Recycling of the Cell Adhesion Molecule L1 in Axonal Growth Cones (original) (raw)
Abstract
The cell adhesion molecule (CAM) L1 plays crucial roles in axon growth in vitro and in the formation of major axonal tracts in vivo. It is generally thought that CAMs link extracellular immobile ligands with retrogradely moving actin filaments to transmit force that pulls the growth cone forward. However, relatively little is known about the fate of CAMs that have been translocated into the central (C)-domain of the growth cone. We have shown previously that L1 is preferentially endocytosed at the C-domain. In the present study, we further analyze the subcellular distribution of endocytic organelles containing L1 at different time points and demonstrate that internalized L1 is transported into the peripheral (P)-domain of growth cones advancing via an L1-dependent mechanism. Internalized L1 is found in vesicles positioned along microtubules, and the centrifugal transport of these L1-containing vesicles is dependent on dynamic microtubules in the P-domain. Furthermore, we show that endocytosed L1 is reinserted into the plasma membrane at the leading edge of the P-domain. Monitoring recycled L1 reveals that it moves retrogradely on the cell surface into the C-domain. In contrast, the growth cone advancing independently of L1 internalizes and recycles L1 within the C-domain. For the growth cone to advance, the leading edge needs to establish strong adhesive interactions with the substrate while attachments at the rear are released. Recycling L1 from the C-domain to the leading edge provides an effective way to create asymmetric L1-mediated adhesion and therefore would be critical for L1-based growth cone motility.
Keywords: neural cell adhesion molecule, L1, axonal growth cone, endocytosis, recycling, microtubule
The motility of nerve growth cones plays a major role in axonal elongation during nervous system development. Growth cones express various cell adhesion molecules (CAMs) that recognize localized guidance cues present on neighboring cells or in the extracellular matrix and translate them into a directed axonal extension (Tessier-Lavigne and Goodman, 1996). One important axonal CAM is L1, which belongs to the Ig superfamily (Moos et al., 1988). L1 serves as both a ligand and a receptor. Homophilic L1–L1 binding between adjacent membranes is probably its most common mode of action in promoting axon growth along a bundle of preexisting axons (Stallcup and Beasley, 1985; Grumet and Edelman, 1988; Landmesser et al., 1988; Lemmon et al., 1989). Humans and mice with L1 mutations have defects in major axonal tracts such as the corticospinal tract and the corpus callosum (Cohen et al., 1997; Dahme et al., 1997; Fransen et al., 1998; Kamiguchi et al., 1998a; Demyanenko et al., 1999).
Growth cone motility depends on cytoskeletal dynamics (Bentley and O'Connor, 1994; Tanaka and Sabry, 1995). The two major cytoskeletal components in growth cones are actin filaments, which are predominantly located in the peripheral (P)-domain, and microtubules in the central (C)-domain (Bridgman, 1992). Spatially localized actin polymerization and depolymerization and actin–myosin interactions generate retrograde movement of actin filaments (Mitchison and Cramer, 1996), which is viewed as a force-generating system to pull the growth cone forward (Lin and Forscher, 1995). CAMs in the P-domain transmit this force by mechanically linking extracellular immobile ligands with the retrograde actin flow, leading to anterograde migration of the growth cone (Lin et al., 1994; Suter et al., 1998). However, relatively little is known about the fate of CAMs that have been translocated into the C-domain by coupling to the retrograde actin flow. It is likely that growth cones have an active mechanism by which CAMs can be recycled from the C-domain to the leading edge. It has been shown that CAMs, such as neural CAM (NCAM) and β1 integrin, undergo bidirectional movement on the cell surface of growth cones (Sheetz et al., 1990;Schmidt et al., 1995; Grabham and Goldberg, 1997), suggesting the centrifugal transport for CAM recycling. In addition to this cell-surface pathway, it is possible that CAM recycling occurs via intracellular vesicular transport, because the plasma membrane is actively retrieved from and reinserted into the growth cone surface (Cheng and Reese, 1987; Dailey and Bridgman, 1993). Indeed, this type of CAM recycling has been observed in migrating cells using integrins as adhesive receptors (Bretscher, 1992; Lawson and Maxfield, 1995;Bretscher and Aguado-Velasco, 1998). We have shown recently that L1 is internalized from the cell surface at the C-domain of axonal growth cones (Kamiguchi et al., 1998b). In the present paper, we demonstrate that endocytosed L1 in the C-domain is transported toward the P-domain followed by reinsertion into the plasma membrane of the leading edge. This is the first demonstration of intracellular trafficking and recycling of CAMs in nerve growth cones, shedding new light on CAM-dependent growth cone motility.
MATERIALS AND METHODS
Cell culture. Dorsal root ganglia (DRGs) were dissected from the lumbar region of embryonic day 10 chicks and dissociated sequentially with 2.4 units/ml dispase II (Boehringer Mannheim, Indianapolis, IN) and 0.1 mg/ml DNase (Boehringer Mannheim) in Ca2+- and Mg2+-free PBS. The dissociated cells were resuspended in DMEM (Life Technologies, Gaithersburg, MD) supplemented with 10% fetal bovine serum (FBS) and 100 ng/ml nerve growth factor (NGF) and then preplated for 1 hr. The neuron-enriched culture was prepared by replating the detached cells on a two-chamber plastic slide (Lab-Tek, Naperville, IL) that had been coated either with laminin (5 μg/cm2; Life Technologies) or with a chimeric protein consisting of the Fc region of human IgG and the whole extracellular domain of human L1 (Fransen et al., 1998). The cultures were maintained in a humid atmosphere of 95% air and 5% CO2 at 37°C.
Immunocytochemistry of internalized L1. Internalized L1 in nerve growth cones was visualized as described previously (Kamiguchi et al., 1998b). Live DRG neurons were incubated with rabbit polyclonal anti-chick L1 Fab (25 μg/ml) (Lemmon and McLoon, 1986) for 15 or 30 min at 37°C to allow for endocytosis of the Fab bound to L1. In some experiments, the bivalent antibody was used instead of the Fab. After rinsing at 4°C, the cells were fixed with 4% formaldehyde for 30 min. Because this fixation protocol did not permeabilize the cells, subsequent incubation with unlabeled anti-rabbit IgG (200 μg/ml; Molecular Probes, Eugene, OR) for 1 hr at 37°C specifically blocked the cell-surface Fab. Then, the cells were fixed again with 4% formaldehyde for 10 min to immobilize the unlabeled secondary antibody. After washing, the cells were permeabilized and blocked with 0.1% Triton X-100 and 10% horse serum in PBS for 1 hr. Internalized L1 was visualized by incubating the cells with Texas Red-X (TxR)-conjugated anti-rabbit IgG (1:100; Molecular Probes) for 1 hr at 20°C.
Immunocytochemistry of recycled L1. Live DRG neurons were incubated with rabbit polyclonal anti-chick L1 Fab (25 μg/ml) for 30 min at 37°C to allow for endocytosis of the L1–Fab complex. The cells were cooled to 4°C to stop further endocytic trafficking and incubated with unlabeled anti-rabbit IgG (200 μg/ml) for 45 min at 4°C to block the cell-surface Fab. After extensive washes at 4°C, the cells were incubated at 37°C for various periods in DMEM that was supplemented with 10% FBS and 100 ng/ml NGF, prewarmed and preequilibrated with 5% CO2. This incubation allowed the cells to recover and proceed with the trafficking of endocytosed L1 that had been tagged with the anti-L1 Fab. The cells were then fixed with 4% formaldehyde for 30 min, and recycled L1 on the cell surface was detected by visualizing the anti-L1 Fab that had not been blocked with the unconjugated secondary antibody. This was done by incubating the unpermeabilized cells with TxR-conjugated anti-rabbit IgG (1:100) for 1 hr at 20°C.
Other immunocytochemical analyses. For microtubule labeling, the cells were fixed with 3.7% formaldehyde and 0.2% glutaraldehyde in 50 mm PIPES buffer, pH 6.9, containing 50 mmKCl, 1 mm MgCl2, 1 mmEGTA, and 2% glycerol. After being permeabilized with 0.1% Triton X-100, the cells were incubated with rat monoclonal antibody against a tyrosinated form of the α-tubulin subunit (YL1/2; 10 μg/ml; Sera-Lab, Sussex, UK). The cells were then incubated with Alexa 594-conjugated anti-rat IgG (1:200; Molecular Probes).
In the experiments designed to visualize internalized L1, recycled L1, or microtubules, the cells were double-labeled for NCAM to outline the growth cone structure. Fixed cells were incubated with either rabbit polyclonal or mouse monoclonal antibody against chick NCAM (a kind gift of Dr. Urs Rutishauser, Memorial Sloan-Kettering Cancer Center, New York, NY) followed by incubation with Oregon Green-conjugated secondary antibody against either rabbit or mouse IgG (1:200; Molecular Probes).
The labeled cells were mounted with SlowFade Light (Molecular Probes). Images of growth cones were taken with a Zeiss LSM 410 confocal laser microscope (Zeiss, Göttingen, Germany), using an argon–krypton laser (excitation lines, 488 and 568 nm) and a 100× Plan-Neofluar (numerical aperture, 1.3) oil objective. Pinhole settings were chosen to give a single optical section of 0.83 μm.
