Production and Characterization of Improved Adenovirus Vectors with the E1, E2b, and E3 Genes Deleted (original) (raw)
Abstract
Adenovirus (Ad)-based vectors have great potential for use in the gene therapy of multiple diseases, both genetic and nongenetic. While capable of transducing both dividing and quiescent cells efficiently, Ad vectors have been limited by a number of problems. Most Ad vectors are engineered such that a transgene replaces the Ad E1a, E1b, and E3 genes; subsequently the replication-defective vector can be propagated only in human 293 cells that supply the deleted E1 gene functions in trans. Unfortunately, the use of high titers of E1-deleted vectors has been repeatedly demonstrated to result in low-level expression of viral genes still resident in the vector. In addition, the generation of replication-competent Ad (RCA) by recombination events with the E1 sequences residing in 293 cells further limits the usefulness of E1-deleted Ad vectors. We addressed these problems by isolating new Ad vectors deleted for the E1, E3, and the E2b gene functions. The new vectors can be readily grown to high titers and have several improvements, including an increased carrying capacity and a theoretically decreased risk for generating RCA. We have also demonstrated that the further block to Ad vector replication afforded by the deletion of both the E1 and E2b genes significantly diminished Ad late gene expression in comparison to a conventional E1-deleted vector, without destabilization of the modified vector genome. The results suggested that these modified vectors may be very useful both for in vitro and in vivo gene therapy applications.
The primary objective of gene therapy is to deliver a functional gene to tissues where the respective gene activity is missing or defective. While the number of human diseases (genetic and nongenetic) that potentially can be treated in such a manner is large, each disease has its own set of parameters that must be fulfilled before a clinical treatment can be realized. All of the diseases, however, have a common denominator: the need for a vector to efficiently deliver the respective therapeutic gene to the affected tissues and/or organs. There are a variety of vectors currently being studied for potential use in vivo. Nonviral vectors (i.e., liposomes) have the advantage of minimizing the immune response directed against the vector and have essentially unlimited carrying capacities, but they have not yet demonstrated efficient, high-level gene transfer in vivo. Viral vector preparations can efficiently transduce a significant number of cells in vivo, but they also have limitations. Retrovirus-based vectors, for example, have limited carrying capacities and are unable to transduce mitotically quiescent cells, and vector titers are several orders of magnitude less than in other viral systems, such as the adenovirus (Ad)-derived vectors.
Ad vectors hold great promise for gene therapy of a number of human disorders, for a variety of reasons. Ad vectors can transduce multiple types of tissues in vivo, including nondividing, differentiated cells such as those found in liver, kidney, muscle (skeletal and cardiac), respiratory, and nervous system tissues (4, 6, 9, 11, 14, 15, 28, 30). Diseases that affect these tissues are therefore potentially amenable to Ad-mediated gene therapy. Conventional [E1−] (E1-lacking) Ad vectors (or first-generation Ad vectors) have a large carrying capacity, limited to 8.0 kb at present. Ad vectors can be concentrated to very high titers (>1013 particles/ml), which contributes to the ability of Ad vectors to infect and transduce a significant number of target cells after a single in vivo administration (30, 41). Ad vectors can express transgenes episomally, unlike retrovirus-based vectors, which are dependent on integration (and cell division) for transgene expression. First-generation Ad vectors have been demonstrated to sustain expression of transduced genes for extended periods in immune-incompetent (and in some cases immune-competent) animals (34, 35). Finally, live Ad preparations have been used for the vaccination of military recruits, and Ad strains 2 and 5 (Ad2 and Ad5; most commonly used for vector development) are not associated with severe disease. Despite these significant attributes, there are major shortcomings of current Ad vectors that must be addressed before their full clinical potential may be realized.
The most difficult problem with Ad vectors is their inability to sustain long-term transgene expression, secondary to host immune responses that eliminate virally transduced cells in immune-competent animals (12, 40, 41, 44). While immune responses have been demonstrated against the transgene-encoded protein product (34), it has also been demonstrated that Ad vector epitopes are major factors in triggering the host immune response (12, 43). In support of this view, it has been repeatedly demonstrated that transgenes such as the bacterial β-galactosidase gene are highly immunogenic when transduced by Ad vectors, in contrast to other delivery systems (e.g., direct DNA injection or adeno-associated virus administration), where an immune response against the immunogenic transgene is lacking and transgene expression persists (36, 37). We postulate that modified Ad vectors may have improved in vivo efficacy, as a result of their decreased abilities to replicate and to express multiple viral functions and/or epitopes (1, 2). Toward this goal, this report describes the isolation, characterization, and efficient large-scale production of an improved Ad vector that incorporated deletions not only in the Ad E1 and E3 genes (absent in most first-generation Ad vectors) but also the Ad polymerase (E2b) gene.
MATERIALS AND METHODS
Construction and isolation of an Ad deleted for the polymerase gene.
