Myosin 2 Is a Key Rho Kinase Target Necessary for the Local Concentration of E-Cadherin at Cell–Cell Contacts (original) (raw)
Abstract
Classical cadherins accumulate at cell–cell contacts as a characteristic response to productive adhesive ligation. Such local accumulation of cadherins is a developmentally regulated process that supports cell adhesiveness and cell–cell cohesion. Yet the molecular effectors responsible for cadherin accumulation remain incompletely understood. We now report that Myosin 2 is critical for cells to concentrate E-cadherin at cell–cell contacts. Myosin 2 is found at cadherin-based cell–cell contacts and its recruitment requires E-cadherin activity. Indeed, both Myosin 2 recruitment and its activation were stimulated by E-cadherin homophilic ligation alone. Inhibition of Myosin 2 activity by blebbistatin or ML-7 rapidly impaired the ability of cells to concentrate E-cadherin at adhesive contacts, accompanied by decreased cadherin-based cell adhesiveness. The total surface expression of cadherins was unaffected, suggesting that Myosin 2 principally regulates the regional distribution of cadherins at the cell surface. The recruitment of Myosin 2 to cadherin contacts, and its activation, required Rho kinase; furthermore, inhibition of Rho kinase signaling effectively phenocopied the effects of Myosin 2 inhibition. We propose that Myosin 2 is a key effector of Rho-Rho kinase signaling that regulates cell–cell adhesion by determining the ability of cells to concentrate cadherins at contacts in response to homophilic ligation.
INTRODUCTION
Simple things are sometimes surprisingly complex. This is exemplified by the relationship between cadherin adhesion and cell–cell contacts. Classical cadherins accumulate and concentrate at contacts between cells in many solid tissues of the body (Takeichi, 1991). At those contacts cadherins mediate cell–cell recognition, organize cells into coherent sheets and populations, and modulate morphogenetic processes such as polarity and locomotion. The ability of cells to concentrate their cadherins at contacts likely supports adhesive strengthening, stabilizes the cohesiveness of contacts and is commonly thought to be necessary for the assembly of adherens junctions. Consistent with its morphogenetic potential, such local concentration of E-cadherin within cell–cell contacts is a developmentally regulated event in the early mouse embryo that coincides with compaction (Vestweber et al., 1987). Despite this, the molecular and cellular mechanisms responsible for concentrating cadherins at contacts remain incompletely understood.
Cadherins function as membrane-spanning macromolecular complexes (Takeichi, 1991) and cooperation between cadherin ectodomains and cytoplasmic elements appears necessary to concentrate cadherins at contacts. The necessary requirement for a functional ectodomain was first suggested by the observation that cadherin mutants lacking a functional ectodomain distributed diffusely when expressed on the surfaces of cadherin-deficient fibroblasts (Fujimori and Takeichi, 1993). Subsequent biochemical and structural studies of cadherin ectodomains raised the possibility that protein-protein interactions within this region itself might themselves support oligomerization. The cadherin ectodomain can engage in both lateral (cis) interactions as well as in homophilic (trans) interactions. It was postulated that the combination of these _cis_- and _trans_-interactions might cause cadherins to coassemble into zipperlike arrays (Shapiro et al., 1995) or more complex three-dimensional structures (Gumbiner, 1996), resulting in their local concentration. However, it is also commonly observed that cadherin mutants lacking their cytoplasmic tails fail to accumulate in cell–cell contacts (Fujimori and Takeichi, 1993), despite retaining demonstrable homophilic binding activity and being expressed at the cell surface (Brieher et al., 1996; Yap et al., 1997). This indicates that, whatever the contribution(s) of the ectodomain (Boggon et al., 2002), cytoplasmic interactions are also necessary to concentrate cadherins at cell–cell contacts.
Key cytoplasmic contributions appear to involve interactions between cadherins, cell signaling pathways, and the actin-based cytoskeleton. Notably, members of the Rho family of small GTPases can exert significant effects on cadherin accumulation. Disruption of Rho signaling, in particular, caused E-cadherin to be rapidly lost from epithelial cell–cell contacts before the cohesive interactions between cells were disrupted (Braga et al., 1997, 2000; Takaishi et al., 1997; Charrasse et al., 2002). Furthermore, effectors of Rho signaling, including mDia and Rho kinase were found to support the integrity of cadherin-based cell contacts in some (Vaezi et al., 2002) studies, but not in all instances (Sahai and Marshall, 2002). Similarly, the integrity of the actin cytoskeleton is necessary for cadherin adhesion and cadherin mutants that cannot interact with α-catenin, the best-understood mechanism that binds cadherins to cortical actin, often fail to accumulate at cell–cell contacts (Sako et al., 1998). Given that Rho GTPases are fundamental regulators of the actin cytoskeleton, it is attractive to postulate that Rho may support local cadherin concentration through an impact on cadherin-actin interactions. However, it is becoming increasingly apparent that multiple dynamic forms of actin activity occur at cadherin adhesive contacts, accompanied by a range of actin regulators and actin-based effector molecules (Kovacs et al., 2002b; Vaezi et al., 2002). The precise targets of Rho that might support local cadherin accumulation, though, remain to be identified.
In the current report, we focused on the potential for Myosin 2 to regulate local cadherin accumulation at cell–cell contacts. Myosin 2 is a well-characterized target of Rho signaling that has been reported to localize to epithelial cell–cell junctions (Bertet et al., 2004; Conti et al., 2004; Zallen and Wieschaus, 2004) as well as to perijunctional actin cables (Krendel et al., 1999). Myosin 2 activity is implicated in remodelling cadherin-based cell–cell contacts during planar cell intercalation in Drosophila embryos (Bertet et al., 2004) and potentially also in cultured mammalian cells (Krendel et al., 1999). Strikingly, mouse embryos deficient in the Myosin 2A isoform, and embryoid bodies derived from such embryos, display decreased cell–cell cohesion and reduced E-cadherin staining at cell–cell contacts (Conti et al., 2004). We now report that homophilic cadherin ligation is sufficient to recruit Myosin 2 to cadherin adhesive contacts in a Rho-kinase-dependent manner, where it is necessary to support the local concentration of E-cadherin and the integrity of adherens junctions.
MATERIALS AND METHODS
Cell Culture, Transfections, and Adhesion Assays
MCF-7 and Chinese hamster ovary (CHO) cells stably transfected with human E-cadherin (hE-CHO) have been described previously (Kovacs et al., 2002a; Paterson et al., 2003). Cells were transfected using either Lipofectamine Plus Reagent or Lipofectamine 2000 according to manufacturer's instructions and analyzed 24–72 h after transfection. hE/Fc was prepared and used as previously described (Kovacs et al., 2002a).
Homophilic adhesion to hE/Fc-coated substrata was measured by resistance to detachment as previously described (Verma et al., 2004). In brief, nitrocellulose-coated six-well plates were incubated with hE/Fc (in Hanks' balanced salt solution [HBSS], containing 5 mM CaCl2) overnight at 4°C and then blocked with bovine serum albumin (10 mg/ml, 2 h at 4°C). Cells were isolated by incubation for 10 min in 0.01% (wt/vol) crystalline trypsin (Sigma, St. Louis, MO) in HBSS containing 5 mM CaCl2. Freshly isolated cells were allowed to attach to substrata for 90 min at 37°C in a CO2 incubator and then subjected to detachment by systematic pipetting. For this, five regions in each well (the 4 quadrants and center) were washed twice with 200 μl HBSS/CaCl2 delivered using a stand-mounted pipette. Cells remaining adherent to the wells were then incubated with MTT (10 mg/ml) dissolved in dimethyl sulfoxide and read at OD595 in a microplate reader. Cellular content in wells after pipetting was compared with the cellular content of wells prepared under identical conditions, but not subjected to pipetting (yielding the total number of cells plated in each well).
Plasmids
pEGFP-NMHC-2A (nonmuscle Myosin 2A heavy chain; GFP-Myosin 2A) was a kind gift from Dr. R. Adelstein (Wei and Adelstein, 2000). Human E-cadherin tagged with YFP at its C-terminus was generated by PCR amplification of the coding sequence for hE-cadherin using primers that introduced a 5′-_Hin_dIII site and 3′-_Sac_II site. This was subsequently subcloned into pEYFP-N1 (Clontech, Palo Alto, CA). CHO cells stably expressing hE-Cad-YFP were made by Lipofectamine transfection and selection with G418.