Drug application. Taxol (Paclitaxel) was purchased from Sigma (St. Louis, MO) and dissolved in dimethylsulfoxide (DMSO). In the experiments designed to examine the effects of taxol on microtubule assembly or L1 trafficking, DRG neurons were treated with 10 nm taxol for 1 hr. The cells were then fixed for microtubule labeling or incubated with anti-L1 antibody to induce L1 endocytosis.
Growth cone selection and image analysis. Growth cones were randomly selected on the basis of NCAM staining under a microscope, and all of the growth cones positively labeled for endocytosed L1 or recycled L1 were photographed and included in this study. For image analysis we wanted to examine 45–60 growth cones that were positively labeled for internalized L1 or recycled L1 at each time point. Often this required repeating the experiment two to five times and pooling the data to obtain a sufficient number of labeled growth cones. Therefore, the data reported in Results represent pooled data sets from all experiments. Classifications of distribution patterns of endocytosed L1 and recycled L1 were done as described (see Figs.2A, 6, respectively). All of the classifications were performed by an observer who was not informed about treatment conditions (a blind observer).
Fig. 2.
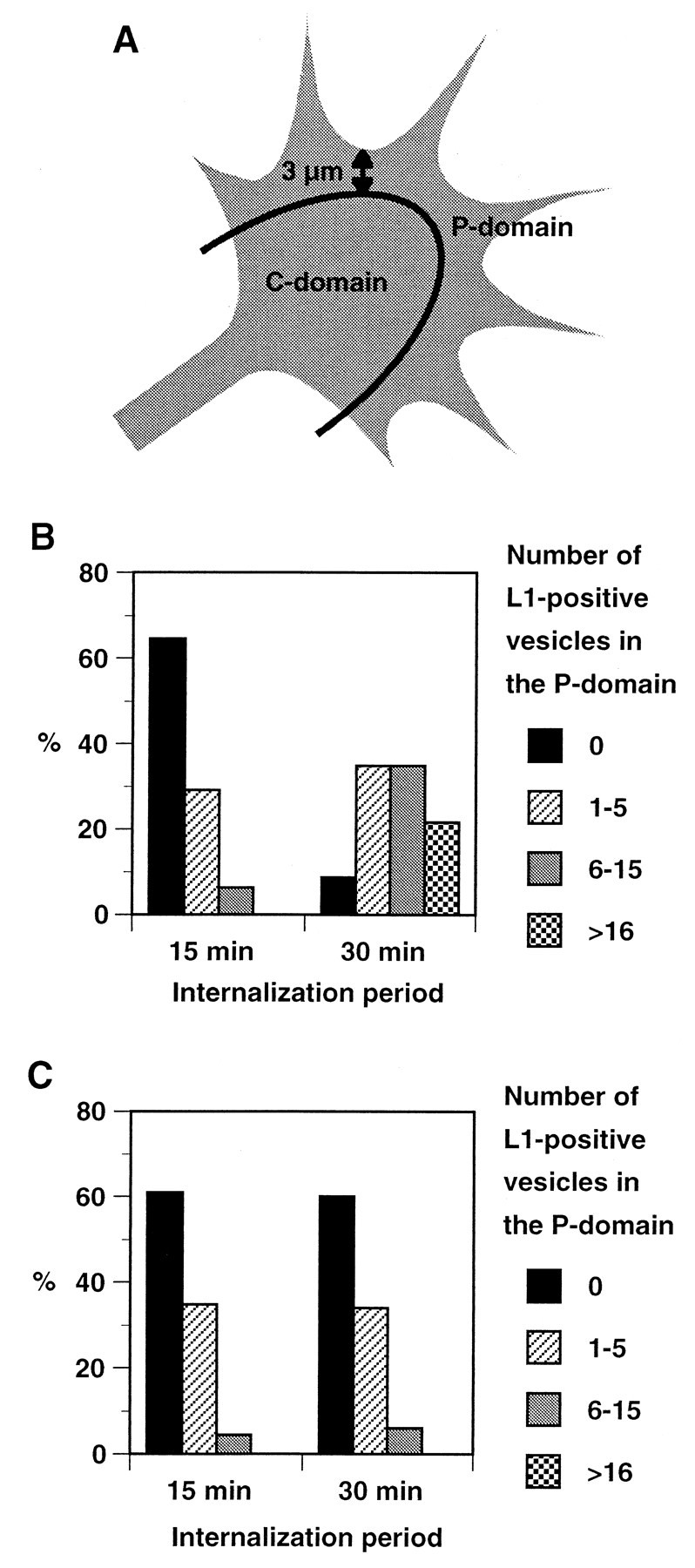
A, Schematic representation of the growth cone showing the P- and C-domains. For analyses of distribution patterns of endocytosed L1, the P-domain was defined as the area consisting of the filopodia and the lamellar regions within 3 μm of the leading edge based on Schmidt et al. (1995). B, Changes of distribution of endocytosed L1 over time in growth cones migrating on L1. Endocytosed L1 was visualized as shown in Figure 1, and the number of L1-positive endocytic organelles in the P-domain was categorized into four groups. Shown are the percentages of growth cones in each group at the 15 min (n = 48) and 30 min (n = 46) time points. There was a statistically significant difference between the two time points (p < 0.0001). C, Changes of distribution of endocytosed L1 over time in growth cones migrating on laminin. Shown are the percentages of growth cones in each group at the 15 min (n = 46) and 30 min (n = 50) time points.
Statistics. Data were analyzed by nonparametric statistics using StatView 4.5 (Abacus Concepts, Calabasas, CA). A statistical difference between two groups was assessed by a Mann–Whitney_U_ test. A comparison among three groups (see Fig. 6) was performed by a Mann–Whitney U test, with a Kruskal–Wallis test confirming that there was a significant difference at the_p_ = 0.0001 level.
Fig. 6.

Changes of distribution of recycled L1 on growth cones over time. Recycled L1 on growth cones was visualized as shown in Figure 5, and the distribution patterns were categorized into three classes: recycled L1 found only along the leading edge (Class 1), on the growth cone body (Class 3), or on both (Class 2). A, Distribution of recycled L1 on growth cones migrating on L1. Shown are the percentages of growth cones in each class at the 30 min (n = 51), 45 min (n = 46), and 60 min (n = 50) time points. There was a statistically significant difference between the 30 and 45 min time points (p < 0.0001) and between the 45 and 60 min time points (p < 0.0001). B, Distribution of recycled L1 on growth cones migrating on laminin. Shown are the percentages of growth cones in each class at the 30 min (n = 50), 45 min (n = 45), and 60 min (n = 50) time points. These data are a compilation of several different experiments that were pooled.
RESULTS
Distribution of endocytosed L1 in axonal growth cones
A traditional method for studying the movement of membrane proteins is to label them with specific antibodies. This approach can be used to follow cell-surface receptors such as the β2-adrenergic receptor (Cao et al., 1999) or v-SNAREs involved in membrane recycling (Teter et al., 1998). Similarly, endocytosed L1 in growth cones can be visualized by incubating live neurons with anti-L1 Fab and allowing for endocytosis of the Fab bound to L1. This experimental paradigm has been shown to visualize endocytosed L1 specifically (Kamiguchi et al., 1998b). If DRG neurons are incubated with anti-L1 Fab for 15 min, the majority of endocytosed L1 labeled by the Fab is restricted to the base and the C-domain of growth cones, an active region of L1 endocytosis where L1 colocalizes with a marker for clathrin-coated pits (Kamiguchi et al., 1998b). In this paper, we aim to analyze further the distribution pattern of endocytosed L1 in growth cones at different time points to gain insight into intracellular L1 trafficking. First, we tested whether anti-L1 Fab remains bound to L1 in endosomal compartments whose pH is slightly acidic: pH 6.0–6.5 in early endosomes and pH 5.0–6.0 in late endosomes (Nixon and Cataldo, 1995). Dot blot analyses using purified L1 from chick brain as an antigen showed that the Fab–antigen binding is unchanged even at pH 4.0 for at least 3 hr (data not shown), indicating that intracellular L1 trafficking can be followed by locating the anti-L1 Fab in growth cones.
The representative distribution of L1 in endocytic pathways in DRG growth cones migrating on an L1 substrate is shown in Figure1A–D. As reported previously, endocytosed L1 was typically confined to the C-domain and was absent from the P-domain after a 15 min incubation with anti-L1 Fab (Fig. 1A,B). In the case of bifurcating growth cones, endocytosed L1 was found in the base of each daughter growth cone (data not shown). After 30 min, anti-L1 Fab labeled a larger number of vesicular compartments distributed throughout the growth cone including the P-domain (Fig. 1C,D). L1-positive endocytic vesicles were also observed in the distal axonal shaft. In some cases, the distal axonal shaft was filled with many vesicular organelles containing endocytosed L1 (data not shown). Next we analyzed the subcellular distribution of endocytosed L1 in DRG growth cones advancing via an L1-independent mechanism on a laminin substrate. The representative images are shown in Figure 1E–H. After a 15 min incubation with anti-L1 Fab, endocytosed L1 was typically confined to the C-domain (Fig.1E,F), suggesting that the preferential internalization of L1 at the C-domain is not affected by the substrates on which the growth cone migrates. However, endocytosed L1 was still restricted to the C-domain of growth cones on laminin after a 30 min incubation with anti-L1 Fab (Fig. 1G,H), which is in strong contrast with L1 endocytic trafficking observed in growth cones on an L1 substrate.
