CHRNB2 Is the Second Acetylcholine Receptor Subunit Associated with Autosomal Dominant Nocturnal Frontal Lobe Epilepsy (original) (raw)
Abstract
Autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) is an uncommon, idiopathic partial epilepsy characterized by clusters of motor seizures occurring in sleep. We describe a mutation of the β2 subunit of the nicotinic acetylcholine receptor, effecting a V287M substitution within the M2 domain. The mutation, in an evolutionary conserved region of CHRNB2, is associated with ADNFLE in a Scottish family. Functional receptors with the V287M mutation are highly expressed in Xenopus oocytes and characterized by an ∼10-fold increase in acetylcholine sensitivity. CHRNB2 is a new gene for idiopathic epilepsy, the second acetylcholine receptor subunit implicated in ADNFLE.
The autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) phenotype (MIM 600513) is clinically distinctive and is relatively homogeneous between families, although seizure severity and specific frontal lobe seizure manifestations vary within families (Scheffer et al. 1995). Site-specific mutations within the α4 subunit of the nicotinic acetylcholine receptor (nAChR) are associated with ADNFLE (Steinlein et al. 1995, 1997, 2000; Hirose et al. 1999; Saenz et al. 1999; Phillips et al. 2000) and affect amino acids that face into the pore of the ion channel, with demonstrated functional effects (Weiland et al. 1996; Kuryatov et al. 1997; Steinlein et al. 1997; Bertrand et al. 1998). The main functional nAChR in the brain is a pentamer comprised of α4 and β2 subunits, and the second transmembrane (M2) domains of these subunits interact to form the wall of the ion channel. Thus, the gene for the β2 subunit (CHRNB2), and its M2 domain in particular, is an obvious candidate for mutation in families and sporadic cases in which there is no mutation in CHRNA4. De Fusco et al. (2000) report such a mutation, at the same valine site at which we now report a different amino acid substitution.
The proband (V-1; fig. 1), a white girl of normal intellect, presented at age 11 years with nocturnal seizures. She would awaken from sleep with a sensation of breathing difficulty. After a few seconds, she would appear to hold her breath and grunt. Sometimes she would recover quickly and cry or scream. On other occasions, tonic extension of her left arm occurred with truncal flexion. Seizures would last up to a few minutes, and stereotyped attacks recurred at around 15-min intervals throughout the night. She had clear recollections of these events. Seizures also occurred during a daytime sleep. Video EEG telemetry showed only muscle artifact during the seizure. Although her seizures were initially well controlled by carbamazepine, relapse occurred, and further control was difficult. Currently, she is seizure free, because of a combination of phenytoin and topiramate.
Figure 1.

Scottish family, with family members carrying the mutated CHRNB2 codon287 indicated by “m”
There are nine other affected family members spanning four generations. Symptoms in the majority either have been so mild that medical attention has not been sought or have been easily controlled by carbamazepine. In some family members, the seizures have remitted spontaneously, but the oldest surviving family member (II-2; fig. 1), at age 81 years, continues to have occasional seizures while on phenytoin. DNA was only available from eight members of this family: six affected individuals, one obligate carrier, and his unaffected wife.
The second transmembrane domains (M2) of the nAChR subunits were screened by direct sequencing. Products for sequencing and restriction enzyme analysis were generated using primers A (TTCACACCTAGTGGTAGTG) and B (TGGCTGCTGCATGAAGAGCA), which amplify a 468-bp segment of the CHRNB2 gene. Products for single-strand conformation analysis (SSCA) were generated using primers C (CTGCTGCTCATCTCCAAGAT) and B, which amplify a 220-bp segment of the gene. All PCR reactions contained 67 mM Tris-HCl (pH 8.8); 16.5 mM (NH4)2SO4; 6.5 μm EDTA; 1.5 mM MgCl2; 200 μM each dNTP; 10% DMSO; 0.17 mg/ml BSA; 10 mM β-mercaptoethanol; 15 μg/ml each primer; 100 U/ml Taq DNA polymerase, and 10 μg/ml genomic DNA. Reactions for SSCA also contained 200 μCi/ml [32P-α-]dCTP. Reactions were performed using 10 cycles of 94°C for 60 s, 60°C for 90 s, and 72°C for 90 s and 25 cycles of 94°C for 60 s, 55°C for 90 s, and 72°C for 90 s, followed by 10 min at 72°C. PCR-amplified templates were purified for sequencing using QiaQuick PCR preps (Qiagen). Primers used for sequencing were the same as those used for PCR. For each reaction, 25 ng of primer and 100 ng of template were used. The BigDye sequencing kit (ABI) was used for sequencing reactions, and the products were analyzed using an ABI 377 Sequencer and the EditView program.