The ∼20-kb _Xba-Bam_HI subfragment of pBHG11 (Microbix, Toronto, Ontario, Canada) was subcloned into pBluescriptKSII+ (Stratagene, La Jolla, Calif.), yielding pAXB. Plasmid pAXB was digested with _Bsp_EI, T4 DNA polymerase end filled, and _Bam_HI digested, and the ∼9.0-kb fragment was isolated. Plasmid pAXB was also digested with _Bsp_HI, T4 DNA polymerase end filled, and _Bam_HI digested, and the ∼13.7-kb fragment was ligated to the previously isolated 9.0-kb fragment, generating pAXB-Δpol. This subcloning strategy deleted 608 bp (Δpol; Ad5 nucleotides [nt] 7274 to 7881) within the amino terminus of the polymerase gene. This deletion also effectively removed open reading frame 9.4, present on the rightward reading strand in this region of the Ad genome. The _Xba-Bam_HI subfragment of pAXB-Δpol was reintroduced into _Xba-Bam_HI-digested pBHG11, to generate pBHG11-Δpol (Fig. 1A). Theoretically, pBHG11-Δpol should have been capable of generating recombinant [E1−,Δpol] Ad vectors after cotransfection of polymerase-transcomplementing cells with a conventional Ad shuttle plasmid; unfortunately, we were never able to generate such a vector with this approach. It is possible that our version of pBHG11 had acquired a cryptic point mutation prohibiting viable vector isolation. We therefore cotransfected plasmid pBHG11-Δpol with _Sca_I-digested _dl_7001-derived genomic DNA into the polymerase-expressing cell line C-7 (Fig. 1B). The Ad _dl_7001 genomic DNA had an ∼3.0-kb deletion within the E3 region of genes and was isolated as an intact virion DNA-terminal protein (TP) complex, _dl_7001-TP (17). The C-7 cell line has been previously described (1, 2). Briefly, these cells are capable of transcomplementing [E1−] Ad, as well as temperature-sensitive Ad mutants defective in both the polymerase and preterminal protein genes. This was accomplished by the stable cointroduction of transgenes constitutively expressing the polymerase and preterminal protein genes into human 293 cells (1, 2).
FIG. 1.
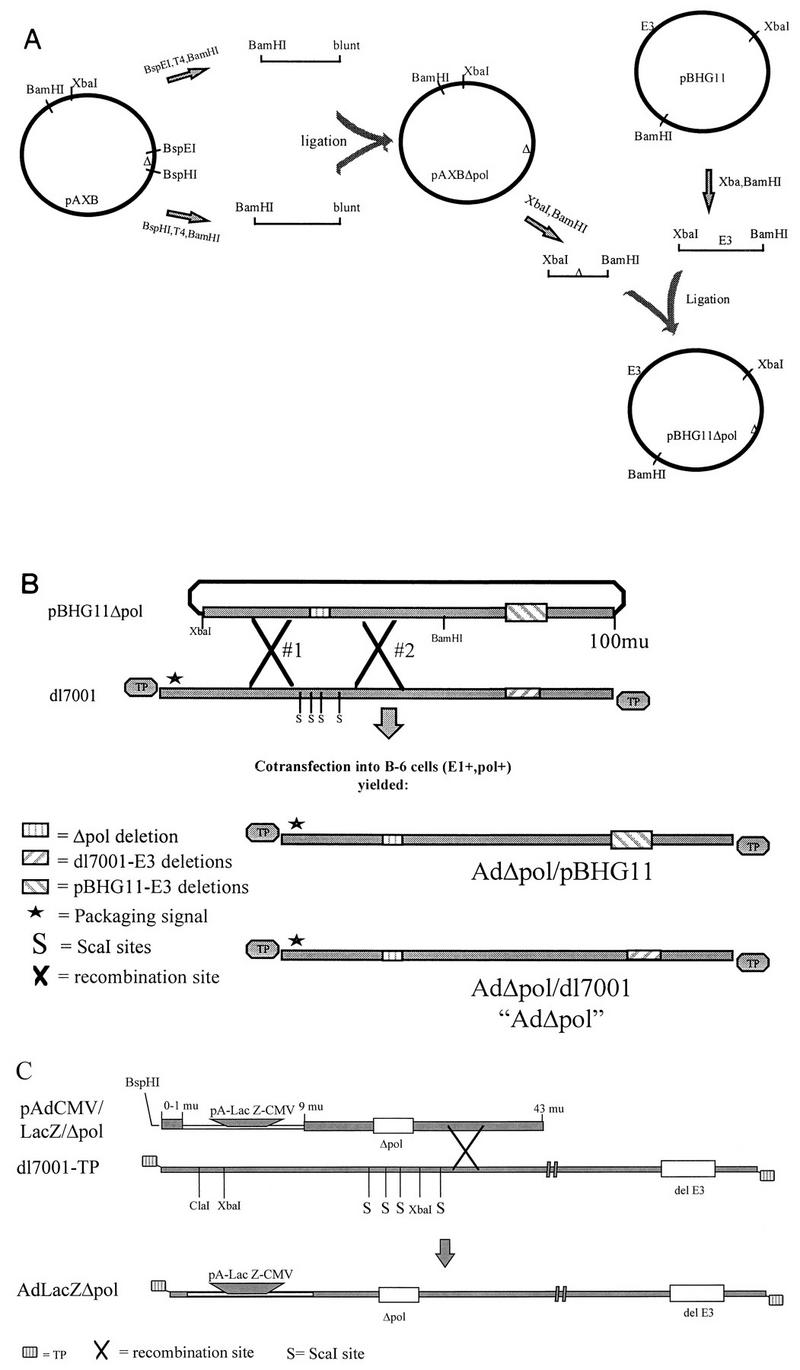
Diagrammatic representation of the steps used to isolate pBHG11Δpol (A), AdΔpol and AdΔpol/pBHG11 (B), and AdLacZΔpol (C). mu, map units.
The cotransfection strategy resulted in the isolation of two viruses (due to either a single or a double recombination event between the two input DNA molecules) that had the Δpol alteration present in either a _dl_7001-derived E3 deletion background (AdΔpol) or a pBHG11-derived E3 deletion background (AdΔpol/pBHG11). While both viruses were viable, the _dl_7001-derived virus demonstrated superior growth characteristics and was therefore used for further studies.
Production of [E1−,Δpol,E3−] Ad vectors.