Fig. 1.
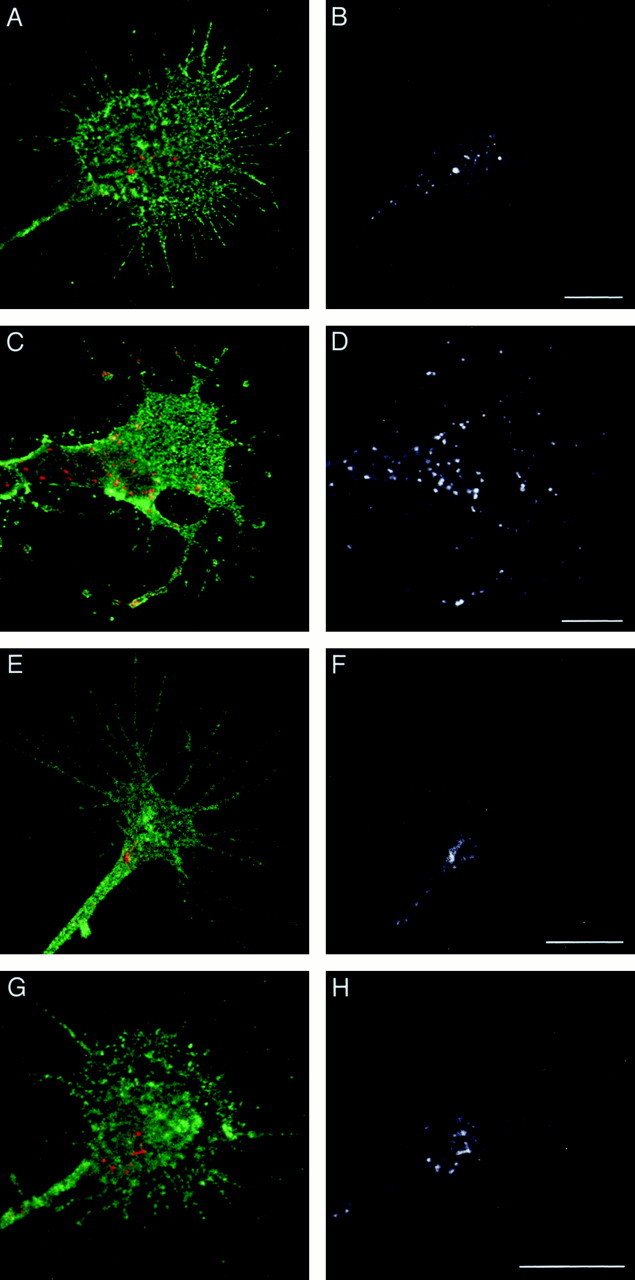
Subcellular distribution of endocytosed L1 in growth cones. DRG neurons cultured on L1 (A–D) or laminin (E–H) were incubated with anti-L1 Fab for 15 min (A, B, E,F) or 30 min (C, D,G, H) to allow for internalization of the Fab bound to L1. The cells were double-labeled for NCAM to outline the growth cone structure. In superimposed images (A, C, E, G), endocytosed L1 is colored red, and NCAM is colored green. To facilitate visualization of endocytosed L1, the red channel only is shown in_black_ and white (B, D, F, H). Scale bars: A, B, 10 μm; C, D, 10 μm; E, F, 10 μm; G, H, 10 μm.
On L1, 48 (42.9%) out of 112 and 46 (47.9%) out of 96 growth cones were positively labeled for endocytosed L1 after 15 and 30 min incubations with anti-L1 Fab, respectively. On laminin, 46 (35.1%) out of 131 and 50 (40.7%) out of 123 growth cones were positively labeled for endocytosed L1 after 15 and 30 min incubations with anti-L1 Fab, respectively. These positive growth cones were analyzed to quantify the difference in the distribution of endocytosed L1 at different time points. In this analysis, a blind observer counted the number of vesicle-like structures labeled by anti-L1 Fab in the P-domain (see Fig. 2A for the definition). Because it was sometimes difficult to count the exact number of vesicles if they were located very close to each other or fusing, we categorized the number of the vesicles into four groups (Fig. 2B; bins, 0, 1–5, 6–15, and >16).
At the 15 min time point on L1, 31 (64.6%) out of 48 growth cones did not show any labeled vesicles in the P-domain, whereas only 3 (6.3%) had more than five (Fig. 2B). In contrast, only 4 (8.7%) out of 46 growth cones at the 30 min time point on L1 were devoid of labeled vesicles in the P-domain, whereas 26 (56.5%) had more than five (Fig. 2B). There was a statistically significant difference between the two time points. A similar result was obtained when DRG growth cones were incubated for 15 or 30 min with bivalent anti-L1 antibody instead of Fab (data not shown). These results indicate that the number of endocytic organelles containing L1 in the P-domain increases with prolonged incubation periods that allow for endocytic trafficking of L1. This suggests two possibilities: (1) endocytosed L1 in the C-domain is transported into the P-domain, or (2) the P-domain is also capable of internalizing L1 at a slower rate than the C-domain. However, the latter possibility is unlikely because our previous studies showed that colocalization of L1 with a marker for clathrin-coated pits is restricted to the C-domain (Kamiguchi et al., 1998b).
At the 15 min time point on laminin, 28 (60.9%) out of 46 growth cones did not show any labeled vesicles in the P-domain, whereas only 2 (4.3%) had more than five (Fig. 2C). A similar distribution pattern was observed at the 30 min time point on laminin; 30 (60.0%) out of 50 growth cones were devoid of labeled vesicles in the P-domain, whereas only 3 (6.0%) had more than five (Fig. 2C). These results indicate that endocytosed L1 in the C-domain is not transported into the P-domain on a laminin substrate, which is in strong contrast with L1 trafficking on an L1 substrate. Therefore, intracellular trafficking of L1 after internalization at the C-domain should be somehow regulated by the substrates on which the growth cone migrates.
Centrifugal transport of endocytosed L1 requires dynamic ends of microtubules in the P-domain
To confirm the idea that endocytosed L1 in the C-domain is transported into the P-domain on an L1 substrate, we set up an experiment to examine the distribution of endocytosed L1 in growth cones when the cytoplasmic transport machinery is disrupted. In neuronal processes, membranous organelles are typically transported along microtubules, driven by microtubule-based molecular motors (Hirokawa, 1998). Many microtubules penetrate into the P-domain and reach near the leading edge in growth cones advancing on an L1 substrate, whereas microtubules are confined to the C-domain on a laminin substrate (Burden-Gulley and Lemmon, 1996). Therefore, microtubules might serve as a rail on which motor proteins convey L1-containing organelles toward the leading edge if the growth cone migrates on L1 but not on laminin. When DRG growth cones cultured on L1 were double-labeled to visualize microtubules and endocytosed L1 (30 min time point), the majority of endocytosed L1 was found in vesicles positioned along the microtubules (Fig.3A). Although it was difficult to assess the exact spatial relationships between microtubules and endocytosed L1 in the C-domain because of the dense distribution of microtubules, endocytosed L1 appeared clearly associated with the microtubules in the P-domain. This observation supports the idea that the transport of endocytosed L1 is guided by microtubules.
Fig. 3.