Antibodies
Primary antibodies were as follows: 1) Mouse monoclonal antibody (mAb) against the cytoplasmic domain of E-cadherin (Transduction Laboratories, Lexington, KY); 2) mouse mAb HECD-1 against the ectodomain of human E-cadherin (a kind gift from Dr. Peggy Wheelock with the permission of Dr. M. Takeichi); 3) rabbit pAb for human nonmuscle Myosin 2A heavy chain (Covance, Madison, WI); 4) rabbit pAb against GFP (Roche, Indianapolis, IN); 5) rabbit pAb raised against Thr18- and Ser19-phosphorylated MLC (ppMLC, a kind gift of Dr. J. Staddon) (Ratcliffe et al., 1999); 6) mouse mAb SHE 78–7 against human E-cadherin (Zymed, Laboratories, South San Francisco, CA); and 7) monoclonal anti-β-tubulin (Sigma). Species-specific secondary antibodies conjugated to Alexa-488 or Alexa-594 were obtained from Molecular Probes (Eugene, OR). F-actin was identified with TRITC-phalloidin.
Trypsin Protection Assay
The surface expression of E-cadherin was measured by sensitivity to surface trypsinization as described previously (Verma et al., 2004). In brief, cells were incubated with crystalline trypsin (0.05% wt/vol) in HBSS in the presence of either 2 mM CaCl2 or 5 mM EDTA for 20 min at 37°C. Cells were collected and lysed directly into Laemmli sample buffer. Equal volumes of the cellular extracts were separated by SDS-PAGE followed by Western analysis with antibodies specific for the ectodomain of E-cadherin (HECD-1) and β-tubulin (as a loading control).
Immunofluorescence Microscopy and Image Analysis
Where indicated, samples were briefly extracted with nonionic detergent before fixation. Briefly, cells were transferred to ice for 3 min, incubated with prepermeabilization buffer (0.5% TX-100, 10 mM Pipes pH 6.8, 50 mM NaCl, 3 mM MgCl2, 300 mM sucrose containing 1× Complete protease inhibitors [Roche]) for 10 min followed by fixation with paraformaldehyde (4% PFA in CSK) for 20 min at room temperature.
Epi-illumination fluorescence microscopy of fixed specimens was performed using IX81 microscopes fitted with 100×, 1.40 NA objectives. Images were acquired with Hamamatsu Orca-1 ER cameras (Bridgewater, NJ) driven by Metamorph imaging software (Version 6.2, Universal Imaging, West Chester, PA). Background correction and contrast manipulation of raw data images were performed in either ImageJ (version 1.30; NIH) or Adobe Photoshop (version 7; San Jose, CA). _Z_-stacks of epifluorescence images were deconvolved using the Autoquant Blind 3D deconvolution program (AutoQuant Imaging, Watervliet, NY).
The intensity of E-cadherin fluorescence at cell contacts was measured using the line scan function in Metamorph (Universal Imaging). A total of 50 lines, 120–160 pixels in length, were drawn through each junction, and the average pixel intensity for each position along the line was determined. A minimum of 20 junctions were analyzed per experiment, and the area under the curves was determined using Prism (v3.0; Graph Pad, Sorrento, CA) and Microsoft Excel (Redmond, WA). Experiments were performed three times. To quantitate cell extension on hE/Fc-coated substrata (Helwani et al., 2004), the outer margins of broad cadherin-based lamellipodia were identified by phalloidin staining. For each cell the lengths of each outer margin were measured and summed, and the total length of the outer margins expressed as a percentage of the perimeter of that cell (Lamellipodial index [LI]).
Live Cell Imaging
E-Cad-YFP cells were plated onto 25-mm round coverslips (Lomb Scientific and Co., Taren Point, Australia) and housed throughout the course of movie capture in a custom-built water-jacketed chamber maintained at 37.4°C. Cells were incubated in phenol red-free HBSS/Ca containing 0.05% fetal calf serum, buffered with 10 mM HEPES (pH 7.4). Epifluorescent live cell imaging was performed using an Olympus IX81 equipped with 60X and 100X PLAN-Apo (NA 1.4) objectives (Lake Success, NY). Images were captured with a Hamamatsu Orca1-ER camera driven by Metamorph software. For live cell imaging of blebbistatin-treated cells, images of cells were taken immediately before, and 1 h after, addition of blebbistatin (10 μM) using identical exposures (600 ms) and camera acquisition settings. Intensity of E-cadherin-YFP at cell–cell contacts was determined using the line-scan analysis function in Metamorph (version 6.2), as described above.
Fluorescence recovery after photobleaching (FRAP) analysis utilizing the aforementioned live cell imaging system has been previously reported (Helwani et al., 2004). Briefly, junctions were identified under low light, and a prebleach image was captured. Immediately afterward the iris field diaphragm was then closed to its minimum diameter and the light path was cleared of filters, enabling exposure of the selected region of interest (ROI) to maximal light from the mercury burner for no more than 25 s. FRAP time series movies were then commenced, with images captured every 30 s for a total of 20–25 min. Where inhibitors were used to assess their impact on junctional E-cadherin accumulation, cells were pretreated for 60 min at 37°C before transfer to the heated slide mount and incubated in media containing the appropriate drug. FRAP analysis was performed in ImageJ as previously described (Helwani et al., 2004). Raw data were normalized as detailed in Rabut and Ellenberg (2005). Briefly, raw data were first adjusted by background subtraction at each time point, corrected to a time-matched ROI that had not been photobleached, and then normalized to the background-subtracted prebleach image. Kinetic modeling was performed in Prism, using the equation detailed in Table 1.
Table 1.
FRAP analysis of E-cadherin dynamic behavior at cell-cell contacts
| Treatment | % Recoverya | Rate constant of recovery (min-1) | _t_1/2 (min) |
|---|---|---|---|
| Control | 64.84 ± 4.7 | 0.069 ± 0.009 | 10.8 ± 1.8 |
| ML-7 (10 μM) | 35.6 ± 4.1b | 0.013 ± 0.002b | 57.0 ± 8.7b |
| Y27632 (10 μM) | 32.4 ± 6.9c | 0.014 ± 0.006b | 42.5 ± 9.6c |
RESULTS
Myosin 2 Accumulates at Cell–Cell Junctions in an E-Cadherin-dependent Manner
We began by comparing the cellular localization of Myosin 2 and E-cadherin in MCF-7 mammary epithelial monolayers (Figure 1). Because similar results were obtained for both the Myosin 2A and Myosin 2B isoforms, only data for Myosin 2A are shown here. Indirect immunofluorescence microscopy revealed that endogenous Myosin 2A distributed extensively throughout the cells, with prominent staining in radial striations in the periphery of the cytoplasm (unpublished data), as observed by others (Vaezi et al., 2002); this radial pattern was characteristically found at the basal regions of the cells. In addition, Myosin 2 also localized in the immediate proximity of E-cadherin-enriched cell–cell contacts, where it stained in puncta that overlapped with some, but not all, areas of cadherin staining (Figure 1A). Preextraction of MCF-7 monolayers with nonionic detergents before fixation substantially reduced the cytoplasmic pool of endogenous Myosin 2A (unpublished data), revealing a residual pool found with E-cadherin at cell–cell contacts. Similarly transiently expressed EGFP-Myosin 2A was clearly visible at cadherin adhesions in preextracted samples (Figure 1B). Together, these data suggested that a subpopulation of Myosin 2 can accumulate at cadherin-based cell–cell contacts, both in the actin-rich perijunctional regions of the contacts and at more limited sites within the contacts themselves.
Figure 1.

Myosin 2 accumulates at cell–cell contacts in an E-cadherin-dependent manner. (A) Endogenous Myosin 2A localizes in puncta at cadherin-based cell–cell contacts. Confluent MCF-7 monolayers were fixed and labeled with antibodies specific for E-cadherin and Myosin 2A. (B) Exogenous GFP-tagged Myosin 2A also localizes in regions within E-cadherin-based cell–cell contacts. MCF-7 cells were transiently transfected with EGFP-tagged Myosin 2A, briefly extracted with TX-100 before fixation and costained both for E-cadherin and the GFP epitope tag (GFP-Myosin 2A). A single image plane is shown after 3D deconvolution. (C) E-cadherin is necessary for Myosin 2A localization at cell–cell contacts. Confluent MCF-7 cell monolayers were incubated in medium alone (No antibody) or in the presence of the E-cadherin function-blocking SHE78–7 mAb (1:50) for 15 min. E-cadherin was detected using the prebound blocking antibody, whereas Myosin 2A was detected using isoform-specific antibodies. Exposure to SHE78–7 substantially reduced and reorganized Myosin 2A staining at contacts that still retained E-cadherin.