Sequencing revealed a G→A transition in the M2 domain of CHRNB2 in the proband and in the other family members where presence of the mutation has been indicated (fig. 1). The G→A transition created a _Nla_III restriction site. Primers A and B amplify a 468-bp fragment containing four normally present _Nla_III sites in addition to the mutant _Nla_III site. Digestion with _Nla_III gives fragments of 318, 78, 54, 9, and 9 bp in the normal allele and fragments of 273, 78, 54, 45, 9, and 9 bp in the mutated allele, confirming the presence of the mutation. Bands of 318 bp and 273 bp are easily detected on 2% agarose gels. The mutation is also detectable by SSCA in family members with the G→A transition. The bandshift detected by SSCA could only be associated with the G→A transition, since no other base changes were detected in the PCR product. SSCA excluded this transition in 102 anonymous Australian blood donors. For SSCA screening of blood donor samples, PCR reactions were mixed with an equal volume of formamide loading buffer (96% formamide, 1 mM EDTA, 0.1% bromophenol blue, 0.1% xylene cyanol) and were heated to 95°C for 3 min before snap-cooling on ice. 5 μl of each sample was loaded on 10% (49:1) polyacrylamide, 5% glycerol and was run on TBE gels. The gels were run at 700 V for 20 h at room temperature, were dried, and were exposed to X-ray film.
The c1025G→A mutation replaces a highly conserved valine with methionine at position 287, using nomenclature on the NCBI database (fig. 2), or at position 262, using the numbering of Rempel et al. (1998). All affected individuals and the unaffected obligate carrier had the mutation (fig. 1). There is much homology between CHRN genes of different species, especially in the four transmembrane domains. CHRNB2, CHRNB3, and CHRNB4 all are expressed in brain. The M2 domain of CHRNB3 has only 59% homology with the other two, and in vitro studies in the rat show that, when coexpressed with the α subunits alone, β3 does not assemble into a functional receptor. The β3 subunits may function in ion channels gated by ligands other than nicotine and acetylcholine (Willoughby et al. 1993). The M2 domains of CHRNB2 and CHRNB4 are almost completely conserved, and, in particular, valine287 is fully conserved (fig. 3). CHRNB1, which is expressed only in muscle, has leucine at this position. This may indicate that valine287 is essential for normal ion-channel function in the brain. The V287M mutation is located near the extracellular end of the M2 domain (dark barrel shown in fig. 4_A_) that lines the ionic pore. Valine287 faces into the pore of the ion channel in both the open and the closed state (Devilliers-Thiery et al. 1993), and, when valine287 is replaced by a bulkier methionine, normal ion flow through the channel might be disrupted.
Figure 2.

DNA sequence of CHRNB2, showing the c1025G→A transition. The upper chromatogram shows the mutation. The lower chromatogram shows the control sequence.
Figure 3.

Similarities within the M2 domain. Codon 287, located near the extracellular end of the M2 domain, is indicated by the box.
Figure 4.

A, left panel, Neuronal α4β2 heteropentameric receptor resulting from the assembly of two α4 and three β2 subunits. Right panel, α4β2 is shown as a pentamer with two potential ACh-binding sites. “β2*” identifies the V287M amino acid substitution. The ACh receptor can assemble without any mutated β2 subunit (α4β2 wild-type receptor) or with one, two, or three mutated β2* subunits. B, upper panel, Current traces elicited by consecutive applications of four or five increasing ACh concentrations (horizontal bars). The values above each bar indicate the concentration of ACh applied to the cell. Lower panel, Differences in ACh affinity are emphasized in a log-log plot. The logarithm of the absolute value of the current (measured in the upper panel, Log [−_I_]) is plotted as a function of the logarithm of ACh concentrations (Log [ACh]). −I and [ACh] are expressed in nA and nM, respectively. Values are mean ± SEM, with _n_=9 (α4β2), _n_=17 (α4β2*), and _n_=10 (α4[β2*+β2]). Solid lines are the best linear regression throughout data points. C, upper panel, Representative macroscopic currents recorded in oocytes expressing either the wild-type or mutated β2 subunit. Currents evoked by four ACh concentrations (0.1, 0.2, 0.8 and 8 μM) are superimposed. Cells were held at −100 mV and challenged with ACh (10 s) once every 120 s. Bars indicate ACh applications. Lower panel, ACh activation curves for α4β2, α4β2* and α4(β2*+β2). Dose-response curves were normalized to the maximal current amplitude of each cell. Values are mean ± SEM. For each ACh concentration, 3–8 independent measurements were averaged. Values of the curve fits are given in the text.