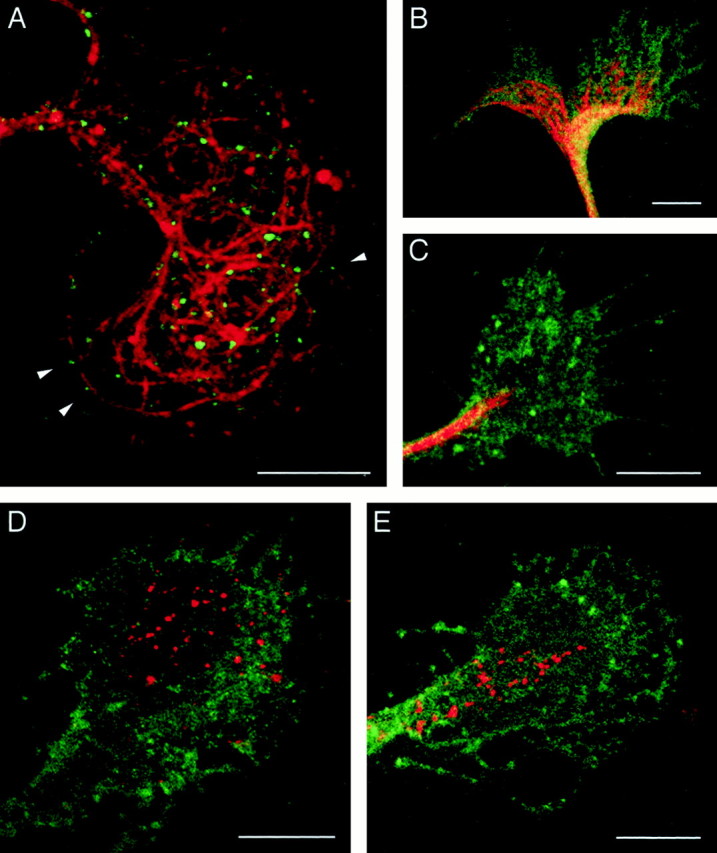
A, Localization of microtubules and endocytosed L1 in a growth cone. DRG neurons cultured on L1 were incubated with anti-L1 antibody for 30 min to allow for internalization of the antibody bound to L1. The cells were fixed and double-labeled for microtubules using an antibody against tyrosinated α-tubulin. Shown is a superimposed image in which endocytosed L1 is colored in_green_ and microtubules are colored in_red_. Arrowheads indicate examples of endocytosed L1 in vesicles positioned along the microtubules. B, C, Effects of taxol on microtubule organization in growth cones. DRG neurons cultured on L1 were pretreated with DMSO (B) or 10 nm taxol (C) for 1 hr and labeled for microtubules using an antibody against tyrosinated α-tubulin (red). The cells were double-labeled for NCAM to outline the growth cone structure (green). D, E, An effect of taxol on the subcellular distribution of endocytosed L1 in growth cones migrating on L1. After pretreatment with DMSO (D) or 10 nm taxol (E) for 1 hr, DRG neurons were incubated with anti-L1 antibody for 30 min to allow for internalization of the antibody bound to L1. The cells were double-labeled for endocytosed L1 (red) and NCAM to outline the growth cone structure (green). Scale bars, 10 μm.
Low concentrations (nanomolar) of taxol, a microtubule-stabilizing compound, have been shown to suppress microtubule dynamics without causing growth cone collapse (Letourneau and Ressler, 1984; Jordan et al., 1993). Consequently, the microtubules in taxol-treated growth cones do not splay out and extend as far distally into the P-domain as they normally do in control cultures (Williamson et al., 1996;Challacombe et al., 1997). We have confirmed this effect of taxol on microtubules in growth cones cultured on an L1 substrate (Fig.3B,C). An antibody against tyrosinated α-tubulin labeled many microtubules that splayed out and extended near the leading edge in DMSO-treated control growth cones. In contrast, growth cones that had been treated with 10 nm taxol for 1 hr contained tightly bundled microtubules that did not extend distally into the P-domain. On the basis of these observations, we were able to examine the effect of taxol on the distribution of endocytosed L1 in growth cones.
DRG growth cones on an L1 substrate were pretreated with either 10 nm taxol or DMSO for 1 hr and incubated with bivalent anti-L1 antibody for 30 min to allow for endocytic trafficking of the antibody. Sixty (37.0%) out of 162 taxol-treated growth cones and 45 (38.1%) out of 118 control growth cones were positively labeled for endocytosed L1, indicating that the taxol treatment did not significantly affect internalization of L1 from the plasma membrane. In the growth cones treated with taxol, endocytosed L1 was confined to the C-domain even after a 30 min incubation (Figs. 3E,4). However, in the control cultures at the same time point, endocytosed L1 was distributed throughout the growth cone including the P-domain (Figs. 3D, 4). The difference was quantified by counting the number of L1-positive vesicular compartments in the P-domain (Fig. 4). Treatment with DMSO alone did not significantly affect the distribution of endocytosed L1 compared with no DMSO treatment (see also Fig. 2B). Because endocytic trafficking from the plasma membrane to early endosomes is not sensitive to taxol at concentrations as high as 4 μm (Sonee et al., 1998), the different distribution pattern of endocytosed L1 after taxol treatment can be attributed to alterations in postendosomal trafficking. Therefore, we conclude that endocytosed L1 in the C-domain is transported toward the leading edge and that this transport is dependent on the dynamic ends of microtubules in the P-domain of growth cones on an L1 substrate.
Fig. 4.

Taxol-induced changes of distribution of endocytosed L1 in growth cones migrating on L1. Endocytosed L1 at the 30 min time points was visualized as shown in Figure 3,D and E. A total of 60 taxol-treated growth cones and 45 DMSO control growth cones were analyzed, and the number of L1-positive endocytic organelles in the P-domain was categorized into four groups. Shown are the percentages of growth cones in each group. There was a statistically significant difference between the taxol-treated and control growth cones (p < 0.0001).
Endocytosed L1 is recycled to the leading edge of growth cones advancing on L1
We next investigated the fate of endocytosed L1 after its trafficking into the P-domain of growth cones on an L1 substrate. The majority of endocytic organelles in growth cones exhibit pH values characteristic of sorting and recycling endosomes (Overly and Hollenbeck, 1996), and growth cones lack late endosomal/lysosomal compartments (Parton et al., 1992). Therefore, endocytosed L1 in growth cones is likely to be recycled back to the plasma membrane rather than to be degraded. We have reported previously that, in L1-transfected NIH-3T3 cells, all of the endocytosed L1 detected by anti-L1 antibody is found in endosomal compartments that contain transferrin receptors (Kamiguchi et al., 1998b). It has also been shown that, in migrating fibroblasts, endocytosed transferrin receptors are recycled to the plasma membrane of the leading lamella (Hopkins et al., 1994). These observations suggest that endocytosed L1 in growth cones is also recycled to their leading edge.
To test this hypothesis, we developed an experimental method of detecting recycled L1 on the cell surface. Growth cones were allowed to internalize anti-L1 Fab bound to L1 for 30 min, and the cell-surface Fab was blocked with unconjugated secondary antibody at 4°C. The cells were then rewarmed to 37°C and incubated for 0, 15, 30, 45, or 60 min to allow for continued membrane trafficking and exocytosis of the L1–Fab complex. Recycled L1 was detected by labeling any unblocked anti-L1 Fab that reappeared on the cell surface with TxR-conjugated secondary antibody. In this experiment, TxR-conjugated secondary antibody did not recognize newly synthesized L1 transported into the growth cone from the soma (Vogt et al., 1996), because it is not bound to the Fab. At the 0 min time point, none of 389 growth cones examined exhibited positive labeling with TxR-conjugated secondary antibody (Fig. 5A,D). The failure of labeling of vesicle-like structures that contain internalized L1 confirms that the cell membrane of growth cones remains unpermeabilized under this experimental protocol. Similarly, we did not observe any positively labeled growth cones out of 562 examined at the 15 min time point (data not shown). However, after resuming membrane trafficking for 30 min, a small percentage of the growth cones (4.0%, 51 out of 1280) was positively labeled for recycled L1 (Fig. 5B,E). After this time point, the percentage of positive growth cones increased with time: 20.6% (46 out of 223) and 41.3% (50 out of 121) at the 45 and 60 min time points, respectively (Fig.5C,F,G,J). Although these positive signals most likely represent recycled L1, it was also possible that the unconjugated secondary antibody used to block the cell-surface anti-L1 Fab became detached during the increasing incubation periods, leaving epitopes of the Fab available for the TxR-conjugated secondary. To exclude this possibility, another control experiment was performed. First, DRG neurons were incubated with anti-L1 Fab at 4°C instead of 37°C for 30 min. This incubation allowed the Fab to bind cell-surface L1 but prevented the Fab from being internalized (data not shown). Then, the cell-surface Fab was blocked with unconjugated secondary antibody at 4°C. After a 60 min incubation at 37°C, the cells were incubated with TxR-conjugated secondary antibody. Under this experimental paradigm, none of 412 growth cones was labeled by TxR-conjugated secondary antibody. This confirms that the positive red-channel signals in Figure 5, B, C, and G, represent reappearance of the L1–Fab complex on the cell surface rather than detachment of the unconjugated secondary. Taken collectively, these findings indicate that endocytosed L1 starts to reappear on the growth cone surface ∼30 min after resumption of membrane trafficking at 37°C. After a 60 min incubation at 37°C, 41.3% of the growth cones showed recycled L1 on their surface, whereas we could detect internalized L1 in 47.9% of the growth cones examined. Therefore, the majority (∼85%) of the growth cones that have internalized L1 are able to recycle it to the surface by the 60 min time point.
Fig. 5.