Furthermore, E-cadherin activity was necessary for Myosin 2 to accumulate at cell–cell contacts. Incubation of MCF-7 monolayers with the function-blocking anti-E-cadherin antibody SHE 78–7 (Kovacs et al., 2002b) caused much Myosin 2 staining to be lost from cell–cell contacts within 15–30 min, when E-cadherin remained still detectable at the contacts (Figure 1C). Longer exposure to blocking antibodies caused cadherin to be lost from contacts and eventually disrupted the cohesiveness of the contacts themselves.
E-Cadherin Homophilic Ligation Is Sufficient to Recruit and Activate Myosin 2A
To extend these observations, we then asked whether E-cadherin homophilic adhesion might be capable of recruiting Myosin 2A to the cell cortex, using a recombinant cadherin ligand (hE/Fc), consisting of the complete ectodomain of human E-cadherin fused to the Fc region of IgG (Kovacs et al., 2002a, 2002b). When adsorbed on latex beads, hE/Fc and similar cadherin ligands (Brieher et al., 1996; Lambert et al., 2002; Niessen and Gumbiner, 2002) can bind to the dorsal surfaces of E-cadherin-expressing cells, thereby presenting spatially confined adhesive platforms that recruit a range of actin-regulatory and signaling molecules, without detectable integrin activation (Yap et al., 1997; Kovacs et al., 2002a). As reported previously, binding of hE/Fc-coated beads is sufficient to induce the E-cadherin molecular complex (as marked by staining for β-catenin) to accumulate at sites of adhesion, where it characteristically stained as a “flare” in the vicinity of the bead (Figure 2A, arrowheads). No comparable β-catenin accumulation was observed where cells bound to latex beads coated with the nonspecific ligand, concanavalin A (ConA). Marked recruitment of EGFP-Myosin 2A was observed at sites where E-cadherin-expressing cells bound to hE/Fc-coated beads, compared with ConA controls (Figure 2A). This suggested strongly that cadherin homophilic ligation was sufficient to recruit Myosin 2 into adhesive contacts.
Figure 2.
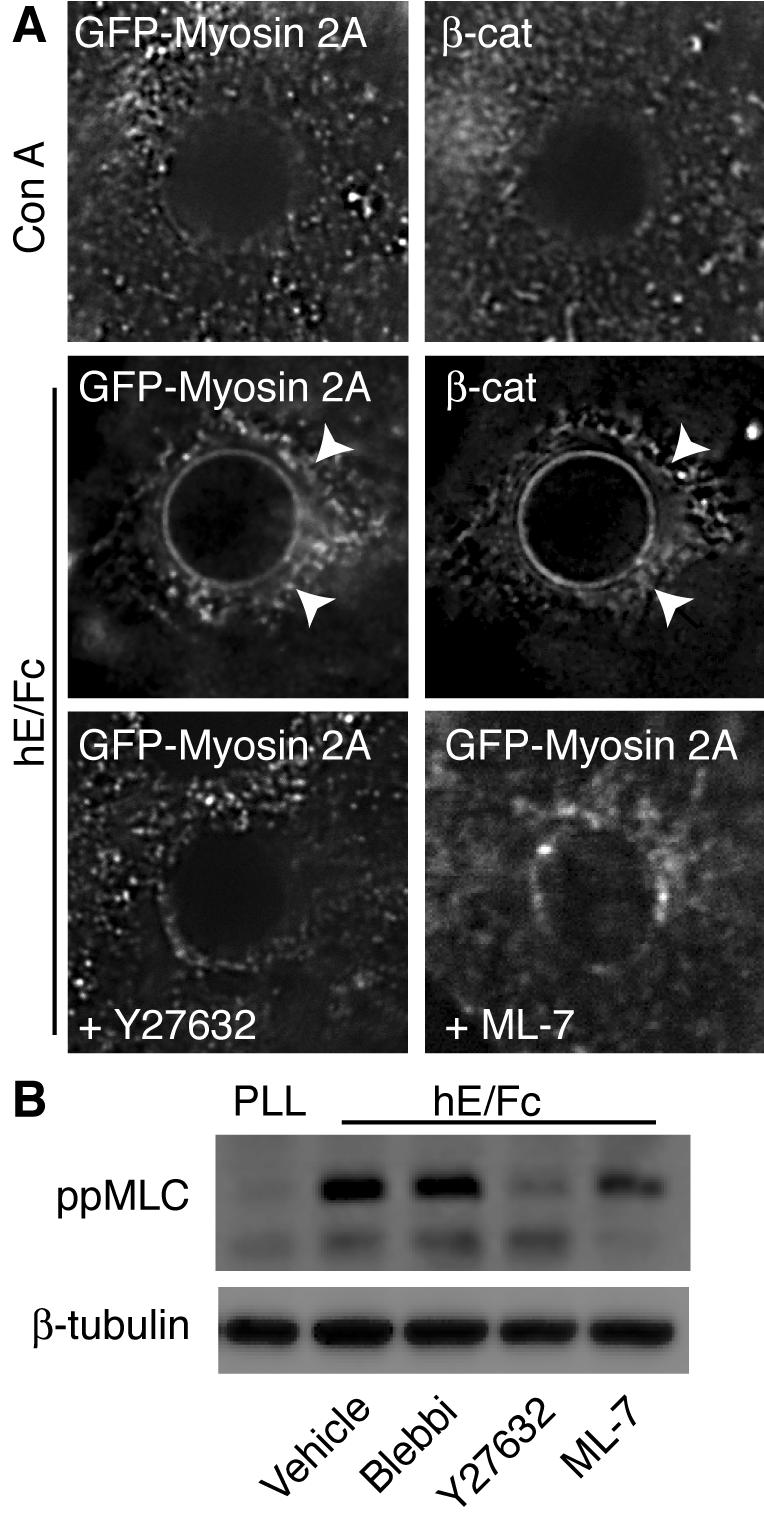
E-cadherin homophilic ligation is sufficient to recruit and activate Myosin 2A. (A) E-cadherin homophilic ligation triggers the recruitment of Myosin 2A to sites of adhesion. Latex beads coated with either hE/Fc or ConA were allowed to adhere for 90 min to the dorsal surface of hE-CHO cells transiently expressing GFP-tagged Myosin 2A (GFP-Myosin 2A). Cells were then immunolabeled with antibodies specific for GFP and β-catenin (marking the cadherincatenin complex). Both GFP-Myosin 2A and the cadherin complex show significantly greater accumulation in flares around the hE/Fc-beads (arrowheads) when compared with ConA-coated control beads. Incubation with either Y-27632 (10 μM) or ML-7 (10 μM) markedly reduced the recruitment of GFP-Myosin 2A to cadherin homophilic adhesions. (B) E-cadherin homophilic ligation activates Myosin 2. Cells were allowed to adhere to dishes coated with either poly-l-lysine (PLL) or hE/Fc in the presence of blebbistatin (Blebbi, 10 μM), Y-27632 (10 μM), or ML-7 (10 μM) for 90 min. Western blots from cell lysates were probed for di-phosphorylated, activated myosin light chain (ppMLC) or β-tubulin (as a loading control).
Myosin 2 is activated by phosphorylation of the regulatory myosin light chain (MLC), an event that responds to both myosin light chain kinase (MLCK) and Rho kinase signaling (Bresnick, 1999; Ueda et al., 2002). Phosphorylated MLC (ppMLC) was detectable at cell–cell contacts by immunofluorescence (see Figure 6B), whereas Myosin 2A recruitment to sites of hE/Fc-induced adhesion was largely abolished in cells treated with either Y-27632 or ML-7 (Figure 2A), inhibitors of Rho kinase and MLCK, respectively. This suggested that activation of Myosin 2 was necessary for its recruitment by cadherin adhesion.
Figure 6.

Myosin 2 and activated MLC localizes to cell–cell contacts in response to Rho kinase signaling. MCF-7 monolayers were treated with 10 μM Y-27632 for 30 min, fixed, and processed for immunolocalization of endogenous Myosin 2A and E-cadherin (A) or ppMLC and E-cadherin (B). Strikingly, Y-27632 treatment reduced significantly the accumulation of both Myosin 2 and ppMLC at cell–cell contacts.