To define the effects of the β2 V287M mutation on the physiological properties of the α4β2 nAChR, we introduced the V287M amino acid substitution into the β2 coding sequence. Mutation V287M was introduced in the CHRNB2 cDNA (Monteggia et al. 1995), according to the PCR-derived strategy described by Nelson and Long (1989). The unique _Nhe_I and _Pml_I restriction sites were used to ligate the mutated fragment into the wild-type CHRNB2 cDNA. Mutation V287M was verified by complete sequencing of the 411-bp mutagenesis cassette.
Stage V or VI Xenopus laevis oocytes were isolated and nuclear-injected with 10 nl of 0.2 ng/nl DNA solution according to a standard procedure (Bertrand et al. 1991). For functional reconstitution of α4β2, α4β2V287M, and α4(β2+β2V287M) receptors, cDNAs coding for CHRNA4 (α4) (Monteggia et al. 1995), CHRNB2 (β2), and _CHRNB2_-V287M (β2V287M) subunits were mixed at molecular ratios of 1:1, 1:1, and 2:1:1, respectively. Oocytes were maintained at 18°C for 2–3 d, in standard Barth’s solution containing (in mM): NaCl 88, KCl 1, NaHCO3 2.4, MgSO4 0.8, Ca(NO3)2 0.3, CaCl2 0.4, HEPES-NaOH 10, pH 7.4, and were complemented with kanamycin (20 μg/ml), penicillin (100 μg/ml), and streptomycin (100 μg/ml).
The α4β2 and α4β2V287M cDNA combinations, injected into oocytes in a 1:1 ratio, will be considered first. Both wild-type and V287M containing receptors (where all three β2 subunits carried the V287M amino acid substitution) yielded robust currents (>5 μA) when challenged with saturating acetylcholine (Ach) concentrations. Macroscopic currents were recorded by a two-electrode voltage clamp, at 18°C, using a GENECLAMP 500 amplifier (Axon Instruments). Borosilicate electrodes were filled with 3 M KCl. Oocytes were continuously perfused with OR2 bath solution (Bertrand et al. 1991) containing (in mM): NaCl 82.5, KCl 2.5, CaCl2 2.5, MgCl2 1, atropine sulfate 0.0005, HEPES-NaOH 5, pH 7.4. ACh (Fluka) was diluted in OR2 from a 100-mM stock solution to the final concentrations used for experiments. Gravity-driven solutions were applied to the recording chamber through computer-controlled electromagnetic valves. For all experiments, holding potential was Vhold = −100 mV.
Distinction between these two types of receptors was unambiguously observed at low concentrations of ACh. As shown in the upper panel of fig. 4B, 3 nM ACh was already sufficient to activate reliable ionic currents with the mutated receptor (81±24 nA, _n_=18). In contrast, 50 nM ACh was required to evoke currents of similar amplitude in wild-type nAChRs (65±22 nA, _n_=9).
To characterize these differences, we first recorded currents evoked by four or five consecutive applications of increasing ACh concentrations that were non- or slightly desensitizing. Representative currents are shown in the upper panel of fig. 4B. A plot of the logarithm of ACh-evoked current (absolute value) versus the logarithm of agonist concentrations exhibited the expected linear relationship (fig. 4B, lower panel). Solid lines correspond to best linear regression fits. The α4β2V287M exhibited ∼1 order of magnitude higher ACh sensitivity than did wild-type receptors. These data confirmed our initial qualitative observation that mutated receptors were more sensitive to ACh.