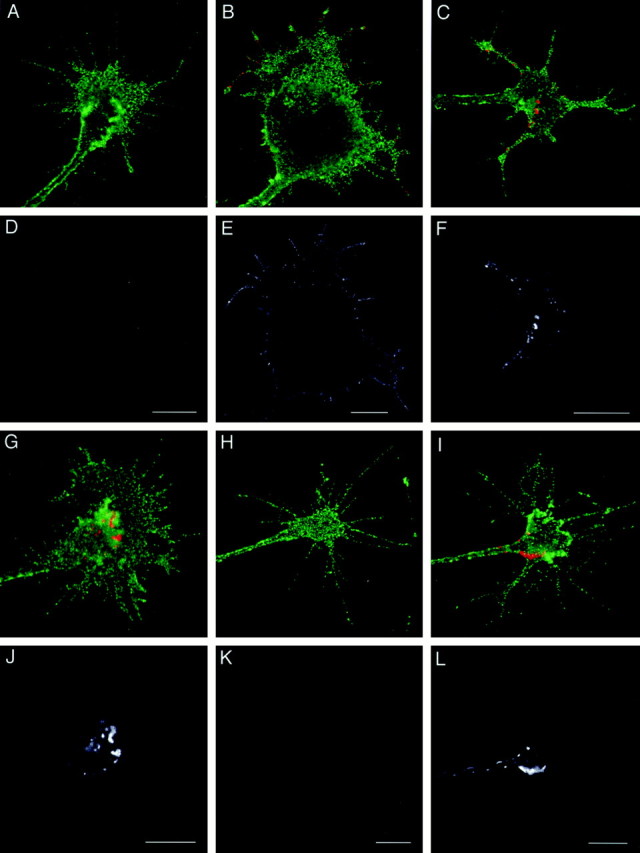
Cell-surface distribution of recycled L1 on growth cones. DRG neurons cultured on L1 (A–G, J) or laminin (H, I, K, L) were allowed to internalize anti-L1 Fab bound to L1 for 30 min, and the cell-surface Fab was blocked. The cells were reincubated for 0 min (A, D, H, K), 30 min (B, E, I, L), 45 min (C, F), or 60 min (G, J) to allow for exocytosis of the L1–Fab complex. Then, recycled L1 was detected by labeling the unblocked Fab that had reappeared on the cell surface. The cells were double-labeled for NCAM to outline the growth cone structure. In superimposed images (A–C, G–I), recycled L1 is colored in red, and NCAM is colored in_green_. To facilitate visualization of recycled L1, the_red_ channel only is shown in black and_white_ (D–F, J–L) below the corresponding superimposed image. Scale bars: A, D, 10 μm; B, E, 10 μm; C, F, 10 μm;G, J, 10 μm; H, K, 10 μm; I, L, 10 μm.
As shown in Figure 5, B and E, recycling L1 starts to reappear on the cell surface at the filopodia and the lamellipodial leading edge. A similar distribution pattern was found in a significant population of the growth cones at the 45 min time point, although they often show recycled L1 on the C-domain as well (Fig.5C,F). In contrast, at the 60 min time point, recycled L1 was found on the growth cone body, most frequently near the border between the P- and C- domains (Fig. 5G,J). In the majority of growth cones on which recycled L1 was found near the C-domain, we could tell by focusing the microscope that recycled L1 was on the apical surface but not on the substrate-facing membrane. This is most likely caused by the greater accessibility of secondary antibodies to the apical growth cone surface versus the substrate-facing surface. To analyze further the localization of recycled L1 at different time points, a blind observer categorized the distribution patterns of recycled L1 into three classes (Fig. 6): recycled L1 found only along the leading edge (Class 1), on the growth cone body (Class 3), or on both (Class 2). After a 30 min recycling period, 45 (88.2%) out of 51 growth cones showed recycled L1 with the Class 1 distribution (Fig.6A). In contrast, at the 60 min time point, 38 (76.0%) out of 50 growth cones exhibited recycled L1 with the_Class 3_ distribution (Fig. 6A). There were statistically significant differences in the localization of recycled L1, indicating that endocytosed L1 is preferentially reinserted into the plasma membrane of the leading edge followed by retrograde movement on the cell surface into the C-domain of growth cones advancing via an L1-dependent mechanism. A similar result was obtained when endocytosed L1was tagged with bivalent anti-L1 antibody instead of Fab (data not shown).
L1 recycling occurs within the C-domain of growth cones advancing on laminin
We next investigated the fate of endocytosed L1 that remains confined to the C-domain of growth cones migrating on a laminin substrate. Recycled L1 on growth cones was monitored for various periods using the same experimental method described above. After 0 and 15 min incubations at 37°C, none was positively labeled for recycled L1 out of 316 and 657 growth cones, respectively (Fig. 5H,K; data not shown). However, after resuming membrane trafficking for 30 min at 37°C, a small percentage of the growth cones (3.8%, 50 out of 1311) was positively labeled for recycled L1, which was typically confined to the C-domain and the distal axonal shaft but was absent from the P-domain (Fig. 5I,L). After this time point, the percentage of positive growth cones increased with time: 18.4% (45 out of 244) and 36.0% (50 out of 139) at the 45 and 60 min time points, respectively. The distribution pattern of recycled L1 at these time points was similar to that at the 30 min time point. As another control, growth cones were incubated with anti-L1 Fab at 4°C instead of 37°C to prevent the Fab from being internalized, followed by the same treatments used in the 60 min time point group. None of 395 growth cones in this control group was positively labeled by TxR-conjugated secondary antibody.
We have analyzed further the localization of recycled L1 at different time points by categorizing into three classes (Fig.6B). At all of the time points examined, the majority of growth cones exhibited the Class 3 distribution of recycled L1: 86.0% (43 out of 50) at 30 min, 82.2% (37 out of 45) at 45 min, and 84.0% (42 out of 50) at 60 min. Taken collectively, these findings indicate that endocytosed L1 in the C-domain is locally recycled within the C-domain and perhaps to the distal axonal shaft when the growth cone migrates on laminin.
DISCUSSION
The nerve growth cone migrates to pull forward and elongate the axon (Lamoureux et al., 1989), which requires coordinated activity of cytoskeletal, membrane, and adhesion systems (Lin et al., 1994). The nerve growth cone and other migrating cells use a similar mechanism for their motility, which is thought to be the result of five consecutive steps (Lauffenburger and Horwitz, 1996; Mitchison and Cramer, 1996;Sheetz et al., 1998): (1) Both actin polymerization and membrane insertion at the cell front generate protrusion of the leading edge. (2) The newly formed leading edge establishes a strong and stable attachment to the substrate via CAMs. (3) Coupling of CAMs to the retrograde actin flow generates traction force to pull the cell body or growth cone forward. (4) The cell rear detaches from the substrate followed by tail retraction, although this tail retraction step is modified in nerve growth cones by the presence of the neurite shaft. (5) CAMs are recycled to the cell front. For the cytoskeletal machinery to pull the cell body or growth cone forward as attachments at the rear are released, a gradient of cell-substrate adhesion (strong adhesion at the front and weak adhesion at the rear) is required. This asymmetric adhesion can be created by different mechanisms, including front-versus-rear asymmetry in CAM-cytoskeletal linkage strength (Schmidt et al., 1993, 1995) or CAM density (Sheetz et al., 1990;Lawson and Maxfield, 1995; Grabham and Goldberg, 1997).
In migrating growth cones, CAMs can be viewed as the “feet” needed to crawl on a relevant substrate. Although newly synthesized CAMs are supplied from the soma to the growth cone (Craig et al., 1995; Vogt et al., 1996), it is not economical to use them for only a single forward step. Therefore, a mechanism would be required to bring the feet from the rear of the growth cone up to the front for reuse. This recycling can occur either by cell-surface transport (Sheetz et al., 1990; Schmidt et al., 1995; Grabham and Goldberg, 1997) or by intracellular vesicular transport as demonstrated in this paper: endocytosis of L1 at the C-domain followed by vesicular transport and recycling to the leading edge. On the basis of this and previously published results by other investigators, we propose a model of L1 trafficking as illustrated in Figure 7. The L1 cytoplasmic domain (L1CD) contains at least two regions that interact with the actin cytoskeleton via ankyrin and an unknown molecule (Davis and Bennett, 1994; Dahlin-Huppe et al., 1997). NrCAM, a member of the L1 family whose cytoplasmic domain is highly homologous to the L1CD, has indeed been shown to couple with the retrograde actin flow in growth cones (Faivre-Sarrailh et al., 1999). So it is likely that L1 transmits traction force to pull the growth cone forward by linking extracellular substrates with the actin cytoskeleton. The cytoskeletal linkage as well as continuous addition of membrane components to the leading edge (Bretscher and Aguado-Velasco, 1998) would translocate L1 into the C-domain. Consistent with this is our observation that recycled L1 moves centripetally on the growth cone with increasing periods of incubation. Although we did not measure the rate of this L1 movement, it seems that L1 moved much slower than actin filaments that flow centripetally at a rate of 3–6 μm/min (Forscher and Smith, 1988). A similar discrepancy has been reported on NrCAM dynamics on growth cones; beads coated with a ligand to NrCAM moved centripetally at ∼5 μm/min, whereas translocation of immunocytochemically labeled NrCAM from the leading edge to the C-domain took 10 min (Faivre-Sarrailh et al., 1999). So it is difficult to infer the actual translocation velocity of CAMs from immunocytochemical approaches. However, our result could suggest that recycled L1 on growth cones, which is capped by either Fab or bivalent antibodies in this case, does not fully engage with the retrograde actin flow. Perhaps, full engagement between L1 and the actin cytoskeleton requires clustering of L1 molecules induced by cell–cell contacts or ligand-coated beads (Dubreuil et al., 1996; Malhotra et al., 1998; Faivre-Sarrailh et al., 1999). Although we observed the behavior of L1 present on the apical surface of growth cones in this paper, it is widely accepted that the retrograde CAM movement triggered by ligand binding on the apical surface reflects the CAM behavior on the substrate-facing surface that actually participates in growth cone migration (Suter and Forscher, 1998).