We then used activation-specific ppMLC antibodies (Ratcliffe et al., 1999) to test whether cadherin homophilic ligation could stimulate Myosin 2 activity. Freshly isolated MCF-7 cells were allowed to adhere for 90 min to substrata coated with either hE/Fc or poly-l-lysine (PLL), before lysis and separation of polypeptides by SDS-PAGE. As shown in Figure 2B, conspicuously higher levels of activated ppMLC were detected in cells adherent to hE/Fc compared with those attached to PLL, indicating that cadherin homophilic adhesion could activate Myosin 2. MLC phosphorylation upon adhesion to hE/Fc was reduced in cells treated with either Y-27632 or ML-7. Although at the concentrations used ML-7 did not reduce ppMLC levels to the same extent as Y-27632, it substantially reduced the amount of Myosin 2 detectable at cell–cell contacts (Supplementary Figure 1), indicating that ML-7 could effectively perturb Myosin 2 activity in cells. In contrast, stimulation of Myosin 2 phosphorylation was not affected by blebbistatin, a specific inhibitor of Myosin 2 ATPase activity (Straight et al., 2003) that does not directly affect the phosphorylation status of MLC. Taken together, these findings indicate that E-cadherin homophilic ligation is sufficient to recruit and activate Myosin 2, processes which involve both MLCK and Rho kinase signaling.
Inhibition of Myosin 2 Motor Activity Perturbs the Local Concentration of E-Cadherin at Cell–Cell Contacts
To assess the functional impact of Myosin 2, we first directly inhibited Myosin 2 motor activity with blebbistatin (Figure 3). Strikingly, blebbistatin (100 μM) rapidly altered the immunofluorescent staining of E-cadherin in confluent MCF-7 monolayers. Instead of accumulating in the characteristic intense circumferential “chicken-wire” pattern seen in the apical regions of control cultures (Figure 3A), E-cadherin staining in the apical regions of drug-treated cells appeared less intense and concentrated, often being interrupted by discontinuities (Figure 3A). In addition, E-cadherin staining in blebbistatin-treated cells often extended away from the apical contacts, in what appeared to represent overlapping cell–cell contacts (Figure 3A, arrow). Quantitation revealed that blebbistatin reduced the fluorescence intensity of E-cadherin staining at cell–cell contacts by ∼50% within 60 min of treatment, a reduction that persisted for several hours (Figure 3A). However, neither the total cellular level of E-cadherin, nor the amount expressed at the cell surface measured using surface trypsin sensitivity assays, were affected by blebbistatin (Figure 3B). This indicated that inhibition of Myosin 2 initially caused E-cadherin to redistribute on the surface away from cell–cell contacts.
Figure 3.

Myosin 2 motor activity is necessary for the local concentration of E-cadherin at cell–cell contacts. (A) Blebbistatin perturbs E-cadherin accumulation in cell–cell contacts. MCF-7 cells were treated with blebbistatin in complete growth media for varying periods of time and then fixed and processed for E-cadherin immunofluorescence. Representative images of cells treated with drug or control for 60 min are shown. Note that although vehicle-treated cells showed continuous chicken-wire E-cadherin staining (arrowheads), in drug-treated cells the apical cadherin staining was less intense and often discontinuous (arrowhead), whereas E-cadherin staining also extended away in overlapping cell–cell contacts. E-cadherin fluorescence intensity at cell–cell contacts (Junctional E-cadherin) was measured by digital image analysis of drug-treated and control cultures (data are means ± SE). (B) Inhibition of Myosin 2 does not affect the surface expression of E-cadherin. MCF-7 cells were treated with blebbistatin (100 μM) for 90 min before exposure to crystalline trypsin for 20 min in the presence of either 2 mM CaCl2 (Ca) or 5 mM EGTA (E). Lysates from these cells and untreated cells (SM, starting material) were subjected to SDS-PAGE and immunoblotted for E-cadherin and β-tubulin (as a loading control).
As a further test of this notion, we used live cell imaging to test how blebbistatin affected cadherin accumulation at contacts between CHO cells stably expressing E-cadherin tagged with YFP at its C-terminus (hE-Cad-YFP-CHO cells; Figure 4). Like other GFP-tagged cadherins (Adams et al., 1998), E-Cad-YFP was efficiently expressed at the surface of CHO cells, typically accumulating in puncta at cell–cell contacts (Figure 4). E-Cad-YFP also associated with catenins and conferred cadherin-specific adhesion on these cells that otherwise lack significant levels of endogenous classical cadherins (unpublished data). Because visible light is reported to have complex interactions with blebbistatin (Kolega, 2004; Sakamoto et al., 2005), we briefly imaged individual contacts immediately before, and 60 min after, treatment with the drug, thereby minimizing cellular exposure to light. E-Cad-YFP puncta were less apparent and fluorescence intensity at contacts was reduced to 58.25 ± 4.7% of baseline in blebbistatin-treated cells, whereas cadherin fluorescence was unchanged in control cultures (Figure 4). E-Cad-YFP fluorescence was also reduced to a similar extent when multiple different contacts were sampled before and after exposure to blebbistatin (ensuring that no individual contact was imaged twice; unpublished data). Trypsin sensitivity assays showed that E-Cad-YFP remained on the cell surface (unpublished data), indicating that the protein was redistributing to nonjunctional regions of the cell surface, consistent with what we observed for endogenous E-cadherin in MCF-7 cells (Figure 3). Taken together, these data indicate that inhibition of Myosin 2 caused E-cadherin to be lost from cell–cell contacts and redistribute to extrajunctional regions of the cell surface.
Figure 4.

Blebbistatin perturbs the accumulation of YFP-tagged E-cadherin at cell–cell contacts. CHO cells stably expressing E-Cad-YFP were examined by live-cell imaging. Individual contacts were imaged (600-ms exposures) before, and 1 h after, addition of blebbistatin (10 μM) using identical camera acquisition settings. Fluorescence intensity at contacts was determined as described in Materials and Methods; fluorescence intensity after blebbistatin (E-Cad-YFP accumulation) was expressed as a percentage of fluorescence intensity before the drug; n = 10–14, p < 0.001 (Student's t test). E-Cad-YFP accumulated in prominent puncta at cell–cell contacts. Puncta were less prominent (arrows), and fluorescence intensity at contacts reduced, after treatment with blebbistatin.
Activation of Myosin 2 by MLCK Is Necessary for E-Cadherin to Accumulate at Cell–Cell Contacts
We then asked whether inhibiting MLCK, the principal kinase responsible for phosphorylating the regulatory MLC (Bresnick, 1999), also affected cadherin-based cell–cell contacts. Immunofluorescent staining revealed that ML-7 (10 μM) significantly reduced the intensity of endogenous E-cadherin staining at contacts between MCF-7 cells (Figure 5A), similar to the effect of blebbistatin. This implied that activation of Myosin 2 by MLCK was necessary for surface E-cadherin to concentrate at cell–cell contacts. To test this notion further we used FRAP analysis to assess the kinetics of steady-state cadherin accumulation at contacts between hE-Cad-YFP-CHO cells (Figure 5, B and C, and Table 1). E-Cad-YFP fluorescence in control cells rapidly recovered after photobleaching of cell–cell contacts, with ∼65% recovery achieved within the 20–25-min observation period (Table 1). In contrast, E-Cad-YFP fluorescence recovery was significantly retarded in cells treated with ML-7 (10 μM): only ∼35% recovery was attained within the 24-min observation period (Figure 5C). Kinetic modeling revealed an approximately fivefold decrease in the rate of recovery of E-Cad-YFP fluorescence at contacts of ML-7-treated cells, when compared with recovery in control cells (Table 1). Together, these data provide further evidence that Myosin 2 participated in the preferential accumulation of E-cadherin at cell–cell contacts.
Figure 5.

Myosin light-chain kinase activity is necessary for local accumulation of E-cadherin at cell–cell contacts. (A) Inhibition of MLCK affects E-cadherin accumulation at cell–cell contacts. MCF-7 monolayers were incubated with ML-7 (10 μM) or vehicle for 60–90 min then processed for E-cadherin immunofluorescence and quantitation. One set of cells (Recovery) were treated with drug for 60 min and then allowed to recover in fresh drug-free medium before fixation. (B) ML-7 affects recovery of E-cadherin-YFP fluorescence after photobleaching of cell–cell contacts. Contacts between CHO cells stably expressing E-cadherin-YFP were photobleached, and the subsequent recovery of cadherin fluorescence was monitored by time-lapse epi-fluorescence microscopy. Cells were incubated with ML-7 (10 μM) or vehicle alone before experiments. Representative images from movies are shown; circles mark the photobleached areas. (C) E-cadherin-YFP fluorescence recovery at cell–cell contacts after photobleaching was calculated and modeled as detailed in Materials and Methods.