To further examine ACh sensitivity, we determined, over a broad range of concentrations, the dose-response relationship for both control and mutated receptors. Figure 4_C_ shows superimposed currents evoked by 0.1, 0.2, 0.8, and 8 μM external ACh. Plotting peak current versus the logarithm of agonist concentration allowed the determination of the apparent ACh sensitivity (fig. 4C, lower panel). The recent work of Covernton and Connolly (2000), as well as observations made in our lab (B. Buisson, personal communication) suggest that α4β2 activation curves are best described by the sum of two Hill equations (eq. [1], solid lines in fig. 4C, lower panel). Most of ACh activation curves appeared biphasic with a high- and low-affinity component (Covernton and Connolly 2000; B. Buisson, personal communication). Consequently, ACh dose–response relationships were best fitted to the sum of two empirical Hill equations:
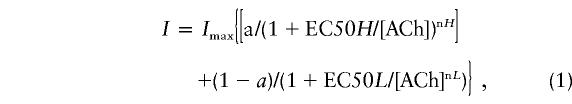
where _I_max is the amplitude of the maximal current elicited by ACh application, EC50 is the ACh concentration for half-maximal current activation, [ACh] is the concentration of ACh, and n is the Hill coefficient. Parameters related to the high- and low-affinity component are identified by H and L, respectively. Parameter a is the relative contribution of the high-affinity component to the total current response over the range of concentrations. a is expressed as the fraction of the high-affinity sites.
Corresponding EC50s, Hill slope values, and the relative contribution of each component, the high-affinity (H) and low-affinity (L) component, are summarized in table 1. Within the limits of the model used for the data fit, the α4β2V287M receptor exhibited both a decrease in the EC50s and an increase in the relative contribution of the high-affinity component. Thus, both log-log plot and dose-response curves accounted for a higher apparent ACh affinity associated with the β2-V287M mutation.
Table 1.
Properties of Wild-Type and β2*-V287M Containing Receptors[Note]
| SubunitCombination | EC50 (H) | EC50 (L) | n (H) | n (L) | % (H) | % (L) | n |
|---|---|---|---|---|---|---|---|
| a4b2 | 3.90 ± 1.30 | 47.6 ± 18.8 | 1.2 ± .2 | 1.3 ± .2 | 26 ± 5 | 74 ± 5 | 8 |
| a4b2* | .25 ± .04 | 2.9 ± .9 | 1.6 ± .1 | 1.9 ± .1 | 77 ± 1.5 | 23 ± 1.5 | 7 |
| a4 (b2*+b2) | .42 ± .14 | 5.3 ± 2.3 | 1.6 ± .1 | 1.0 ± .1 | 65 ± 3 | 35 ± 3 | 7 |
Determination of the peak-versus-plateau current ratio revealed no major differences between the wild-type and mutated receptors. This indicates that the V287M mutation causes no significant alteration of desensitization properties. Although the slowness of drug application on oocytes may prevent detection of very fast components in the Ach-evoked current, we believe that our data faithfully reflect the major response components.
Early studies (Anand et al. 1991; Cooper et al. 1991) suggest that nAchRs are heteropentamers resulting from the assembly of two α4 and three β2 subunits. Data discussed thus far relates to receptors where all three β2 subunits carried the V287M amino acid substitution. Since patients are heterozygous for the β2 gene, all cells probably express both the wild-type and mutated subunits. Therefore, it is mandatory to coinject wild-type and mutated β2 subunits. If the two β2 promoters are equipotent, the first experimental approach would be to inject equal amounts of wild-type and mutated β2 in the same oocyte. Within the same batch of oocytes, currents evoked by saturating ACh concentrations were indistinguishable on the basis of their amplitude. In contrast, an obvious difference was observed for the apparent ACh affinity. As is shown in table 1, both the high and low values of EC50 were comparable to receptors containing three V287M β2 subunits. Additionally, the relative contribution of high- versus low-affinity component was similar. Like α4β2V287M, α4(β2+β2V287M) exhibited a higher affinity in the log-log plot (triangles, fig. 4_B_).
These findings are consistent with the autosomal dominant mode of transmission of ADNFLE. As schematized in the right panel of figure 4A, four distinct subunit compositions are possible when both wild-type and mutated subunits are expressed in a cell. According to this assembly model, 75% of the receptors contain a mutated β2 subunit. Thus, the dominant effect of the mutation can be easily accounted for. Interestingly, oocytes expressing heterozygote or homozygote combination display very similar properties. This suggests that presence of a single β2 mutation within a receptor complex may be sufficient to confer the properties associated with the V287M substitution.