The AdΔpol virus was grown to high titer, and viral DNA was isolated as previously described (1), digested with _Asc_I, T4 polymerase end filled, and _Bst_1107I digested. The ∼9.3-kb blunt-ended Δpol-containing fragment was subcloned into the _Bst_1107I-digested shuttle plasmid pAdAscI (a shuttling plasmid used to generate traditional [E1−] Ad vectors [13a]). This subcloning strategy yielded pAdAscL-Δpol, a new shuttling plasmid specifically designed for the rapid isolation of recombinant Ad vectors deleted for both the Ad E1 and polymerase genes. The pAdAscL-Δpol plasmid contained nts 1 to 15671 of the left end of the Ad5 genome but was effectively deleted for the E1 genes (Ad nt 358 to 3328, replaced by the _Asc_I site) and the 608-bp polymerase deletion. A nucleus-targeted bacterial β-galactosidase transgene (lacZ) flanked by a minimal cytomegalovirus promoter/enhancer element, the MINX intron (27), and a simian virus 40-derived polyadenylation signal was subcloned into the _Asc_I site of pAdAscL-Δpol, generating the shuttle plasmid pAdCMV/LacZ/Δpol (Fig. 1C). Ten micrograms of pAdCMV/LacZ/Δpol linearized with _Bsp_HI (restriction site within 60 bp of the left end of Ad) was CaPO4 cotransfected with 500 ng of _Xba_I-, _Cla_I-, and _Sca_I-digested _dl_7001-TP virion DNA onto three 60-mm-diameter dishes containing 2 × 106 Ad polymerase-expressing C-7 cells (Fig. 1C). The multiple restriction enzyme digestion of _dl_7001 virion DNA significantly reduced the isolation of nonrecombinant viruses after transfection (13a). Sixteen hours after transfection, the cells were harvested and mixed with ∼8 × 106 C-7 cells (nontransfected). The cell mixture was distributed to nine 24-well tissue culture cluster plates and incubated at 37.0°C for 5 to 9 days. Individual wells demonstrating viral cytopathic effects were harvested, and the isolated virus was amplified by repeated infection of either B-6 or C-7 cells. Isolation of the AdLacZΔpol recombinant vector was subsequently confirmed by (i) β-galactosidase conversion of the chromogenic substrate 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) in cells transduced by the vector, (ii) DNA restriction mapping of the vector genome, and (iii) multiple functional studies (see below).
Ad vector genome replication studies.
The respective cell lines were infected at the indicated multiplicity of infection (MOI) with either AdΔpol, Adsub360LacZ (E1 deletion alone), or AdLacZΔpol and incubated at 37.0°C for the indicated times. Infected cells were then harvested, and DNA was prepared and analyzed as previously described (1).
Ad vector kinetics and one-step burst assays. (i) Kinetics assay.
Tissue culture dishes (60-mm diameter) containing 2 × 106 293 or B-6 cells were infected with AdLacZΔpol at an MOI of 0.01 β-galactosidase-forming units (BFU) per cell. Cells and media were harvested from the dishes after incubation at 37.0°C for the indicated times. The number of BFU produced was then determined by limiting dilution infection of C-7 cells or LP-293 cells. Eighteen hours later, infected cells were stained for β-galactosidase activity, and the number of transducing particles was quantitated by visual inspection of blue-staining cells. The BFU generated in the original lysate was then determined by multiplying the number of stained nuclei by the appropriate dilution factor.
(ii) One-step burst assay.
A total of 2 × 106 293 or B-6 cells were infected with AdLacZΔpol or Adsub360LacZ (in triplicate) at an MOI of 5 and incubated at 37.0°C for 40 h, and the total BFU generated was determined by limiting dilution assay as described for the kinetic assays.
Ad vector late gene expression studies.
LP-293 cells, B-6 cells, or HeLa cells were infected with either AdΔpol, Adsub360LacZ, or AdLacZΔpol at the indicated MOIs and incubated at 37.0°C for 40 to 44 h. Infected cells were harvested, rinsed with phosphate-buffered saline (PBS), and lysed in Tris-Cl (pH 6.8)–4% sodium dodecyl sulfate (SDS)–10% glycerol–10% β-mercaptoethanol. Protein extracts were freeze-thawed three times, DNA was sheared, and protein concentrations were determined via the Bradford assay, using the Coomassie Plus staining reagent (Pierce, Rockford, Ill.). Equivalent amounts of each protein lysate were heated to 100.0°C for 2 min and electrophoretically separated in an SDS–10% polyacrylamide gel. The separated proteins were wet-transferred to a Biotrace NT membrane (Gelman Sciences, Ann Arbor, Mich.) and probed with a rabbit polyclonal antibody (supplied by R. Gerard, University of Texas Southwestern, Dallas) generated against the knob portion of the 66-kDa Ad fiber protein monomer. Bound antibody was detected with the ECL detection system (Amersham Life Sciences, Arlington Heights, Ill.).
In vivo administration of AdLacZΔpol.
Sixty 150-mm-diameter tissue culture plates containing ∼2.5 × 107 C-7 cells were infected with the AdLacZΔpol at an approximate MOI of 5 and incubated at 37.0°C for 40 h. The infected cells were harvested, resuspended in 10 mM Tris-Cl (pH 8.0), and sonicated, and the virus was purified by two rounds of cesium chloride density centrifugation. The virus containing band was desalted over a Sephadex CL-6B column (Pharmacia Biotech, Piscataway, N.J.), glycerol was added to a concentration of 12%, and aliquots were stored at −80°C. The titer of this stock was 6 × 1010 BFU per ml. The total number of particles in this stock was 1.2 × 1012, as determined by measurement of the optical density at 260 nm of an aliquot of the virus after SDS lysis (24); therefore, the bioactivity of the preparation was at a minimum of 0.05 (6 × 1010/1.2 × 1012), a value similar to that achieved after isolation of first-generation Ad vectors (24). Seven- to nine-week-old BALB/c mice were injected in the left tibialis anterior muscle or via the tail vein with a PBS solution containing 109 BFU of AdLacZΔpol. Five to six days after infection, the mice were sacrificed, and the muscle or liver specimens removed and frozen in OCT compound. Cryosections were obtained, briefly fixed in a 3.7% formaldehyde–PBS solution, stained overnight for β-galactosidase activity, rinsed in PBS, and briefly postfixed in 3.7% formaldehyde–0.5% glutaraldehyde in PBS. Sections were then eosin counterstained and photographed.