Myosin 2 Supports Local Accumulation of E-Cadherin as an Effector of Rho Kinase Signaling
Myosin 2 is a well-documented effector of Rho signaling, whose influence is mediated by Rho kinase. Both Rho and Rho kinase activity have been observed to affect cadherin organization at cell–cell contacts (Fukata and Kaibuchi, 2001; Vaezi et al., 2002), suggesting that the observed impact of Myosin 2 on local cadherin accumulation might reflect its role as an effector of Rho kinase. To test this, we first examined whether inhibiting Rho kinase affected the localization of Myosin 2 at cell–cell contacts. As shown in Figure 6A, endogenous Myosin 2A at cadherin-based cell–cell contacts was clearly reduced in cells acutely treated with Y-27632. Moreover, whereas activation-specific ppMLC staining was readily identified at cadherin-based contacts between control cells, this was largely abolished by Y-27632 (Figure 6B). Taken with our observation that Rho kinase inhibition inhibited the recruitment and activation of Myosin 2 by E-cadherin homophilic ligation (Figure 2B), this indicated that Myosin 2 at cell–cell contacts responds to a Rho kinase-dependent signaling pathway.
If Myosin 2 regulates the local concentration of E-cadherin as an effector of Rho kinase signaling, we predicted that inhibition of Rho kinase should mimic the effects of inhibiting Myosin 2. Indeed, the intensity of endogenous E-cadherin staining at contacts between MCF-7 cells was significantly reduced by Y-27632 (Figure 7A). Similarly, recovery of E-Cad-YFP fluorescence after photobleaching was reduced by Y-27632 to a comparable degree as ML-7 (Figure 7B, Table 1). But despite these changes in local accumulation of cadherin, Y-27632 did not affect the surface expression of E-cadherin in MCF-7 cells as measured using surface trypsin sensitivity assays (Figure 7C). Therefore inhibiting Rho kinase signaling had effects on the surface distribution of E-cadherin that were identical to those seen when Myosin 2 activity was perturbed.
Figure 7.

Rho kinase signaling is necessary for the local accumulation of E-cadherin at cell–cell contacts. (A) Accumulation of endogenous E-cadherin at contacts between MCF-7 cells treated with Y-27632 (10 μM) for 0–6 h. E-cadherin accumulation at cell–cell contacts was measured by quantitative immunofluorescence microscopy. Data are means ± SE. (B) FRAP analysis of E-cadherin-YFP accumulation at contacts between hE-YFP-CHO cells treated with either Y-27632 (10 μM) or vehicle alone. (C) Surface expression of E-cadherin in MCF-7 cells treated with Y-27632 (10 μM) or vehicle alone was measured by surface trypsin protection assays (SM, starting material, E, trypsinization in the presence of EGTA). All the cadherin remained accessible to surface trypsinization in both control and drug-treated cells. These data were representative of three independent experiments.
Myosin 2 Activity Is Necessary for Cadherin Adhesion and Contact Formation in Response to Homophilic Ligation
These findings pointed to a positive contribution of Myosin 2 to cadherin-based cell–cell contacts. In contrast, other studies identified a role for myosin-based contractility in disrupting contacts between cancer cells in monolayer culture (Avizienyte et al., 2004). Those studies, like ours, used global inhibitors of myosin activity, which carry the potential interpretive caveat that it is difficult to distinguish the effects that this ubiquitous motor may have on cadherin function from those potentially associated with integrin-based adhesion, transcellular contractility, or trafficking. To further test the notion that Myosin 2 activity can positively affect cadherin function, we therefore examined the impact of perturbing Myosin 2 function in cadherin-specific assays using planar substrata coated with hE/Fc (Figure 8). This allowed us to isolate effects on cadherin function independent of contractile events linked to cell-matrix adhesion.
Figure 8.

Myosin 2 activity is necessary for cadherin homophilic adhesion. (A) Inhibition of Myosin 2 activity affects E-cadherin-mediated adhesion measured by resistance to detachment from cadherin-coated substrata. hE-CHO cells were plated onto substrata coated with hE/Fc and allowed to adhere in the presence or absence of blebbistatin (10 μM), Y-27632 (10 μM), or ML-7 (10 μM) for 90 min. Cells were then detached and residual adherent cells measured as described in Materials and Methods. The numbers of adherent cells in drug-treated samples were normalized to controls subjected to the same experimental manipulations. Experiments were performed in triplicate on at least three separate occasions. (B) Myosin activity is necessary for E-cadherin-mediated contact zone extension. Freshly isolated hE-CHO cells were allowed to adhere to and extend contacts upon hE/Fc-coated substrata for 90 min in the presence of blebbistatin (10 μM), Y-27632 (10 μM), or ML-7 (10 μM). The ability of cells to extend contacts (Lamellipodial Index) was measured on phalloidin-stained samples as described in Materials and Methods. Data are means ± SE (n = 30–35). Insets show representative images of phalloidin-stained control and blebbistatin-treated cells; the arrowhead indicates a broad cadherin-based lamella.
As previously described (Kovacs et al., 2002b; Verma et al., 2004), freshly isolated cadherin-containing cells rapidly adhered to, and extended broad lamellipodia upon, hE/Fc-coated substrata (Figure 8B, arrowhead). Formation of lamellipodia in this assay system is inhibited by function-blocking anti-cadherin antibodies (Kovacs et al., 2002b), indicating that these are cadherin-dependent lamellipodia. Addition of blebbistatin (10 μM), ML-7 (10 μM), or Y-27632 (10 μM) to freshly isolated cells at the beginning of the assays significantly decreased the strength of cadherin adhesion as measured by the resistance of cells to detachment from hE/Fc-coated substrata (Figure 8A). This was associated with a marked reduction in the ability of cells to extend their adhesive contact zones, as measured by the formation of cadherin-based lamellipodia (Figure 8B). Together, these data indicated that Myosin 2 activity contributes positively to cadherin adhesive function, independently of other effects that it has in cells.
Myosin 2 Activity Preferentially Supports Actin Bundles at Cadherin Adhesive Contacts
We then sought further insight into the potential mechanism by which Myosin 2 might affect the ability of cadherins to accumulate within adhesive contacts. Because Myosin 2 is an actin-based motor that mediates cytoskeletal contractility, we first examined the effect of perturbing Myosin 2 activity in hE-CHO cells adherent to hE/Fc-coated substrata (Figure 9). Phalloidin staining revealed two patterns of actin organization in control cells: broad bands of F-actin found at the outer margins of cadherin-based lamellipodia and prominent bundles that traversed the cells (Figure 9A). Cellular E-cadherin stained throughout the adhesive interfaces, typically being found as discontinuous linear streaks at the very outer margins of cadherin-based lamellipodia and as lateral clusters of varying size within the contact zones. The lateral clusters appeared to exist as two qualitatively distinct types: large streaklike clusters (“macroclusters”) that typically formed the termini of prominent actin bundles (Figure 9, A and B, arrowheads) and finer puncta that were often independent of actin bundles (Figure 9A). To assess which of these structures might require Myosin 2, we allowed cells to first adhere and spread on hE/Fc-coated substrata for 60 min, sufficient for the cells to form cadherin clusters and their associated F-actin bundles (Figure 9B). Adherent cells were then treated with blebbistatin, Y-27632, or ML-7 for a further 30 min before fixation. All drugs largely abolished both the streaklike macroclusters and the actin bundles associated with them (Figure 9B), although the F-actin staining at the leading edges of lamellipodia was preserved.
Figure 9.

Myosin 2 activity supports cadherin-based actin bundles in homophilic adhesion assays. (A) Two patterns of F-actin organization are evident in hE-CHO cells adherent to hE/Fc-coated substrata. Cells were allowed to adhere to hE/Fc for 90 min before fixation and immunostaining for cellular E-cadherin using antibodies directed against the cytoplasmic tail and F-actin (phalloidin). Phalloidin staining revealed dense bands of F-actin at the outer margins of cadherin-dependent lamellipodia and also prominent bundles that typically terminated in large cadherin clusters (macroclusters, arrowheads). (B) Lateral organization of cellular E-cadherin and formation of actin bundles at cadherin adhesive interfaces requires Myosin 2. hE-CHO cells that had adhered to hE/Fc-coated coverslips for 60 min were then treated with the indicated drugs (all at 10 μM) for a further 30 min. Cadherin macroclusters (arrowheads) and the actin bundles they demarcate were largely abolished in drug-treated cells.