Fig. 7.

A model of L1 trafficking in the axonal growth cone migrating via an L1-dependent mechanism. L1 is internalized from the plasma membrane at the C-domain via clathrin-mediated pathways. Subsequently, endocytosed L1 is transported into the P-domain via sorting and recycling endosomes, a process that is dependent on the dynamic ends of microtubules (not shown in this figure). Then, trafficking L1 is reinserted into the plasma membrane at the leading edge. Recycled L1 on the cell surface moves toward the C-domain most likely by coupling to the retrogradely moving actin filaments via ankyrin or other linker molecules. The L1CD has at least two different states depending on conformation or phosphorylation. L1's interaction with ankyrin is regulated by phosphorylation (Garver et al., 1997), as is its ability to interact with clathrin adaptors (see last paragraph of Discussion for details).
Thus, L1 enters the C-domain of axonal growth cones where it is endocytosed via the clathrin-mediated pathway and sorted into early endosomes (Kamiguchi et al., 1998b). Endocytic organelles containing L1 are transported either retrogradely toward the soma or anterogradely toward the growth cone periphery. Although the fate of retrogradely moving L1 remains to be determined, it might be degraded or transmit signals to the soma (Itoh et al., 1995). Consistent with the general idea of a role of microtubules in organelle transport (Hirokawa, 1998), our results indicate that dynamic microtubules in the P-domain are required for transport of L1-containing endocytic organelles toward the leading edge. However, involvement of the actin-based centrifugal transport mechanism is also possible (Evans and Bridgman, 1995). In any case, these organelles carrying L1 probably correspond to a subpopulation of vesicles that were observed moving centrifugally in live growth cones by differential interference contrast microscopy (Goldberg and Burmeister, 1986). After this vesicular transport, L1 is reinserted into the plasma membrane of the leading edge, most likely participating in the formation of new adhesive sites. These observations on L1 likely apply to other L1 family members such as neurofascin and NrCAM and perhaps members of the Tag-1/Axonin-1 CAM family because growth cones growing on these substrates exhibit very similar morphologies and behaviors. However, if growth cones are migrating on extracellular matrix components, then this model may not completely apply. The organization of the cytoskeleton in growth cones is substantially different with microtubules failing to reach far into the P-domain. Consequently integrins may not be transported intracellularly far into the P-domain. The failure of L1 to recycle to the P-domain when the growth cones were on laminin is probably caused by the lack of microtubules in the P-domain, although a more complex explanation involving specific regulation of recycling dependant on the substrate is possible. Immunohistochemical studies over the years of L1 expression on growth cones on different substrates have not suggested an accumulation of L1 in the C-region on non-L1 substrates. So rates of internalization of L1 must be matched by insertion into the plasma membrane from newly synthesized L1 and recycled L1.
To demonstrate that L1 trafficking is required for L1-mediated axon growth, it would be essential to conduct an experiment in which L1 trafficking is disrupted and then motility of the growth cones is analyzed. Although we have identified molecular mechanisms for L1 trafficking to some extent (Kamiguchi et al., 1998b), they are involved not only in L1 trafficking but also in other biological events critical for growth cone motility. For example, suppression of dynamic microtubules by pharmacological treatments has been shown to inhibit neurite elongation even when the neurons are growing independently of L1 (Letourneau and Ressler, 1984; Tanaka et al., 1995). Alternatively if we mutate the L1CD to block L1 endocytosis, L1 is no longer targeted to the growth cone because L1's endocytic signal overlaps with its axonal sorting signal (Kamiguchi and Lemmon, 1998; Kamiguchi et al., 1998b). For these reasons, we have not been able to disrupt specifically L1 trafficking in the growth cone and, therefore, to prove that L1 trafficking is required for L1-mediated axon growth. However, a large amount of cell biological data and mathematical models of cell migration (DiMilla et al., 1991; Lauffenburger and Horwitz, 1996) strongly suggest a critical role of L1 trafficking in growth cone advance. This concept is also supported by the observation that L1 recycling from the C-domain to the leading edge is specific to the growth cone advancing via an L1-dependent mechanism. Furthermore, L1 trafficking could play a role in growth cone bifurcation or turning. For example, internalizing L1 near the central leading edge of bifurcating growth cones (data not shown) might decrease adhesion of this area to the substrate, which has been shown to facilitate growth cone bifurcation (Wessells and Nuttall, 1978). It is also intriguing to speculate that reorienting the direction of L1 recycling could be coupled to growth cone turning. In other words, L1 is likely to be recycled asymmetrically, being guided by the microtubules that extend preferentially in the direction toward which the growth cone will steer (Bentley and O'Connor, 1994; Tanaka and Sabry, 1995).
In addition to the initial idea that CAMs regulate axon growth on the basis of their ability to mediate adhesive interactions, it is now clear that CAM-associated intracellular signals are also critical. For example, L1-dependent axon growth has been shown to involve calcium signaling (Williams et al., 1992), the fibroblast growth factor receptor (FGFr) (Saffell et al., 1997), L1-associated kinases (Wong et al., 1996; Schaefer et al., 1999), and the nonreceptor tyrosine kinase pp60c-src (Ignelzi et al., 1994). How do these signaling events cooperate with the L1 trafficking to regulate growth cone motility? It has been shown that tyrosine phosphorylation of neurofascin, a member of the L1 family of CAMs, abolishes its ankyrin-binding activity (Garver et al., 1997), suggesting that L1-associated kinases or phosphatases regulate its interaction with the actin cytoskeleton. We have found that internalization of L1 is required for ERK2 phosphorylation of L1 (Schaefer et al., 1999). We have also found that dephosphorylation of another tyrosine allows the L1CD to interact with the endocytic machinery (A. W. Schaefer, S. Storms, I. Kamiguchi, M. Pendergast, I. Rapoport, G. Landreth, T. Kirchhausen, and V. Lemmon, unpublished observations). Thus the phosphorylation state of the L1CD is likely to determine the pathway of L1 trafficking. Furthermore, activation of the FGFr by L1 produces a localized increase of calcium influx (Archer et al., 1999) that could then trigger vesicle endocytosis and/or exocytosis (De Camilli and Takei, 1996; Geppert and Südhof, 1998) and influence actin dynamics (Lankford and Letourneau, 1991). In this way, L1-associated signals could be involved in growth cone motility by modulating the pathway and the rate of L1 trafficking.
Footnotes
This study was supported by a grant from the Institute of Physical and Chemical Research (RIKEN), by Health Sciences Research Grants for Specific Diseases 1999-SD-17, Intractable Hydrocephalus, from the Ministry of Health and Welfare, Japan (H.K.), and by National Institutes of Health Grants EY-5285 and P30-EY11373 (V.L.). We acknowledge the excellent technical assistance of Maryanne Pendergast. We also thank Tsui Chern Cheah and Fumie Yoshihara for their assistance in image analyses and Drs. Sandra Lemmon, Susann Brady-Kalnay, and Susan Burden-Gulley for helpful comments on this manuscript.
Correspondence should be addressed to Dr. Vance Lemmon, Department of Neurosciences, Case Western Reserve University, 2109 Adelbert Road, Cleveland, OH 44106-4975. E-mail: vxl@po.cwru.edu.
