaborative Study on the Genetics of Alcoholism (H.B., State University of New York HSCB Principal Investigator, and T.R., Washington University, Co-Principal Investigator) includes nine different centers at which data collection, analysis, and/or storage takes place. The nine sites, Principal Investigators, and Co-Investigators are: Indiana University, T.-K.L., J. Nurnberger, Jr., P.M.C., and H.J.E.; University of Iowa, R.R.C. and S.K.; University of California at San Diego, M.A.S.; University of Connecticut, V.H.; State University of New York, Health Sciences Center at Brooklyn, B.P. and H.B.; Washington University at St. Louis, T.R., C. R. Cloninger, J.P.R., and A.G.; Howard University, R. Taylor; Rutgers University, J.A.T.; and Southwest Foundation, L.A. This national collaborative study is supported by the National Institutes of Health Grant U10AA08403 from the National Institute on Alcohol Abuse and Alcoholism. Sequential Oligogenic Linkage Analysis Routines development is supported by National Institutes of Health Grant MH59490.
Abbreviations
EEG
electroencephalogram
GABA
γ-aminobutyric acid
GABAA
GABA type A
QTL
quantitative trait locus
lod
logarithm of odds
References
- 1.Berger H. Arch Psychiatr Nervenkr. 1929;87:527–570. [Google Scholar]
- 2.Crick F, Koch C. Semin Neurosci. 1990;2:263–275. [Google Scholar]
- 3.Llinas R, Pare D. Neuroscience. 1991;44:521–535. doi: 10.1016/0306-4522(91)90075-y. [DOI] [PubMed] [Google Scholar]
- 4.Llinas R, Ribary U. Proc Natl Acad Sci USA. 1993;90:2078–2081. doi: 10.1073/pnas.90.5.2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haenschel C, Baldeweg T, Croft R J, Whittington M, Gruzelier J. Proc Natl Acad Sci. 2000;97:7645–7650. doi: 10.1073/pnas.120162397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nunez P L. In: Neocortical Dynamics and Human EEG Rhythms. Nunez P L, editor. New York: Oxford Univ. Press; 1995. pp. 3–67. [Google Scholar]
- 7.Neidermeyer E. In: Electroencephalography: Basic Principles, Clinical Applications, and Related Fields. 4th Ed. Neidermeyer E, Lopes dS, editors. Baltimore: Williams & Wilkins; 1999. pp. 149–173. [Google Scholar]
- 8.Stassen H, Bomben G, Propping P. Electroencephalogr Clin Neurophysiol. 1987;66:489–501. doi: 10.1016/0013-4694(87)90095-2. [DOI] [PubMed] [Google Scholar]
- 9.Pollock V E, Schneider L, Lyness S. Electroencephalogr Clin Neurophysiol. 1991;79:20–26. doi: 10.1016/0013-4694(91)90152-t. [DOI] [PubMed] [Google Scholar]
- 10.Salinsky M, Oken B, Morehead L. Electroencephalogr Clin Neurophysiol. 1991;79:382–392. doi: 10.1016/0013-4694(91)90203-g. [DOI] [PubMed] [Google Scholar]
- 11.Vogel F. Dtsch Z Nervenheilkd. 1962;184:105–111. [Google Scholar]
- 12.Vogel F. Dtsch Z Nervenheilkd. 1962;184:137–173. [PubMed] [Google Scholar]
- 13.Dumermuth G. Helv Paediatr Acta. 1969;24:45–54. [PubMed] [Google Scholar]
- 14.Lykken D T, Tellegen A, Thorkelson K. Biol Psychiatry. 1974;1:245–249. doi: 10.1016/0301-0511(74)90001-5. [DOI] [PubMed] [Google Scholar]