These observations suggested that Myosin 2 might be especially necessary to support actin cables that are found at cadherin adhesive contacts. To pursue this, we then examined the effect of blebbistatin on F-actin organization at native cell–cell contacts. Analogous to what we had seen in hE/Fc adhesion assays, two patterns of F-actin were identifiable at cell–cell contacts in confluent MCF-7 monolayers: prominent perijunctional actin cables that often terminated in puncta of E-cadherin, and finer cortical staining at the very cadherin adhesions themselves (Figure 10A, arrows). Both patterns were most readily evident at the very apical regions of cell–cell contacts. Characteristically, Myosin 2 staining at cell–cell contacts demarcated actin bundles as well as the more diffuse F-actin staining found immediately at the cell–cell contacts (unpublished data). Blebbistatin (10 μM, 90 min) substantially reduced the perijunctional cables, but cortical F-actin staining was still detectable at the adhesive contacts (Figure 10B, arrowheads). Similar effects were seen with Y-27632 and to a lesser extent with ML-7 (unpublished data). Taken together, these observations suggest that Myosin 2 may preferentially support the actin cables that form at cadherin adhesive contacts.
Figure 10.

Blebbistatin perturbs perijunctional actin cables. Confluent MCF-7 monolayers were stained for E-cadherin or F-actin as indicated. In control cells F-actin was found in prominent perijunctional actin cables (arrowheads) as well as in fine cortical bands (arrows) that localized with E-cadherin itself (arrows). Blebbistatin (10 μM, 90 min) abolished many of the perijunctional actin bundles, but residual cortical F-actin staining was evident at the cell–cell contacts (arrows).
DISCUSSION
Classical cadherins do not distribute passively on the cell surface; instead, they accumulate at cell–cell contacts as a characteristic response to productive adhesive ligation. Such focal concentration of cadherin requires that the intrinsic binding activity of the cadherin ectodomain be functionally linked to essential cytoplasmic determinants, whose nature is incompletely understood. Our current data now identify Myosin 2 as a critical regulator of cadherin accumulation that is coupled to homophilic adhesive ligation through a Rho kinase signaling pathway. Myosin 2 is thus centrally placed to mediate the contributions of cell signaling and the actin cytoskeleton previously implicated in supporting cadherin accumulation at cell–cell contacts.
Myosin 2 Is Necessary for E-Cadherin to Accumulate at Cell–Cell Contacts
We found that the ability of cells to concentrate cadherins at cell–cell contacts was consistently impaired when Myosin 2 activity was either inhibited directly with blebbistatin, or indirectly, by using ML-7 to block MLCK, which is immediately responsible for phosphorylating MLC. Instead of staining as intense bands confined to the apical regions of MCF-7 cell–cell contacts, E-cadherin expression at contacts was reduced in cells treated with either drug, often being replaced by more diffuse punctate staining when contacts were viewed en face. Similarly, in live cell imaging studies the intensity of E-Cad-YFP accumulation at contacts was significantly reduced by blebbistatin. Characteristically this occurred within 30–60 min of treatment with drugs, suggesting that ongoing Myosin 2 activity is necessary to maintain cadherins at cell–cell contacts. Similar effects on endogenous E-cadherin were also seen in cells expressing a dominant negative Myosin 2 mutant (unpublished data). These findings are consistent with the recent report that embryos lacking Myosin 2A showed an adhesive defect accompanied by a loss of cadherin concentration at cell–cell contacts (Conti et al., 2004).
Furthermore we found that ML-7 reduced significantly the rate at which E-cadherin-YFP fluorescence recovered at cell–cell contacts after photobleaching. Note that in these experiments we used FRAP as an independent kinetic index of local cadherin accumulation, rather than to measure the diffusional properties of the cadherin. We reasoned that when cells are at steady state, differences in the rates of E-Cad-YFP fluorescence recovery would reflect differences in the ability of cadherin to preferentially accumulate at the contacts. The reduced fluorescence recovery that we observed might be due to changes in either the surface movement of cadherin into the photobleached contact area and/or in its retention there. Irrespective of the precise mechanism, the impaired recovery of E-cadherin-YFP fluorescence implies a defect in the capacity of cells to locally accumulate cadherin at contacts. Thus, measured by two independent assays, our data indicate that Myosin 2 activity is necessary for E-cadherin to locally accumulate at cell–cell contacts.
Formally, the ability of cells to concentrate cadherins at contacts might reflect traffic of cadherins between the plasma membrane and the cytoplasm and/or mechanisms that control the regional distribution of cadherins once it is at the cell surface. Myosin 2 is certainly implicated in a range of cell trafficking pathways that potentially affect cadherin expression (Stow et al., 1998; Togo and Steinhardt, 2004). These include a role in supporting E-cadherin endocytosis when cell–cell contacts are broken or being remodeled (Ivanov et al., 2004). Directed transport of cadherins to cell–cell contacts has also been reported in some (Mary et al., 2002; Chen et al., 2003), but not all, studies (Adams et al., 1998), and Myosin 2 participates in several steps during exocytic transport (Stow et al., 1998), although whether this affects cadherin transport has not yet been tested. However, if Myosin 2 were principally determining the local accumulation of E-cadherin through cadherin trafficking, then we would predict that inhibiting Myosin 2 should affect the surface expression of E-cadherin. Specifically, inhibition of E-cadherin endocytosis should increase E-cadherin levels at the cell surface, whereas disruption of exocytosis would reduce surface cadherin expression. However, we detected no changes in surface E-cadherin expression, by either trypsin-sensitivity or surface biotinylation assays (unpublished data), under conditions where local concentration of cadherin at contacts was substantially reduced. These data suggest instead that surface cadherins redistribute away from cell–cell contacts as the dominant early consequence of inhibiting Myosin 2. Thus, although we cannot exclude subtle effects on cadherin trafficking, our data indicate that Myosin 2 exerts a major influence to control the regional localization and accumulation of E-cadherin when it is at the cell surface.
Myosin 2 Is an Effector of Rho Kinase Signaling at Cadherin Contacts
The effect of Myosin 2 inhibition that we observed resembled closely the rapid loss of E-cadherin from cell–cell contacts that occurs in a variety of cell types when Rho is inhibited (Braga et al., 1997, 2000; Takaishi et al., 1997; Charrasse et al., 2002). Although it has been recognized for several years that Rho signaling can critically support cadherin-based cell–cell contacts (Fukata and Kaibuchi, 2001), the molecular targets responsible for this activity have remained obscure. Of note, though, Myosin 2 is a major effector for Rho signaling in cells. Rho acts through Rho kinase, which stimulates Myosin 2 by inhibiting myosin phosphatase and possibly also by phosphorylating MLC itself (Amano et al., 1996; Kimura et al., 1996). The final outcome is activation of myosin binding to actin filaments after the phosphorylation of the regulatory myosin light chain.
Three lines of data indicate that this canonical Rho kinase pathway determined the ability of Myosin 2 to support local concentration of cadherins in our experiments. First, inhibition of Rho kinase signaling blocked the recruitment of Myosin 2 both to cadherin-based cell–cell contacts and to sites of homophilic E-cadherin adhesion. Second, activation of Myosin 2 at cadherin contacts was sensitive to Rho kinase signaling. Treatment of cells with Y-27632, a reasonably specific and widely used inhibitor of Rho kinase activity (Uehata et al., 1997), blocked both the phosphorylation of MLC in response to homophilic ligation and the accumulation of ppMLC at cell–cell contacts. Finally, inhibition of Rho kinase signaling produced functional effects very similar to those seen with Myosin 2 inhibitors, including reduced accumulation of cadherin at cell–cell contacts (measured with quantitative IF and FRAP) and reduced adhesion to cadherin-coated substrata. Taken together, these findings suggest strongly that Myosin 2 mediates the effect of Rho signaling to concentrate cadherins locally at cell–cell contacts.
Furthermore, our data indicate that Rho kinase cooperates with an instructive effect of cadherin homophilic ligation itself to recruit and activate Myosin 2 at cadherin adhesive contacts. Thus, not only was E-cadherin adhesion necessary for Myosin 2 to localize at cell–cell contacts, but homophilic adhesion induced using recombinant cadherin ligands was sufficient to recruit and activate Myosin 2. However, recruitment of Myosin 2 by homophilic adhesion also required Rho kinase. One possibility is that Myosin 2 is recruited as an early response to cadherin-activated Rho signaling itself, which has been observed in some (Charrasse et al., 2002), but not all studies (Noren et al., 2001). Alternatively, cadherin homophilic ligation may cooperate with other cell signaling pathways that stimulate Rho kinase activity.