REFERENCES
- 1.Archer F, Doherty P, Collins D, Bolsover S. CAMs and FGF cause a local submembrane calcium signal promoting axon outgrowth without a rise in bulk calcium concentration. Eur J Neurosci. 1999;11:3565–3573. doi: 10.1046/j.1460-9568.1999.00773.x. [DOI] [PubMed] [Google Scholar]
- 2.Bentley D, O'Connor TP. Cytoskeletal events in growth cone steering. Curr Opin Neurobiol. 1994;4:43–48. doi: 10.1016/0959-4388(94)90030-2. [DOI] [PubMed] [Google Scholar]
- 3.Bretscher MS. Circulating integrins: α5β1, α6β4 and Mac-1, but not α3β1, α4β1 or LFA-1. EMBO J. 1992;11:405–410. doi: 10.1002/j.1460-2075.1992.tb05068.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bretscher MS, Aguado-Velasco C. Membrane traffic during cell locomotion. Curr Opin Cell Biol. 1998;10:537–541. doi: 10.1016/s0955-0674(98)80070-7. [DOI] [PubMed] [Google Scholar]
- 5.Bridgman PC. Functional anatomy of the growth cone in relation to its role in locomotion and neurite assembly. In: Letourneau PC, Kater SB, Macagno ER, editors. The nerve growth cone. Raven; New York: 1992. pp. 39–53. [Google Scholar]
- 6.Burden-Gulley SM, Lemmon V. L1, N-cadherin, and laminin induce distinct distribution patterns of cytoskeletal elements in growth cones. Cell Motil Cytoskeleton. 1996;35:1–23. doi: 10.1002/(SICI)1097-0169(1996)35:1<1::AID-CM1>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 7.Cao TT, Deacon HW, Reczek D, Bretscher A, von Zastrow M. A kinase-regulated PDZ-domain interaction controls endocytic sorting of the β2-adrenergic receptor. Nature. 1999;401:286–290. doi: 10.1038/45816. [DOI] [PubMed] [Google Scholar]
- 8.Challacombe JF, Snow DM, Letourneau PC. Dynamic microtubule ends are required for growth cone turning to avoid an inhibitory guidance cue. J Neurosci. 1997;17:3085–3095. doi: 10.1523/JNEUROSCI.17-09-03085.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng TPO, Reese TS. Recycling of plasmalemma in chick tectal growth cones. J Neurosci. 1987;7:1752–1759. doi: 10.1523/JNEUROSCI.07-06-01752.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cohen NR, Taylor JSH, Scott LB, Guillery RW, Soriano P, Furley AJW. Errors in corticospinal axon guidance in mice lacking the neural cell adhesion molecule L1. Curr Biol. 1997;8:26–33. doi: 10.1016/s0960-9822(98)70017-x. [DOI] [PubMed] [Google Scholar]
- 11.Craig AM, Wyborski RJ, Banker G. Preferential addition of newly synthesized membrane protein at axonal growth cones. Nature. 1995;375:592–594. doi: 10.1038/375592a0. [DOI] [PubMed] [Google Scholar]
- 12.Dahlin-Huppe K, Berglund EO, Ranscht B, Stallcup WB. Mutational analysis of the L1 neuronal cell adhesion molecule identifies membrane-proximal amino acids of the cytoplasmic domain that are required for cytoskeletal anchorage. Mol Cell Neurosci. 1997;9:144–156. doi: 10.1006/mcne.1997.0608. [DOI] [PubMed] [Google Scholar]
- 13.Dahme M, Bartsch U, Martini R, Anliker B, Schachner M, Mantei N. Disruption of the mouse L1 gene leads to malformations of the nervous system. Nat Genet. 1997;17:346–349. doi: 10.1038/ng1197-346. [DOI] [PubMed] [Google Scholar]
- 14.Dailey ME, Bridgman PC. Vacuole dynamics in growth cones: correlated EM and video observations. J Neurosci. 1993;13:3375–3393. doi: 10.1523/JNEUROSCI.13-08-03375.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davis JQ, Bennett V. Ankyrin binding activity shared by the neurofascin/L1/NrCAM family of nervous system cell adhesion molecules. J Biol Chem. 1994;269:27163–27166. [PubMed] [Google Scholar]
- 16.De Camilli P, Takei K. Molecular mechanisms in synaptic vesicle endocytosis and recycling. Neuron. 1996;16:481–486. doi: 10.1016/s0896-6273(00)80068-9. [DOI] [PubMed] [Google Scholar]
- 17.Demyanenko G, Tsai A, Maness P. Abnormalities in neuronal process extension, hippocampal development, and the ventricular system of L1 knockout mice. J Neurosci. 1999;19:4907–4920. doi: 10.1523/JNEUROSCI.19-12-04907.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DiMilla PA, Barbee K, Lauffenburger DA. Mathematical model for the effects of adhesion and mechanics on cell migration speed. Biophys J. 1991;60:15–37. doi: 10.1016/S0006-3495(91)82027-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dubreuil RR, MacVicar G, Dissanayake S, Liu C, Homer D, Hortsch M. Neuroglian-mediated cell adhesion induces assembly of the membrane skeleton at cell contact sites. J Cell Biol. 1996;133:647–655. doi: 10.1083/jcb.133.3.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Evans LL, Bridgman PC. Particles move along actin filament bundles in nerve growth cones. Proc Natl Acad Sci USA. 1995;92:10954–10958. doi: 10.1073/pnas.92.24.10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Faivre-Sarrailh C, Falk J, Pollerberg E, Schachner M, Rougon G. NrCAM, cerebellar granule cell receptor for the neuronal adhesion molecule F3, displays an actin-dependent mobility in growth cones. J Cell Sci. 1999;112:3015–3027. doi: 10.1242/jcs.112.18.3015. [DOI] [PubMed] [Google Scholar]
- 22.Forscher P, Smith SJ. Actions of cytochalasins on the organization of actin filaments and microtubules in a neuronal growth cone. J Cell Biol. 1988;107:1505–1516. doi: 10.1083/jcb.107.4.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fransen E, D'Hooge R, Van Camp G, Verhoye M, Sijbers J, Reyniers E, Soriano P, Kamiguchi H, Willemsen R, Koekkoek SKE, De Zeeuw CI, De Deyn PP, Van der Linden A, Lemmon V, Kooy RF, Willems PJ. L1 knockout mice show dilated ventricles, vermis hypoplasia and impaired exploration patterns. Hum Mol Genet. 1998;7:999–1009. doi: 10.1093/hmg/7.6.999. [DOI] [PubMed] [Google Scholar]
- 24.Garver TD, Ren Q, Tuvia S, Bennett V. Tyrosine phosphorylation at a site highly conserved in the L1 family of cell adhesion molecules abolishes ankyrin binding and increases lateral mobility of neurofascin. J Cell Biol. 1997;137:703–714. doi: 10.1083/jcb.137.3.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Geppert M, Südhof TC. Rab3 and synaptotagmin: the yin and yang of synaptic membrane fusion. Annu Rev Neurosci. 1998;21:75–95. doi: 10.1146/annurev.neuro.21.1.75. [DOI] [PubMed] [Google Scholar]
- 26.Goldberg DJ, Burmeister DW. Stages in axon formation: observations of growth of Aplysia axons in culture using video-enhanced contrast-differential interference contrast microscopy. J Cell Biol. 1986;103:1921–1931. doi: 10.1083/jcb.103.5.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grabham PW, Goldberg DJ. Nerve growth factor stimulates the accumulation of β1 integrin at the tips of filopodia in the growth cones of sympathetic neurons. J Neurosci. 1997;17:5455–5465. doi: 10.1523/JNEUROSCI.17-14-05455.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grumet M, Edelman GM. Neuron-glia cell adhesion molecule interacts with neurons and astroglia via different binding mechanisms. J Cell Biol. 1988;106:487–503. doi: 10.1083/jcb.106.2.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hirokawa N. Kinesin and dynein superfamily proteins and the mechanism of organelle transport. Science. 1998;279:519–526. doi: 10.1126/science.279.5350.519. [DOI] [PubMed] [Google Scholar]
- 30.Hopkins CR, Gibson A, Shipman M, Strickland DK, Trowbridge IS. In migrating fibroblasts, recycling receptors are concentrated in narrow tubules in the pericentriolar area, and then routed to the plasma membrane of the leading lamella. J Cell Biol. 1994;125:1265–1274. doi: 10.1083/jcb.125.6.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ignelzi MA, Miller DR, Soriano P, Maness PF. Impaired neurite outgrowth of src-minus cerebellar neurons on the cell adhesion molecule L1. Neuron. 1994;12:873–884. doi: 10.1016/0896-6273(94)90339-5. [DOI] [PubMed] [Google Scholar]
- 32.Itoh K, Brackenbury R, Akeson RA. Induction of L1 mRNA in PC12 cells by NGF is modulated by cell–cell contact and does not require the high-affinity NGF receptor. J Neurosci. 1995;15:2504–2512. doi: 10.1523/JNEUROSCI.15-03-02504.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jordan MA, Toso RJ, Thrower D, Wilson L. Mechanism of mitotic block and inhibition of cell proliferation by taxol at low concentrations. Proc Natl Acad Sci USA. 1993;90:9552–9556. doi: 10.1073/pnas.90.20.9552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kamiguchi H, Lemmon V. A neuronal form of the cell adhesion molecule L1 contains a tyrosine-based signal required for sorting to the axonal growth cone. J Neurosci. 1998;18:3749–3756. doi: 10.1523/JNEUROSCI.18-10-03749.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kamiguchi H, Hlavin ML, Lemmon V. Role of L1 in neural development: what the knockouts tell us. Mol Cell Neurosci. 1998a;12:48–55. doi: 10.1006/mcne.1998.0702. [DOI] [PubMed] [Google Scholar]