- 15.Whitton J L, Elgie S M, Kugel H, Moldofsky H. Electroencephalogr Clin Neurophysiol. 1985;60:293–298. doi: 10.1016/0013-4694(85)90002-1. [DOI] [PubMed] [Google Scholar]
- 16.van Beijsterveldt C E M, Boomsma D I. Hum Genet. 1994;94:319–330. doi: 10.1007/BF00201587. [DOI] [PubMed] [Google Scholar]
- 17.van Beijsterveldt C E M, Molenaar P C M, de Geus E J C, Boomsma D I. Am J Hum Genet. 1996;58:562–573. [PMC free article] [PubMed] [Google Scholar]
- 18.Begleiter H, Reich T, Hesselbrock V, Porjesz B, Li T-K, Schuckit M A, Edenberg H J, Rice J P. Alcohol Health Res World. 1995;19:228–236. [Google Scholar]
- 19.Bucholz K K, Cadoret R, Cloninger C R, Dinwiddie S H, Hesselbrock V M, Nurnberger J I, Jr, Reich T, Schmidt I, Schuckit M A. J Stud Alcohol. 1994;55:149–158. doi: 10.15288/jsa.1994.55.149. [DOI] [PubMed] [Google Scholar]
- 20.Begleiter H, Porjesz B, Reich T, Edenberg H J, Goate A, Blangero J, Almasy L, Foroud T, Van Eerdewegh P, Polich J, et al. Electroencephalogr Clin Neurophysiol. 1998;108:244–250. doi: 10.1016/s0168-5597(98)00002-1. [DOI] [PubMed] [Google Scholar]
- 21.Reich T, Edenberg H J, Goate A, Williams J T, Rice J P, Van Eerdewegh P, Foroud T, Hesselbrock V, Schuckit M A, Bucholz K, et al. Am J Med Genet. 1998;81:207–215. [PubMed] [Google Scholar]
- 22.Boehnke M. Am J Hum Genet. 1991;48:22–25. [PMC free article] [PubMed] [Google Scholar]
- 23.Green P, Lange K, Cox D R. Documentation for CRIMAP. St. Louis: Department of Genetics, School of Medicine, Washington University; 1990. , Version 2.4. [Google Scholar]
- 24.Bruce A, Gao H. S+ Wavelets User's Manual. Seattle: Mathsoft; 1994. [Google Scholar]
- 25.Strang G, Nguyen T. Wavelets and Filter Banks. Wellesley, MA: Wellesley–Cambridge; 1996. [Google Scholar]
- 26.Gasser T, Sroka L, Mocks J. Psychophysiology. 1986;23:704–712. doi: 10.1111/j.1469-8986.1986.tb00697.x. [DOI] [PubMed] [Google Scholar]
- 27.Gasser T, Sroka L, Mocks J. Electroencephalogr Clin Neurophysiol. 1987;61:181–193. doi: 10.1016/0013-4694(85)91058-2. [DOI] [PubMed] [Google Scholar]
- 28.Nunez P L, Srinivasan R, Westdorp A F, Wijesinghe R S, Tucker D M, Silberstein R B, Cadusch P J. Electroencephalogr Clin Neurophysiol. 1997;103:499–515. doi: 10.1016/s0013-4694(97)00066-7. [DOI] [PubMed] [Google Scholar]
- 29.Cook I A, O'Hara R, Uijtdehaage S H, Mandelkern M, Leuchter A F. Electroencephalogr Clin Neurophysiol. 1998;107:408–414. doi: 10.1016/s0013-4694(98)00092-3. [DOI] [PubMed] [Google Scholar]
- 30.Hamming R. Digital Filters. Englewood Cliffs, NJ: Prentice–Hall; 1983. [Google Scholar]
- 31.Mocks J. IEEE Trans Biomed Eng. 1988;35:482–484. doi: 10.1109/10.2119. [DOI] [PubMed] [Google Scholar]
- 32.Wang K, Begleiter H, Porjesz B. Brain Topogr. 2000;12:263–271. doi: 10.1023/a:1023455404934. [DOI] [PubMed] [Google Scholar]
- 33.Almasy L, Blangero J. Am J Hum Genet. 1998;62:1198–1211. doi: 10.1086/301844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blangero J, Williams J T, Almasy L. Adv Genet. 2001;42:151–181. doi: 10.1016/s0065-2660(01)42021-9. [DOI] [PubMed] [Google Scholar]