Irrespective of the precise molecular mechanism, Myosin 2 appears then to be one of a range of actin-regulatory proteins that can be rapidly recruited to cadherin contacts in response to homophilic ligation alone. In this case, we suggest that Myosin 2 acts as part of a positive feedback pathway that stabilizes the surface localization of cadherins following homophilic ligation. We envisage that the recruitment of Myosin 2 in response to cadherin homophilic ligation would support the local accumulation of cadherin, ultimately leading to cohesive adhesion. Myosin 2 would then play a key role in the stabilization of nascent cell–cell contacts and in maintaining established contacts.
Myosin 2 Exerts a Positive Influence on Cadherin Function
How then might Myosin 2 affect the surface distribution of cadherins? One mechanism might be through lateral clustering, which occurs in response to homophilic cadherin ligation (Yap et al., 1997). When cells adhere to cadherin-coated substrata, we characteristically observe two distinct patterns of cadherin clusters, fine puncta, and larger streak-like macroclusters, that resemble the distinction between local contacts and focal adhesions seen at integrin adhesions. Strikingly, inhibition of Myosin 2 activity appeared to preferentially abolish the macrocluster pool, suggesting that Myosin 2 participated in the generation of these larger cadherin clusters. Interestingly, those cadherin macroclusters typically formed the termini for prominent actin bundles, which were also reduced substantially when Myosin 2 activity was blocked, both in planar adhesion assays and at native cell–cell contacts. Myosin 2 has commonly been implicated in generating actin bundles, consistent with its role as an actin-based motor. It is therefore tempting to postulate that Myosin 2-based contractility may support large cadherin clusters by bundling associated actin filaments, analogous to the mechanism by which actin stress fibers may support integrin-based focal adhesions (Chrzanowska-Wodnicka and Burridge, 1996).
It is important to note that whereas our data imply a positive contribution of Myosin 2 to cadherin function, other studies have implicated Myosin 2 in processes that negatively affect cadherin activity (Avizienyte et al., 2004). The latter include contractile events activated by Rho kinase that disrupt cell–cell contacts (Sahai and Marshall, 2002) and also the endocytosis of E-cadherin after contacts are broken (Ivanov et al., 2004). The apparent discrepancies between our results and those of earlier studies may reflect differences in cell types, growth conditions, and in the assays used to perturb and measure cadherin activity. Moreover, it is perhaps not surprising that this ubiquitous motor participates in a range of cellular processes that directly or indirectly affect E-cadherin. For example, contractile events elsewhere in the cell may affect the integrity of cell–cell contacts if they are mechanically coupled. Thus, the balance between Myosin 2 activities in different regions of the cell is likely to critically determine the functional consequences of Myosin 2 inhibition. All current maneuvers to perturb Myosin 2 activity are predicted, however, to act globally within cells, rather than selectively ablating its activity in specific subcellular pools or regions. In this regard, it is interesting to note that the integrity of cell–cell contacts was not disrupted when monolayers were treated with drugs for up to 6 h (unpublished data). It is possible that the residual cadherin found at cell–cell contacts was sufficient to maintain cohesion. Alternatively, global inhibition of Myosin 2 activity is likely to have also blocked the contractile forces that tend to disrupt contacts by pulling cells apart from one another.
Nonetheless, our studies using hE/Fc-based adhesion assays confirm that Myosin 2 contributes positively to cadherin function when it is examined in isolation from other adhesive or transcellular interactions that occur when cells grow as confluent sheets. Thus, we found that inhibition of Myosin 2 activity reduced both cell adhesion and the capacity of cells to extend adhesive contacts upon attachment to cadherin-coated substrata. Indeed, changes in clustering and macroscopic concentration of cadherins, both of which can increase adhesive strength, might account for this positive contribution to adhesion. Taken with our other data, we therefore conclude that Myosin 2 can contribute positively to cadherin function, consistent with observations made in Myosin 2A-deficient mouse embryos (Conti et al., 2004). This emphasizes that, ultimately, the overall contribution of myosin activity to epithelial morphology and organization is likely to reflect the balance between this positive effect on cadherin function and local concentration and the actions of other pools of myosin elsewhere in the cell.
Supplementary Material
[Supplemental Figure 1]
Acknowledgments
We thank our aforementioned colleagues for their kind gifts of reagents, Teresa Munchow for assistance with tissue culture, and Dr. Glen Baxter for his assistance with construction of the hE-Cad-YFP plasmid. Most importantly, we remain indebted to all our colleagues in the lab for their generous support, encouragement, and unflagging enthusiasm. This research was funded by the Human Frontiers Science Program and a Deutsche Forschungsgemeinschaft postdoctoral fellowship (A.K.). A.S.Y. was a Wellcome Trust Senior International Medical Research Fellow and is currently a Senior Research Fellow of the National Health and Medical Research Council of Australia. A.S.Y. is also a Research Affiliate of the ARC Special Research Center for Functional and Applied Genomics, which provided infrastructure support for this work.
References
- Adams, C. L., Chen, Y.-T., Smith, S. J., and Nelson, W. J. (1998). Mechanisms of epithelial cell–cell adhesion and cell compaction revealed by high-resolution tracking of E-cadherin-green fluorescent protein. J. Cell Biol. 142, 1105–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano, M., Ito, M., Kimura, K., Fukata, Y., Chihara, K., Nakano, T., Matsuura, Y., and Kaibuchi, K. (1996). Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase). J. Biol. Chem. 271, 20246–20249. [DOI] [PubMed] [Google Scholar]
- Avizienyte, E., Fincham, V. J., Brunton, V. G., and Frame, M. C. (2004). Src SH3/2 domain-mediated peripheral accumulation of Src and phospho-myosin is linked to deregulation of E-cadherin and the epithelial-mesenchymal transition. Mol. Biol. Cell 15, 2794–2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertet, C., Sulak, L., and Lecuit, T. (2004). Myosin-dependent junction remodelling controls planar cell intercalation and axis elongation. Nature 429, 667–671. [DOI] [PubMed] [Google Scholar]
- Boggon, T. J., Murray, J., Chappuis-Flament, S., Wong, E., Gumbiner, B. M., and Shapiro, L. (2002). C-cadherin ectodomain structure and implications for cell adhesion mechanisms. Science 296, 1308–1313. [DOI] [PubMed] [Google Scholar]
- Braga, V. M., Betson, M., Li, X., and Lamarche-Vane, N. (2000). Activation of the small GTPase Rac is sufficient to disrupt cadherin-dependent cell–cell adhesion in normal human keratinocytes. Mol. Biol. Cell 11, 3703–3721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braga, V. M. M., Machesky, L. M., Hall, A., and Hotchin, N. A. (1997). The small GPTases rho and rac are required for the formation of cadherin-dependent cell–cell contacts. J. Cell Biol. 137, 1421–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bresnick, A. R. (1999). Molecular mechanisms of nonmuscle myosin-II regulation. Curr. Opin. Cell Biol. 11, 26–33. [DOI] [PubMed] [Google Scholar]
- Brieher, W. M., Yap, A. S., and Gumbiner, B. M. (1996). Lateral dimerization is required for the homophilic binding activity of C-cadherin. J. Cell Biol. 135, 487–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charrasse, S., Meriane, M., Comunale, F., Blangy, A., and Gauthier-Rouviere, C. (2002). N-cadherin-dependent cell–cell contact regulates Rho GTPases and beta-catenin localization in mouse C2C12 myoblasts. J. Cell Biol. 158, 953–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, X., Kojima, S., Borisy, G. G., and Green, K. J. (2003). p120 catenin associates with kinesin and facilitates the transport of cadherin-catenin complexes to intercellular junctions. J. Cell Biol. 163, 547–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrzanowska-Wodnicka, M., and Burridge, K. (1996). Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J. Cell Biol. 133, 1403–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti, M. A., Even-Ram, S., Liu, C., Yamada, K. M., and Adelstein, R. S. (2004). Defects in cell adhesion and the visceral endoderm following ablation of nonmuscle myosin heavy chain II-A in mice. J. Biol. Chem. 279, 41263–41266. [DOI] [PubMed] [Google Scholar]