RESULTS
Deletion of polymerase activity from an Ad isolate.
We have previously described the isolation of cells that coexpress the Ad E1 and polymerase genes, based on their ability to support the growth of Ad polymerase temperature-sensitive mutants (1). We next wanted to demonstrate the ability of the cell lines to support the growth of Ads deleted for subregions of the polymerase gene as well as the E3 genes. Therefore, we constructed the virus AdΔpol, which contained a 608-bp deletion within the polymerase gene (Fig. 1B). The polymerase deletion also effectively deleted open reading frame 9.4, with no consequence in regard to the growth potential of the resultant viruses. Confirmation of the AdΔpol genomic structure was confirmed by restriction enzyme digestion (Fig. 2A). AdΔpol had a severe replication defect when not grown in cells expressing the Ad polymerase, confirming the lack of polymerase activity secondary to the introduced 608-bp deletion (Fig. 2B). This experiment also demonstrated that despite high levels of E1 activity (both from the AdΔpol genome and from the E1-expressing 293 cells), AdΔpol was incapable of significant replication, in contrast to first-generation Ad vectors in vitro and in vivo (22, 42). With these results, we then turned our attention to the isolation of an Ad vector deleted for the E1, E3, and polymerase gene functions.
FIG. 2.

(A) DNA of each of the indicated viruses was digested with _Hin_dIII, electrophoresed, blotted, and probed with a 32P-labeled _dl_7001 genomic probe. The recombinant viruses (AdΔpol/dl7001 and AdΔpol/pBHG11) both contained the Δpol alteration. This was verified by the demonstration that the 5.3-kb polymerase-encoding fragment normally present in _dl_7001 (location indicated by the solid arrowheads) migrated as a 4.7-kb DNA fragment in the Δpol-containing viruses (new location depicted by the open arrowhead). Note also that the pBHG11-derived virus contained a larger E3 deletion, (×), in contrast to the _dl_7001-derived E3 deletion (⋆). The indicated cell lines (2 × 106 cells) were identically infected with AdΔpol/dl7001 at an MOI of ∼1.0 PFU/cell. Twenty-four hours after infection, total DNA was isolated, 4 μg of each was digested with _Hin_dIII; the fragments were electrophoretically separated, blotted onto a nylon membrane, and probed with 32P-labeled _dl_7001 genomic DNA. Note the complete lack of replication of AdΔpol/dl7001 in 293 cells.
Isolation and growth kinetics of AdLacZΔpol.
To facilitate the production of [E1−,E3−,Δpol] Ad vectors, we engineered a shuttling system for their construction (Fig. 1C). Cotransfection of linearized pAdCMV/LacZ/Δpol with multiply digested _dl_7001 DNA-TP complex resulted in the successful isolation of AdLacZΔpol. The genomic structure of AdLacZΔpol was confirmed by restriction enzyme analysis (Fig. 3). The kinetics of AdLacZΔpol growth was then determined in 293 cells and B-6 cells (Fig. 4). Note the dramatic lack of production of infectious AdLacZΔpol in LP-293 cells, despite the presence of high levels of E1 activity in this cell line. Furthermore, one-step burst assays of B-6 cells infected with AdLacZΔpol or Adsub360LacZ clearly demonstrated that the AdLacZΔpol vector could be produced to as high (or higher) a titer as the Adsub360LacZ vector. For example, when 2 × 106 B-6 cells were infected at an MOI of 5 BFU with AdLacZΔpol or Adsub360LacZ (each in triplicate), the total BFU released after a 40-h infection were 1.18 × 108 ± 4.2 × 107 for AdLacZΔpol and 1.78 × 107 ± 8.9 × 106 for Adsub360LacZ. High-titer growth of the AdLacZΔpol vector in the B-6 cell line is important, not only for clinical grade production but also because previously described cell lines designed to allow the growth of modified Ad vectors are sometimes inefficient, likely due to the toxicity of the coexpressed Ad genes (46).
FIG. 3.

B-6 cells were infected with AdLacZΔpol; total DNA was harvested 36 h later and digested with _Not_I and _Eco_RI, and the pattern of DNA fragments obtained was compared with that for _Not_I- and _Eco_RI-digested _dl_7001 DNA. The 6.5-kb left inverse terminal repeat (L-ITR)-E1-containing fragment of _dl_7001 (⋆) was replaced by the lacZ minigene cassette that generates three fragments of 3,879, 3,050 (), and 708 bp (not shown). The normal polymerase-coding region of _dl_7001 is contained within a 5,100-bp fragment (○) and is decreased in size to 4,492 bp (×) in Ad LacZΔpol. Std., size standards (positions are indicated in kilobases); CMV, cytomegalovirus.
FIG. 4.