- 35.Beaty T H, Liang K Y. Genet Epidemiol. 1987;4:203–210. doi: 10.1002/gepi.1370040305. [DOI] [PubMed] [Google Scholar]
- 36.Beaty T H, Liang K Y, Seery S, Cohen B H. Genet Epidemiol. 1987;4:211–221. doi: 10.1002/gepi.1370040306. [DOI] [PubMed] [Google Scholar]
- 37.Amos C I, Zhu D K, Boerwinkle E. Ann Hum Genet. 1996;60:143–160. doi: 10.1111/j.1469-1809.1996.tb01184.x. [DOI] [PubMed] [Google Scholar]
- 38.Allison D B, Neale M C, Zannolli R, Schork N J, Amos C I, Blangero J. Am J Hum Genet. 1999;65:531–544. doi: 10.1086/302487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Almasy L, Dyer T D, Blangero J. Genet Epidemiol. 1997;14:953–958. doi: 10.1002/(SICI)1098-2272(1997)14:6<953::AID-GEPI65>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 40.Almasy L, Williams J T, Dyer T D, Blangero J. Genet Epidemiol. 1999;17:S31–S36. doi: 10.1002/gepi.1370170706. [DOI] [PubMed] [Google Scholar]
- 41.Lander E S, Lauren M L, Birren B, Nusbaum C, Zody M C, Baldwin J, Devon K, Dewar K. Nature (London) 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 42.Tobler I, Kopp C, Deboer T, Rudolph U. Proc Natl Acad Sci USA. 2001;98:6464–6469. doi: 10.1073/pnas.111055398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kopell N, Ermentrout G B, Whittington M A, Traub R D. Proc Natl Acad Sci USA. 2000;97:1867–1872. doi: 10.1073/pnas.97.4.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Faulkner H J, Traub R D, Whittington M A. Br J Pharmacol. 1999;128:1813–1825. doi: 10.1038/sj.bjp.0702948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Whittington M A, Traub R D, Faulkner H J, Stanford I M, Jeffreys J G R. Proc Natl Acad Sci USA. 1997;94:12198–12203. doi: 10.1073/pnas.94.22.12198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rodolph U, Crestani F, Mohler H. Trends Pharmacol Sci. 2001;22:188–194. doi: 10.1016/s0165-6147(00)01646-1. [DOI] [PubMed] [Google Scholar]
- 47.Traub R D, Whittington M A, Collins S B, Buzsaki G, Jefferys J G R. J Physiol (London) 1996;493:471–484. doi: 10.1113/jphysiol.1996.sp021397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Whittington M A, Jefferys J G R, Traub R D. Br J Pharmacol. 1996;118:1977–1986. doi: 10.1111/j.1476-5381.1996.tb15633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Faulkner H J, Traub R D, Whittington M A. Br J Pharmacol. 1998;125:483–492. doi: 10.1038/sj.bjp.0702113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lindstrom S, Wrobel A. Exp Brain Res. 1990;79:313–318. doi: 10.1007/BF00608240. [DOI] [PubMed] [Google Scholar]
- 51.Traub R D, Whittington M A, Stanford I M, Jefferys J G R. Nature (London) 1996;383:621–624. doi: 10.1038/383621a0. [DOI] [PubMed] [Google Scholar]
- 52.Traub R D, Jefferys J G R, Whittington M A. Fast Oscillations in Cortical Circuits. Cambridge, MA: MIT Press; 1999. [Google Scholar]
- 53.Whittington M A, Traub R D, Kopell N, Ermentrout B, Buhl E H. Int J Psychophysiol. 2000;38:315–336. doi: 10.1016/s0167-8760(00)00173-2. [DOI] [PubMed] [Google Scholar]