- Fujimori, T., and Takeichi, M. (1993). Disruption of epithelial cell–cell adhesion by exogenous expression of a mutated nonfunctional N-cadherin. Mol. Biol. Cell 4, 37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukata, M., and Kaibuchi, K. (2001). Rho-family GTPases in cadherin-mediated cell–cell adhesion. Nat. Rev. Mol. Cell. Biol. 2, 887–897. [DOI] [PubMed] [Google Scholar]
- Gumbiner, B. M. (1996). Cell adhesion: the molecular basis of tissue architecture and morphogenesis. Cell 84, 345–357. [DOI] [PubMed] [Google Scholar]
- Helwani, F. M., Kovacs, E. M., Paterson, A. D., Verma, S., Ali, R. G., Fanning, A. S., Weed, S. A., and Yap, A. S. (2004). Cortactin is necessary for E-cadherin-mediated contact formation and actin reorganization. J. Cell Biol. 164, 899–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov, A. I., Nusrat, A., and Parkos, C. A. (2004). Endocytosis of epithelial apical junctional proteins by a clathrin-mediated pathway into a unique storage compartment. Mol. Biol. Cell 15, 176–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura, K. et al. (1996). Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science 273, 245–248. [DOI] [PubMed] [Google Scholar]
- Kolega, J. (2004). Phototoxicity and photoinactivation of blebbistatin in UV and visible light. Biochem. Biophys. Res. Commun. 320, 1020–1025. [DOI] [PubMed] [Google Scholar]
- Kovacs, E. M., Ali, R. G., McCormack, A. J., and Yap, A. S. (2002a). E-cadherin homophilic ligation directly signals through Rac and phosphatidylinositol 3-kinase to regulate adhesive contacts. J. Biol. Chem. 277, 6708–6718. [DOI] [PubMed] [Google Scholar]
- Kovacs, E. M., Goodwin, M., Ali, R. G., Paterson, A. D., and Yap, A. S. (2002b). Cadherin-directed actin assembly: E-cadherin physically associates with the Arp2/3 complex to direct actin assembly in nascent adhesive contacts. Curr. Biol. 12, 379–382. [DOI] [PubMed] [Google Scholar]
- Krendel, M., Gloushankova, N. A., Bonder, E. M., Feder, H. H., Vasiliev, J. M., and Gelfand, I. M. (1999). Myosin-dependent contractile activity of the actin cytoskeleton modulates the spatial organization of cell–cell contacts in cultured epitheliocytes. Proc. Natl. Acad. Sci. USA 96, 9666–9670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert, M., Choquet, D., and Mege, R. M. (2002). Dynamics of ligand-induced, Rac1-dependent anchoring of cadherins to the actin cytoskeleton. J. Cell Biol. 157, 469–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mary, S., Charrasse, S., Meriane, M., Comunale, F., Travo, P., Blangy, A., and Gauthier-Rouviere, C. (2002). Biogenesis of N-cadherin-dependent cell–cell contacts in living fibroblasts is a microtubule-dependent kinesin-driven mechanism. Mol. Biol. Cell 13, 285–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niessen, C. M., and Gumbiner, B. M. (2002). Cadherin-mediated cell sorting not determined by binding or adhesion specificity. J. Cell Biol. 156, 389–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noren, N. K., Niessen, C. M., Gumbiner, B. M., and Burridge, K. (2001). Cadherin engagement regulates Rho family GTPases. J. Biol. Chem. 276, 33305–33308. [DOI] [PubMed] [Google Scholar]
- Paterson, A. D., Parton, R. G., Ferguson, C., Stow, J. L., and Yap, A. S. (2003). Characterization of E-cadherin endocytosis in isolated MCF-7 and CHO cells: the initial fate of unbound E-cadherin. J. Biol. Chem. 278, 21050–21057. [DOI] [PubMed] [Google Scholar]
- Rabut, G., and Ellenberg, J. (2005). Photobleaching techniques to study mobility and molecular dynamics of proteins in live cells: FRAP, iFRAP, and FLIP. In: Live Cell Imaging: A Laboratory Manual, ed. R. D. Goldman and D. L. Spector, Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press, 101–126. [DOI] [PubMed]
- Ratcliffe, M. J., Smales, C., and Staddon, J. M. (1999). Dephosphorylation of the catenins p120 and p100 in endothelial cells in response to inflammatory stimuli. Biochem. J. 338(Pt 2), 471–478. [PMC free article] [PubMed] [Google Scholar]
- Sahai, E., and Marshall, C. J. (2002). ROCK and Dia have opposing effects on adherens junctions downstream of Rho. Nat. Cell Biol. 4, 408–415. [DOI] [PubMed] [Google Scholar]
- Sakamoto, T., Limouze, J., Combs, C. A., Straight, A. F., and Sellers, J. R. (2005). Blebbistatin, a myosin II inhibitor, is photoinactivated by blue light. Biochemistry 44, 584–588. [DOI] [PubMed] [Google Scholar]
- Sako, Y., Nagafuchi, A., Tsukita, S., Takeichi, M., and Kusumi, A. (1998). Cytoplasmic regulation of the movement of E-cadherin on the free cell surface as studied by optical tweezers and single particle tracking: corralling and tethering by the membrane skeleton. J. Cell Biol. 140, 1227–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro, L., Fannon, A. M., Kwong, P. D., Thompson, A., Lehmann, M. S., Grubel, G., Legrand, J.-F., Als-Nielden, J., Colman, D. R., and Hendrickson, W. A. (1995). Structural basis of cell–cell adhesion by cadherins. Nature 374, 327–337. [DOI] [PubMed] [Google Scholar]
- Stow, J. L., Fath, K. R., and Burgess, D. R. (1998). Budding roles for myosin II on the Golgi. Trends Cell Biol. 8, 138–141. [DOI] [PubMed] [Google Scholar]
- Straight, A. F., Cheung, A., Limouze, J., Chen, I., Westwood, N. J., Sellers, J. R., and Mitchison, T. J. (2003). Dissecting temporal and spatial control of cytokinesis with a myosin II Inhibitor. Science 299, 1743–1747. [DOI] [PubMed] [Google Scholar]
- Takaishi, K., Sasaki, T., Kotani, H., Nishioka, H., and Takai, Y. (1997). Regulation of cell–cell adhesion by Rac and Rho small G proteins in MDCK cells. J. Cell Biol. 139, 1047–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeichi, M. (1991). Cadherin cell adhesion receptors as a morphogenetic regulator. Science 251, 1451–1455. [DOI] [PubMed] [Google Scholar]
- Togo, T., and Steinhardt, R. A. (2004). Nonmuscle myosin IIA and IIB have distinct functions in the exocytosis-dependent process of cell membrane repair. Mol. Biol. Cell 15, 688–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda, K., Murata-Hori, M., Tatsuka, M., and Hosoya, H. (2002). Rho-kinase contributes to diphosphorylation of myosin II regulatory light chain in nonmuscle cells. Oncogene 21, 5852–5860. [DOI] [PubMed] [Google Scholar]
- Uehata, M. et al. (1997). Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature 389, 990–994. [DOI] [PubMed] [Google Scholar]
- Vaezi, A., Bauer, C., Vasioukhin, V., and Fuchs, E. (2002). Actin cable dynamics and Rho/Rock orchestrate a polarized cytoskeletal architecture in the early steps of assembling a stratified epithelium. Dev. Cell 3, 367–381. [DOI] [PubMed] [Google Scholar]
- Verma, S., Shewan, A. M., Scott, J. A., Helwani, F. M., den Elzen, N. R., Miki, H., Takenawa, T., and Yap, A. S. (2004). Arp2/3 activity is necessary for efficient formation of E-cadherin adhesive contacts. J. Biol. Chem. 279, 34062–34070. [DOI] [PubMed] [Google Scholar]
- Vestweber, D., Gossler, A., Boller, K., and Kemler, R. (1987). Expression and distribution of cell adhesion molecule uvomorulin in mouse preimplantation embryos. Dev. Biol. 124, 451–456. [DOI] [PubMed] [Google Scholar]
- Wei, Q., and Adelstein, R. S. (2000). Conditional expression of a truncated fragment of nonmuscle myosin II-A alters cell shape but not cytokinesis in HeLa cells. Mol. Biol. Cell 11, 3617–3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap, A. S., Brieher, W. M., Pruschy, M., and Gumbiner, B. M. (1997). Lateral clustering of the adhesive ectodomain: a fundamental determinant of cadherin function. Curr. Biol. 7, 308–315. [DOI] [PubMed] [Google Scholar]
- Zallen, J. A., and Wieschaus, E. (2004). Patterned gene expression directs bipolar planar polarity in Drosophila. Dev. Cell 6, 343–355. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
[Supplemental Figure 1]