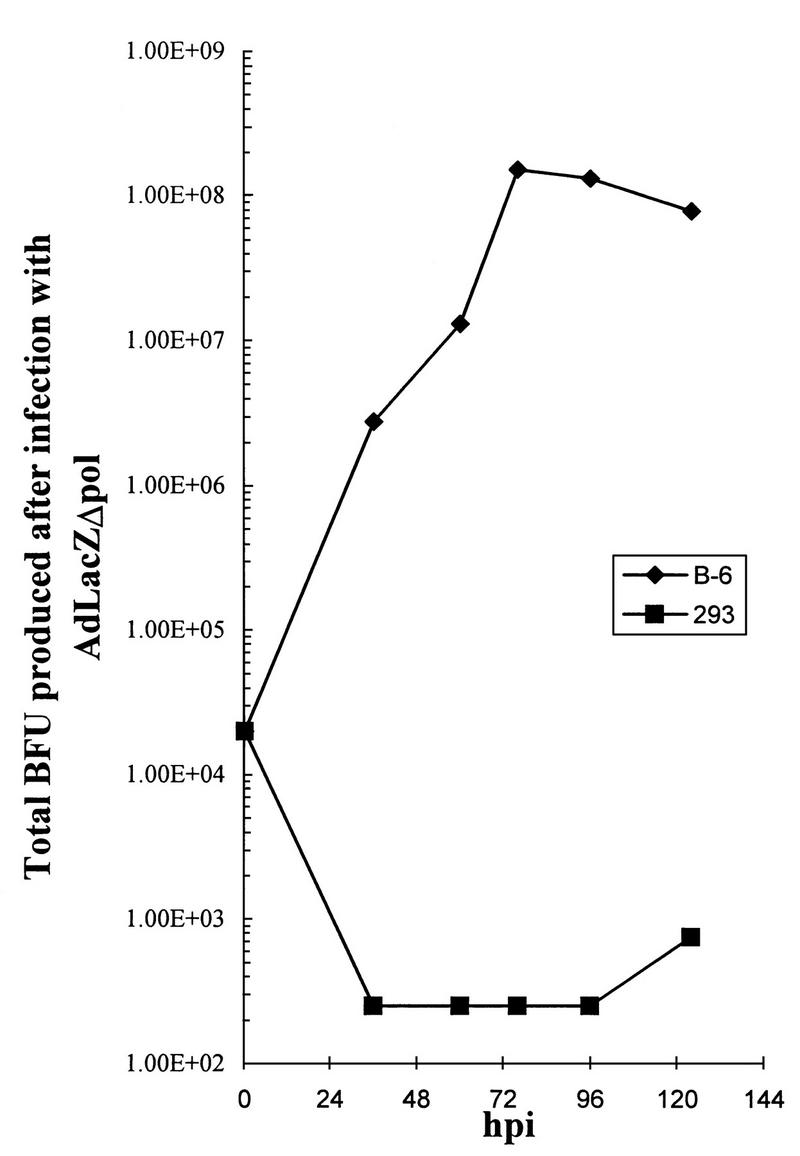
293 or B-6 cells (2 × 106) were infected at an MOI of 0.01 BFU with Ad LacZΔpol, and the total BFU generated was assessed after limiting dilution infection and X-Gal staining of C-7 cells.
AdLacZΔpol is blocked in replication.
Infection of B-6 cells allowed high-level replication of the AdLacZΔpol genomes; however, an identical infection of 293 cells demonstrated a dramatic block of replication (Fig. 5A). This result confirmed that even in the presence of high levels of E1 activity, the Δpol modification conveys upon the vector a severe replication blockade. Despite a lack of significant replication, the AdΔpol and AdLacZΔpol genomes were still present at near input levels 24 h postinfection (hpi) in 293 cells and decreased only after 48 hpi. Supporting these observations, high-titer infection of Hela cells (lacking both E1 and polymerase activities) with AdLacZΔpol demonstrated a significantly greater replication block than displayed by the first-generation Adsub360LacZ vector (Fig. 5B). Despite this lack of replication, the AdLacZΔpol persisted to at least 75% of input virus levels within 24 h of HeLa cell infection and dropped to 50% of input virus by 48 hpi (Fig. 5B). This result is in contrast to a study using “gutted” Ad vectors that are devoid of much of the Ad genome (22). In the latter report, 50% of the gutted Ad vector genomes were degraded within 5 h of transduction of cells both in vitro and in vivo, and the genomes were essentially undetectable by 12 to 24 h posttransduction (22). In summary, our replication studies demonstrated that Ad vectors deleted for polymerase gene activities were severely blocked in the ability to replicate (even in the presence of excessive levels of E1 activity), a blockade that did not simultaneously result in a rapid loss (destabilization) of their genomes.
FIG. 5.

(A) 293 or B-6 cells (2 × 106) were infected at an MOI of 1.5 BFU with Ad LacZΔpol, and total DNA was harvested after the indicated incubation times. DNA was digested with _Hin_dIII, electrophoresed, blotted, and probed with 32P-labeled _dl_7001 DNA. (B) HeLa cells (2 × 106) were infected at the indicated MOIs with AdLacZΔpol or Adsub360LacZ and probed with 32P-labeled _dl_7001 DNA as described for panel A. Densitometric analysis of the image was done with the NIH Image software package.
AdLacZΔpol late gene expression is blocked in noncomplementing cell lines.
Cell types 293 and B-6 were infected with AdLacZΔpol and assessed for viral late gene expression, as determined by fiber protein accumulation. Figure 6A demonstrated that there was at least a 10,000-fold reduction in the ability of the AdLacZΔpol vector to produce the fiber protein after infection of 293 cells, in contrast to infection of the polymerase-complementing B-6 cell line. A further demonstration of the late gene expression blockade was demonstrated after HeLa cells were infected with either the Adsub360LacZ or AdLacZΔpol vector (Fig. 6B). Fiber expression was readily detected after infection of HeLa cells with the Adsub360LacZ vector; however, infection with the AdLacZΔpol vector (at an MOI of 500) did not result in detectable fiber expression. Similar results were obtained at lower MOIs (data not shown). Together, these results demonstrated another benefit of the AdLacZΔpol vector, a significantly decreased expression of viral late genes, secondary to the severe replication blockade afforded by the presence of the Δpol in the modified vector. Ad late gene products such as the fiber protein are potent antigenic epitopes in vivo; therefore, decreased expression of the late, E1, and polymerase gene products may result in a greater efficacy for AdΔpol vectors in vivo.