- 36.Kamiguchi H, Long KE, Pendergast M, Schaefer AW, Rapoport I, Kirchhausen T, Lemmon V. The neural cell adhesion molecule L1 interacts with the AP-2 adaptor and is endocytosed via the clathrin-mediated pathway. J Neurosci. 1998b;18:5311–5321. doi: 10.1523/JNEUROSCI.18-14-05311.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lamoureux P, Buxbaum RE, Heidemann SR. Direct evidence that growth cones pull. Nature. 1989;340:159–162. doi: 10.1038/340159a0. [DOI] [PubMed] [Google Scholar]
- 38.Landmesser L, Dahm L, Schultz K, Rutishauser U. Distinct roles for adhesion molecules during innervation of embryonic chick muscle. Dev Biol. 1988;130:645–670. doi: 10.1016/0012-1606(88)90358-2. [DOI] [PubMed] [Google Scholar]
- 39.Lankford KL, Letourneau PC. Roles of actin filaments and three second-messenger systems in short-term regulation of chick dorsal root ganglion neurite outgrowth. Cell Mot Cytoskeleton. 1991;20:7–29. doi: 10.1002/cm.970200103. [DOI] [PubMed] [Google Scholar]
- 40.Lauffenburger DA, Horwitz AF. Cell migration: a physically integrated molecular process. Cell. 1996;84:359–369. doi: 10.1016/s0092-8674(00)81280-5. [DOI] [PubMed] [Google Scholar]
- 41.Lawson MA, Maxfield FR. Ca2+- and calcineurin-dependent recycling of an integrin to the front of migrating neutrophils. Nature. 1995;377:75–79. doi: 10.1038/377075a0. [DOI] [PubMed] [Google Scholar]
- 42.Lemmon V, McLoon S. The appearance of an L1-like molecule in the chick primary visual pathway. J Neurosci. 1986;6:2987–2994. doi: 10.1523/JNEUROSCI.06-10-02987.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lemmon V, Farr K, Lagenaur C. L1-mediated axon outgrowth occurs via a homophilic binding mechanism. Neuron. 1989;2:1597–1603. doi: 10.1016/0896-6273(89)90048-2. [DOI] [PubMed] [Google Scholar]
- 44.Letourneau PC, Ressler AH. Inhibition of neurite initiation and growth by taxol. J Cell Biol. 1984;98:1355–1362. doi: 10.1083/jcb.98.4.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin CH, Forscher P. Growth cone advance is inversely proportional to retrograde f-actin flow. Neuron. 1995;14:763–771. doi: 10.1016/0896-6273(95)90220-1. [DOI] [PubMed] [Google Scholar]
- 46.Lin CH, Thompson CA, Forscher P. Cytoskeletal reorganization underlying growth cone motility. Curr Opin Neurobiol. 1994;4:640–647. doi: 10.1016/0959-4388(94)90004-3. [DOI] [PubMed] [Google Scholar]
- 47.Malhotra JD, Tsiotra P, Karagogeos D, Hortsch M. Cis-activation of L1-mediated ankyrin recruitment by TAG-1 homophilic cell adhesion. J Biol Chem. 1998;273:33354–33359. doi: 10.1074/jbc.273.50.33354. [DOI] [PubMed] [Google Scholar]
- 48.Mitchison TJ, Cramer LP. Actin-based cell motility and cell locomotion. Cell. 1996;84:371–379. doi: 10.1016/s0092-8674(00)81281-7. [DOI] [PubMed] [Google Scholar]
- 49.Moos M, Tacke R, Scherer H, Teplow D, Fruh K, Schachner M. Neural adhesion molecule L1 as a member of the immunoglobulin superfamily with binding domains similar to fibronectin. Nature. 1988;334:701–703. doi: 10.1038/334701a0. [DOI] [PubMed] [Google Scholar]
- 50.Nixon RA, Cataldo AM. The endosomal-lysosomal system of neurons: new roles. Trends Neurosci. 1995;18:489–496. doi: 10.1016/0166-2236(95)92772-i. [DOI] [PubMed] [Google Scholar]
- 51.Overly CC, Hollenbeck PJ. Dynamic organization of endocytic pathways in axons of cultured sympathetic neurons. J Neurosci. 1996;16:6056–6064. doi: 10.1523/JNEUROSCI.16-19-06056.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Parton RG, Simons K, Dotti CG. Axonal and dendritic endocytic pathways in cultured neurons. J Cell Biol. 1992;119:123–137. doi: 10.1083/jcb.119.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Saffell JL, Williams EJ, Mason IJ, Walsh FS, Doherty P. Expression of a dominant negative FGF receptor inhibits axonal growth and FGF receptor phosphorylation stimulated by CAMs. Neuron. 1997;18:231–242. doi: 10.1016/s0896-6273(00)80264-0. [DOI] [PubMed] [Google Scholar]
- 54.Schaefer AW, Kamiguchi H, Wong EV, Beach CM, Landreth G, Lemmon V. Activation of the MAPK signal cascade by the neural cell adhesion molecule L1 requires L1 internalization. J Biol Chem. 1999;274:37965–37973. doi: 10.1074/jbc.274.53.37965. [DOI] [PubMed] [Google Scholar]
- 55.Schmidt CE, Horwitz AF, Lauffenburger DA, Sheetz MP. Integrin-cytoskeletal interactions in migrating fibroblasts are dynamic, asymmetric, and regulated. J Cell Biol. 1993;123:977–991. doi: 10.1083/jcb.123.4.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schmidt CE, Dai J, Lauffenburger DA, Sheetz MP, Horwitz AF. Integrin-cytoskeletal interactions in neuronal growth cones. J Neurosci. 1995;15:3400–3407. doi: 10.1523/JNEUROSCI.15-05-03400.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sheetz MP, Baumrind NL, Wayne DB, Pearlman AL. Concentration of membrane antigens by forward transport and trapping in neuronal growth cones. Cell. 1990;61:231–241. doi: 10.1016/0092-8674(90)90804-n. [DOI] [PubMed] [Google Scholar]
- 58.Sheetz MP, Felsenfeld DP, Galbraith CG. Cell migration: regulation of force on extracellular-matrix-integrin complexes. Trends Cell Biol. 1998;8:51–54. doi: 10.1016/s0962-8924(98)80005-6. [DOI] [PubMed] [Google Scholar]
- 59.Sonee M, Barrón E, Yarber FA, Hamm-Alvarez SF. Taxol inhibits endosomal-lysosomal membrane trafficking at two distinct steps in CV-1 cells. Am J Physiol. 1998;275:C1630–C1639. doi: 10.1152/ajpcell.1998.275.6.C1630. [DOI] [PubMed] [Google Scholar]
- 60.Stallcup WB, Beasley L. Involvement of the nerve growth factor-inducible large external glycoprotein (NILE) in neurite fasciculation in primary cultures of rat brain. Proc Natl Acad Sci USA. 1985;82:1276–1280. doi: 10.1073/pnas.82.4.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Suter D, Forscher P. An emerging link between cytoskeletal dynamics and cell adhesion molecules in growth cone guidance. Curr Opin Neurobiol. 1998;8:106–116. doi: 10.1016/s0959-4388(98)80014-7. [DOI] [PubMed] [Google Scholar]
- 62.Suter DM, Errante LD, Belotserkovsky V, Forscher P. The Ig superfamily cell adhesion molecule, apCAM, mediates growth cone steering by substrate-cytoskeletal coupling. J Cell Biol. 1998;141:227–240. doi: 10.1083/jcb.141.1.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tanaka E, Sabry J. Making the connection: cytoskeletal rearrangements during growth cone guidance. Cell. 1995;83:171–176. doi: 10.1016/0092-8674(95)90158-2. [DOI] [PubMed] [Google Scholar]
- 64.Tanaka E, Ho T, Kirschner MW. The role of microtubule dynamics in growth cone motility and axonal growth. J Cell Biol. 1995;128:139–155. doi: 10.1083/jcb.128.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tessier-Lavigne M, Goodman CS. The molecular biology of axon guidance. Science. 1996;274:1123–1133. doi: 10.1126/science.274.5290.1123. [DOI] [PubMed] [Google Scholar]
- 66.Teter K, Chandy G, Quinones B, Pereyra K, Machen T, Moore HPH. Cellubrevin-targeted fluorescence uncovers heterogeneity in the recycling endosomes. J Biol Chem. 1998;273:19625–19633. doi: 10.1074/jbc.273.31.19625. [DOI] [PubMed] [Google Scholar]
- 67.Vogt L, Giger RJ, Ziegler U, Kunz B, Buchstaller A, Hermens WTJMC, Kaplitt MG, Rosenfeld MR, Plaff DW, Verhaagen J, Sonderegger P. Continuous renewal of the axonal pathway sensor apparatus by insertion of new sensor molecules into the growth cone membrane. Curr Biol. 1996;6:1153–1158. doi: 10.1016/s0960-9822(02)70682-9. [DOI] [PubMed] [Google Scholar]
- 68.Wessells NK, Nuttall RP. Normal branching, induced branching, and steering of cultured parasympathetic motor neurons. Exp Cell Res. 1978;115:111–122. doi: 10.1016/0014-4827(78)90408-1. [DOI] [PubMed] [Google Scholar]
- 69.Williams EJ, Doherty P, Turner G, Reid RA, Hemperly JJ, Walsh FS. Calcium influx into neurons can solely account for cell contact-dependent neurite outgrowth stimulated by transfected L1. J Cell Biol. 1992;119:883–892. doi: 10.1083/jcb.119.4.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Williamson T, Gordon-Weeks PR, Schachner M, Taylor J. Microtubule reorganization is obligatory for growth cone turning. Proc Natl Acad Sci USA. 1996;93:15221–15226. doi: 10.1073/pnas.93.26.15221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wong EV, Schaefer A, Landreth G, Lemmon V. Involvement of p90rsk in neurite outgrowth mediated by the cell adhesion molecule L1. J Biol Chem. 1996;271:18217–18223. doi: 10.1074/jbc.271.30.18217. [DOI] [PubMed] [Google Scholar]