Thus far, two of the α4 mutations associated with ADNFLE (Steinlein et al. 1995, 1997) have been examined for their effect on channel properties. Both mutations introduce an amino acid modification in the M2 domain. Functional characterization of S248F in oocytes reveals both a significant decrease in currents evoked by saturating ACh concentrations and a small shift of the ACh dose-response toward a lower sensitivity. In contrast, mutation 776insGCT, which introduces an additional leucine residue into the M2 domain (259insL), does not affect the amplitude of ionic currents but causes an apparent increase of the ACh affinity of ∼10-fold. Consequently, although affecting different subunits, mutations α4 259insL and β2 V287M result in similar alterations of the functional properties of the receptor. Interestingly, although β2 V287M causes no significant alteration of desensitization properties, the major effect of β2 V287L is retardation of channel desensitization (De Fusco et al. 2000).
CHRNB2 is a new gene for idiopathic epilepsy and the second nAChR subunit implicated in ADNFLE. This confirms genetic heterogeneity for ADNFLE established by linkage analysis (Oldani et al. 1998; Phillips et al. 1998). Although only two mutations have so far been detected within the β2 subunit, the fact that both affect different amino acid substitutions at the same site suggests the possibility of site-specific mutation similar to that now established for the α4 subunit (Phillips et al. 2000). Questions yet to be answered include how all the various mutations identified so far in α4 and β2 subunits can lead to similar epileptic seizures and which other neurotransmitter systems might be involved for NFLE cases with normal nAChR subunits.
Acknowledgments
We thank the family for their participation. The study was supported by the National Health and Medical Research Council of Australia, Bionomics Ltd., and the Swiss National Foundation. We thank S. Bertrand for her help.
Footnotes
*
These authors contributed equally to this work.
Electronic-Database Information
Accession numbers and URLs for data in this article are as follows:
- CHRNB2 protein sequence, http://www.ncbi.nlm.nih.gov:80/entrez/query.fcgi (accession number 3766451)
- Genbank, http://www.ncbi.nlm.nih.gov/blast/blast.cgi (for human CHRNB2 reference sequence [X53179])
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim (for ADNFLE [MIM 600513])
References
- Anand R, Conroy WG, Schoefer R, Whiting P, Lindstrom J (1991) Neuronal nicotinic acetylcholine receptors expressed in Xenopus oocytes have a pentameric quaternary structure. J Biol Chem 266:11192–11198 [PubMed] [Google Scholar]
- Bertrand D, Cooper E, Valera S, Rungger D, Ballivet M (1991) Electrophysiology of neuronal nicotinic acetylcholine receptors expressed in Xenopus oocytes following nuclear injection of genes or cDNAs. Academic Press, New York [Google Scholar]
- Bertrand S, Weiland S, Berkovic SF, Steinlein OK, Bertrand D (1998) Properties of neuronal nicotinic acetylcholine receptor mutants from humans suffering from autosomal dominant nocturnal frontal lobe epilepsy. Br J Pharmacol 125:751–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper E, Couturier S, Ballivet M (1991) Pentameric structure and subunit stoichiometry of a neuronal nicotinic acetylcholine receptor. Nature 350:235–238 [DOI] [PubMed] [Google Scholar]
- Covernton PJ, Connolly JG (2000) Multiple components in the agonist concentration-response relationships of neuronal nicotinic acetylcholine receptors. J Neurosci Methods 96:63–70 [DOI] [PubMed] [Google Scholar]
- De Fusco M, Becchetti A, Patrignani A, Annesi G, Gambardella A, Quattrone A, Ballabio A, Wanke E, Casari G (2000) The nicotinic receptor β2 subunit is mutant in nocturnal frontal lobe epilepsy. Nat Genet 26:275–276 [DOI] [PubMed] [Google Scholar]
- Devilliers-Thiery A, Galzi JL, Eisele JL, Bertrand S, Bertrand D, Changeux JP (1993) Functional architecture of the nicotinic acetylcholine receptor: a prototype of ligand-gated ion channels. J Membr Biol 136:97–112 [DOI] [PubMed] [Google Scholar]