FIG. 6.

(A) 293 or B-6 cells (2 × 106) were infected at an MOI of 1.5 BFU with Ad LacZΔpol, and protein lysates isolated from the infected cells were electrophoresed, blotted, probed with a fiber polyclonal antibody, and visualized with the ECL system. Dilutions of the B-6 cell-derived lysates are indicated. (B) HeLa cells (2 × 106) were infected at an MOI of 500 with AdLacZΔpol or Adsub360LacZ, and the fiber protein was visualized as described for panel A. Six micrograms of protein lysate from each infection was loaded per well. Each arrow indicates the location of the fiber-specific band, which correctly migrated as a 66-kDa protein based on comparison with the molecular weight standards included in the gel electrophoresis. To further verify that the bands indicated represent fiber protein monomer, the control lysate lane contained a portion of a protein lysate derived from the productive infection of B-6 cells with AdLacZΔpol. Note the lack of fiber expression in the lysate derived from AdLacZΔpol infection of HeLa cells.
In vivo transduction with the AdLacZΔpol vector.
We were able to readily generate high-titer stocks of the AdLacZΔpol vector; therefore, 109 BFU of AdLacZΔpol was injected intravenously (for liver transduction) or into the left tibialis anterior muscle of 7- to 9-week-old BALB/c mice. Five to six days later, the tissues were excised and stained for β-galactosidase activity. As demonstrated in Fig. 7, the AdLacZΔpol vector is capable of extensive transduction and expression of the bacterial β-galactosidase gene in liver tissues. The same result was achieved after intramuscular administration of AdLacZΔpol (data not shown). Therefore, despite the additional replication blockade provided by the deletion of both the E1 and polymerase genes in the AdLacZΔpol vector, efficient transduction and transgene expression occurred in these tissues. This again is in contrast to some recently described helper virus-dependent Ad vector systems, whose modified minigenomes were rapidly eliminated in vivo, before significant transgene expression occurred (22).
FIG. 7.
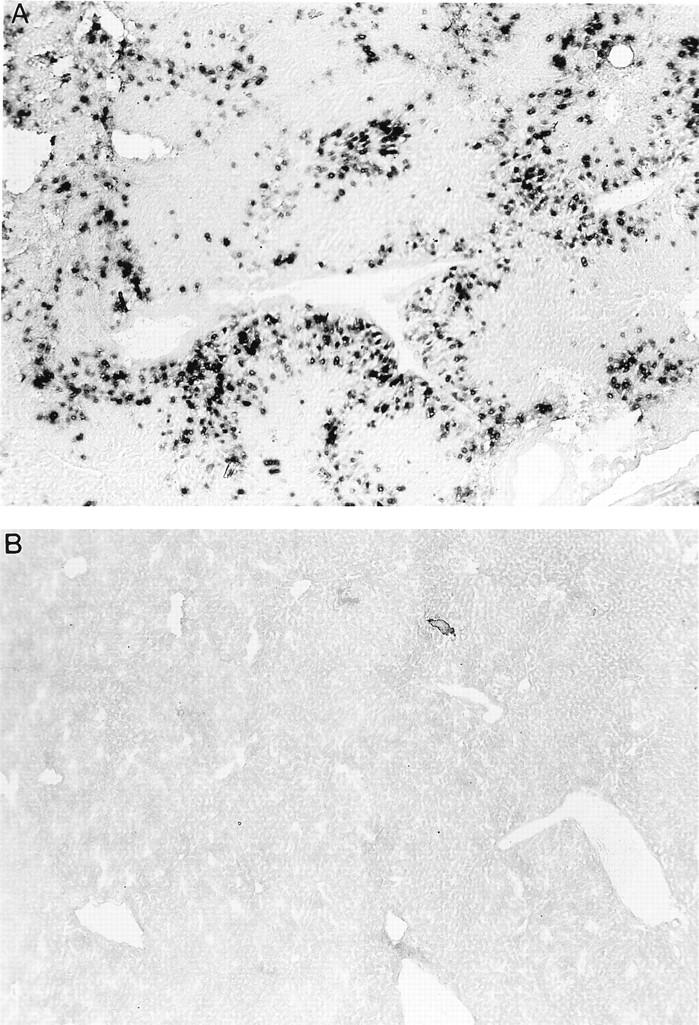
AdLacZΔpol (109 BFU) was injected into the tail veins of BALB/c mice. Six days later, liver sections were harvested and stained for β-galactosidase activity. Note the stained nuclei present only in the infected cells (A) and the complete lack of β-galactosidase from a mock-infected age-matched control animal (B), confirming successful transduction and sustained transgene expression in vivo.
DISCUSSION
The successful isolation and high-level production of [E1−,Δpol,E3−] Ad vectors has several important implications regarding clinically relevant gene therapy applications. The carrying capacity of the [E1−,Δpol,E3−] Ad vector production system described in this report can currently allow insertion of transgene constructs up to 9.0 kb in size. Carrying capacity of these vectors could theoretically approach 11.0 kb by further deletion of subportions of the polymerase gene (2). Increased carrying capacity is critically important when one considers (i) the transfer of larger cDNA minigene constructs (e.g., dystrophin), (ii) the use of larger tissue-specific promoter/enhancer elements (e.g., the muscle creatine kinase enhancer), and (iii) the reintroduction into vectors of Ad genes, which may minimize immune recognition of Ad infected cells in vivo (8, 16, 21). Studies to determine the maximum carrying capacity of the AdΔpol vectors are currently in progress. AdΔpol vectors also have a theoretically decreased potential to generate replication-competent Ad (RCA), since multiple recombination events are required to regenerate a viable virus containing both the E1 and polymerase gene functions. Therefore, the likelihood of RCA contamination of AdΔpol vector preparations will be lessened, an important attribute when one is producing clinical-grade vector preparations.
The [E1−,Δpol,E3−] Ad vector may be advantageous for a number of additional reasons. It has been known for 15 years that in tissue culture systems, [E1−] Ad is not completely replication defective, and at higher MOIs, the E2, E3, and E4 promoters are active and allow viral replication and late gene expression to proceed (this report and references 25 and 26). Not surprisingly [E1−] Ad vectors have also been demonstrated to express the Ad early genes, undergo genome replication, and express the L1- to L5-encoded structural genes when used in vivo (38, 41, 42). Importantly, we have demonstrated that the AdΔpol vectors isolated in this study have a significantly decreased potential to express viral late genes such as the Ad fiber protein compared with first-generation Ad vectors. Isolation of an Ad vector with a decreased potential to express viral epitopes may improve gene transfer in vivo (10). In addition, prolonging transgene expression by modifications such as those introduced into the AdΔpol vectors may eliminate the need for nonspecific (and potentially toxic) immunosuppressive agents (19, 23, 31–33, 39, 44, 45). Studies to test this hypothesis are not straightforward, however, since it has become increasingly evident that after conventional [E1−] Ad vector administration, the host immune response (cytotoxic T lymphocyte mediated) can be directed against both transgene- and Ad-encoded epitopes and can be influenced by (i) the background strain of the animal tested, (ii) the promoter/enhancer elements used to drive expression of the transgene, and (iii) the viral backbone itself (3, 5, 10, 18, 34). Therefore, determination of the in vivo efficacy of the AdΔpol vectors will require evaluation of a variety of parameters (i.e., long-term persistence of transgene expression, acute versus chronic inflammatory reactions, and use of various promoter elements to drive transgene expression) in a number of animal models.
Finally, the AdΔpol vectors do not require any type of helper virus for their high-titer growth, in contrast to some recently described systems relying on a helper virus for the propagation of so-called gutted Ad vectors (7, 13, 20–22, 29). The latter systems may benefit from the isolation of the AdΔpol viruses as well. For example, helper virus contamination of gutted Ad vector preparations is a significant production issue; contamination of such preparations with the AdΔpol virus may elicit less of an in vivo immune response than if the same preparation were contaminated with an equivalent amount of a first-generation Ad vector. In addition, during the propagation of the gutted Ad vector, the risk of producing an RCA would also be lessened with the use of an AdΔpol vector as a helper virus. Interestingly, gutted Ad vectors may in fact be dependent on low levels of helper virus contamination (and low-level expression of helper virus-encoded genes) in order to stabilize their modified genomes after transduction (22). If gutted Ad vectors are dependent on helper virus contamination for stable gene transduction, we would suggest that the use of an AdΔpol contaminant may be of benefit, for the multiple reasons outlined in this report.
ACKNOWLEDGMENTS
We thank Samuel George’s laboratory at Duke University for assistance in the isolation of high-titer Ad vector preparations for use in vivo and the University of Michigan Adenovirus Core Facility for aliquots of the _dl_7001 and Adsub360lacZ viruses.
Support was provided to A.A. by an award to Duke University Medical Center from the Howard Hughes Medical Institute under the Research Resources Program for Medical Schools and from the Muscular Dystrophy Association (USA) to J.S.C. M.A.H. was supported by a postdoctoral fellowship from the Muscular Dystrophy Association.
REFERENCES
- 1.Amalfitano A, Begy C R, Chamberlain J S. Improved adenovirus packaging cell lines to support the growth of replication-defective gene-delivery vectors. Proc Natl Acad Sci USA. 1996;93:3352–3356. doi: 10.1073/pnas.93.8.3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amalfitano A, Chamberlain J S. Isolation and characterization of packaging cell lines that coexpress the adenovirus DNA polymerase and preterminal proteins: implications for gene therapy. Gene Ther. 1997;4:258–263. doi: 10.1038/sj.gt.3300378. [DOI] [PubMed] [Google Scholar]
- 3.Armentano D, Zahner J, Sacks C, Soukden C C, Smith M P, Stgeorge J A, Wadsworth S C, Smith A E, Gregory R J. Effect of the E4 region on the persistence of transgene expression from adenovirus vectors. J Virol. 1997;71:2408–2416. doi: 10.1128/jvi.71.3.2408-2416.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Askari F K, Hitomi Y, Mao M, Wilson J M. Complete correction of hyperbilirubinemia in the Gunn rat model of Crigler-Najjar syndrome type I following transient in vivo adenovirus-mediated expression of human bilirubin UDP-glucuronosyltransferase. Gene Ther. 1996;3:381–388. [PubMed] [Google Scholar]
- 5.Barr D, Tubb J, Ferguson D, Scarla A, Lieber A, Wilson C, Perkins J, Kay M A. Strain related variations in adenovirally mediated transgene expression from mouse hepatocytes in vivo: comparisons between immunocompetent and immunodeficient inbred strains. Gene Ther. 1995;2:151–155. [PubMed] [Google Scholar]