- Hirose S, Iwata H, Akiyoshi H, Kobayashi K, Ito M, Wada K, Kaneko S, Mitsudome A (1999) A novel mutation of CHRNA4 responsible for autosomal dominant nocturnal frontal lobe epilepsy. Neurology 53:1749–1753 [DOI] [PubMed] [Google Scholar]
- Kuryatov A, Gerzanich V, Nelson M, Olale F, Lindstrom J (1997) Mutation causing autosomal dominant nocturnal frontal lobe epilepsy alters Ca2+ permeability, conductance, and gating of human α4-2 nicotinic acetylcholine receptors. J Neurosci 17:9035–9047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteggia LM, Gopalakrishnan M, Touma E, Idler KB, Nash N, Arneric SP, Sullivan JP, Giordano T (1995) Cloning and transient expression of genes encoding the human alpha 4 and beta 2 neuronal nicotinic acetylcholine receptor (nAChR) subunits. Gene 155:189–193 [DOI] [PubMed] [Google Scholar]
- Nelson RM, Long GL (1989) A general method of site-specific mutagenesis using a modification of the Thermus aquaticus polymerase chain reaction. Anal Biochem 180:147–151 [DOI] [PubMed] [Google Scholar]
- Oldani A, Zucconi M, Asselta R, Modugno M, Bonati MT, Dalpra L, Malcovati M, Tenchini ML, Smirne S, Serini-Strambi L (1998) Autosomal dominant nocturnal frontal lobe epilepsy. A video-polysomnographic and genetic appraisal of 40 patients and delineation of the epileptic syndrome. Brain 121:205–223 [DOI] [PubMed] [Google Scholar]
- Phillips HA, Marini C, Scheffer IE, Sutherland GR, Mulley JC, Berkovic SF (2000) A de novo mutation in sporadic nocturnal frontal lobe epilepsy. Ann Neurol 48:264–267 [PubMed] [Google Scholar]
- Phillips HA, Scheffer IE, Crossland KM, Bhatia KP, Fish DR, Marsden CD, Howell SJL, Stephenson JBP, Tolmie J, Plazzi G, Eeg-Olofsson O, Singh R, Lopes-Cendes I, Andermann E, Andermann F, Berkovic SF, Mulley JC (1998) Autosomal dominant nocturnal frontal-lobe epilepsy: genetic heterogeneity and evidence for a second locus at 15q24. Am J Hum Genet 63:1108–1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rempel N, Heyers S, Engels H, Sleegers E, Steinlein OK (1998) The structures of the human neuronal nicotinic acetylcholine receptor β2- and α3-subunit genes (CHRNB2 and CHRNA3). Hum Genet 103:645–653 [DOI] [PubMed] [Google Scholar]
- Saenz A, Galan J, Caloustian C, Lorenzo F, Marquez C, Rodriguez N, Jimenez MD, Poza JJ, Cobo AM, Grid D, Prudhomme JF, de Munain AL (1999) Autosomal dominant nocturnal frontal lobe epilepsy in a Spanish family with a Ser252Phe mutation in the CHRNA4 gene. Arch Neurol 56:1004–1009 [DOI] [PubMed] [Google Scholar]
- Scheffer IE, Bhatia KP, Lopes-Cendes I, Fish DR, Marsden CD, Andermann F, Andermann E, Desbiens R, Keene D, Cendes F, Manson JI, Contantinou J, McIntosh A, Berkovic SF (1995) Autosomal dominant nocturnal frontal lobe epilepsy: a distinctive clinical disorder. Brain 118:61–73 [DOI] [PubMed] [Google Scholar]
- Steinlein OK, Magnusson A, Stoodt J, Bertrand S, Weiland S, Berkovic SF, Nakken KO, Propping P, Bertrand D (1997) An insertion mutation of the CHRNA4 gene in a family with autosomal dominant nocturnal frontal lobe epilepsy. Hum Mol Genet 6:943–948 [DOI] [PubMed] [Google Scholar]
- Steinlein OK, Mulley JC, Propping P, Wallace RH, Phillips HA, Sutherland GR, Scheffer IE, Berkovic SF (1995) A missense mutation in the neuronal nicotinic acetylcholine receptor α4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet 11:201–203 [DOI] [PubMed] [Google Scholar]
- Steinlein OK, Stoodt J, Mulley J, Berkovic S, Scheffer IE, Brodtkorb E (2000) Independent occurrence of the CHRNA4 Ser248Phe mutation in a Norwegian family with nocturnal frontal lobe epilepsy. Epilepsia 41:529–535 [DOI] [PubMed] [Google Scholar]
- Weiland S, Witzemann V, Villarrael A, Propping P, Steinlein O (1996) An amino acid exchange in the second transmembrane segment of a neuronal nicotinic receptor causes partial epilepsy by altering its desensitization kinetics. FEBS Lett 398:91–96 [DOI] [PubMed] [Google Scholar]
- Willoughby JJ, Ninkina NN, Beech MM, Latchman DS, Wood JN (1993) Molecular cloning of a human neuronal nicotinic acetylcholine receptor beta-3-like subunit. Neurosci Lett 155:136–139 [DOI] [PubMed] [Google Scholar]