- 6.Barr E, Carroll J, Kalynych A M, Tripathy S K, Kozarsky K, Wilson J M, Leiden J M. Efficient catheter-mediated gene transfer into the heart using replication-defective adenovirus. Gene Ther. 1994;1:51–58. [PubMed] [Google Scholar]
- 7.Clemens P R, Kochanek S, Sunada Y, Chan S, Chen H H, Campbell K P, Caskey C T. In vivo muscle gene transfer of full-length dystrophin with an adenoviral vector that lacks all viral genes. Gene Ther. 1996;3:965–972. [PubMed] [Google Scholar]
- 8.Cox G A, Cole N M, Matsumura K, Phelps S F, Hauschka S D, Campbell K P, Faulkner J A, Chamberlain J S. Overexpression of dystrophin in transgenic mdx mice eliminates dystrophic symptoms without toxicity. Nature. 1993;364:725–729. doi: 10.1038/364725a0. [DOI] [PubMed] [Google Scholar]
- 9.Engelhardt J F, Simon R H, Yang Y, Zepeda M, Weber-Pendleton S, Doranz B, Grossman M, Wilson J M. Adenovirus-mediated transfer of the CFTR gene to lung of nonhuman primates: biological efficacy study. Hum Gene Ther. 1993;4:759–769. doi: 10.1089/hum.1993.4.6-759. [DOI] [PubMed] [Google Scholar]
- 10.Gao G P, Yang Y P, Wilson J M. Biology of adenovirus vectors with E1 and E4 deletions for liver-directed gene therapy. J Virol. 1996;70:8934–8943. doi: 10.1128/jvi.70.12.8934-8943.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ghadge G D, Roos R P, Kang U J, Wollmann R, Fishman P S, Kalynych A M, Barr E, Leiden J M. CNS gene delivery by retrograde transport of recombinant replication-defective adenoviruses. Gene Ther. 1995;2:132–137. [PubMed] [Google Scholar]
- 12.Gilgenkrantz H, Duboc D, Juillard V, Couton D, Pavirani A, Guillet J G, Briand P, Kahn A. Transient expression of genes transferred in vivo into heart using first-generation adenoviral vectors—role of the immune response. Hum Gene Ther. 1995;6:1265–1274. doi: 10.1089/hum.1995.6.10-1265. [DOI] [PubMed] [Google Scholar]
- 13.Hardy S, Kitamura M, Harris-Stansil T, Dai Y M, Phipps M L. Construction of adenovirus vectors through Cre-lox recombination. J Virol. 1997;71:1842–1849. doi: 10.1128/jvi.71.3.1842-1849.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13a.Hauser, M. A., A. Amalfitano, and J. S. Chamberlain. Unpublished data.
- 14.Heikkila P, Parpala T, Lukkarinen O, Weber M, Tryggvason K. Adenovirus-mediated gene transfer into kidney glomeruli using an ex vivo and in vivo kidney perfusion system—first steps towards gene therapy of Alport syndrome. Gene Ther. 1996;3:21–27. [PubMed] [Google Scholar]
- 15.Horellou P, Vigne E, Castel M N, Barneoud P, Colin P, Perricaudet M, Delaere P, Mallet J. Direct intracerebral gene transfer of an adenoviral vector expressing tyrosine hydroxylase in a rat model of Parkinson’s disease. Neuroreport. 1994;6:49–53. doi: 10.1097/00001756-199412300-00014. [DOI] [PubMed] [Google Scholar]
- 16.Ilan Y, Droguett G, Chowdhury N R, Li Y A, Sengupta K, Thummala N R, Davidson A, Chowdhury J R, Horwitz M S. Insertion of the adenoviral E3 region into a recombinant viral vector prevents antiviral humoral and cellular immune responses and permits long-term gene expression. Proc Natl Acad Sci USA. 1997;94:2587–2592. doi: 10.1073/pnas.94.6.2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jones N, Shenk T. Isolation of deletion and substitution mutants of adenovirus type 5. Cell. 1978;13:181–188. doi: 10.1016/0092-8674(78)90148-4. [DOI] [PubMed] [Google Scholar]
- 18.Kaplan J M, Armentano D, Sparer T E, Wynn S G, Peterson P A, Wadsworth S C, Couture K K, Pennington S E, St. George J A, Gooding L R, Smith A E. Characterization of factors involved in modulating persistence of transgene expression from recombinant adenovirus in the mouse lung. Hum Gene Ther. 1997;8:45–56. doi: 10.1089/hum.1997.8.1-45. [DOI] [PubMed] [Google Scholar]
- 19.Kay M A, Holterman A X, Meuse L, Gown A, Ochs H D, Linsley P S, Wilson C B. Long-term hepatic adenovirus-mediated gene expression in mice following CTLA4Ig administration. Nat Genet. 1995;11:191–197. doi: 10.1038/ng1095-191. [DOI] [PubMed] [Google Scholar]
- 20.Kochanek S, Clemens P R, Mitani K, Chen H H, Chan S, Caskey C T. A new adenoviral vector—replacement of all viral coding sequences with 28 kb of DNA independently expressing both full-length dystrophin and beta-galactosidase. Proc Natl Acad Sci USA. 1996;93:5731–5736. doi: 10.1073/pnas.93.12.5731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kumarsingh R, Chamberlain J S. Encapsidated adenovirus minichromosomes allow delivery and expression of a 14 kb dystrophin cDNA to muscle cells. Hum Mol Genet. 1996;5:913–921. doi: 10.1093/hmg/5.7.913. [DOI] [PubMed] [Google Scholar]
- 22.Lieber A, He C Y, Kirillova I, Kay M A. Recombinant adenoviruses with large deletions generated by Cre-mediated excision exhibit different biological properties compared with first-generation vectors in vitro and in vivo. J Virol. 1996;70:8944–8960. doi: 10.1128/jvi.70.12.8944-8960.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lochmuller H, Petrof B J, Pari G, Larochelle N, Dodelet V, Wang Q, Allen C, Prescott S, Massie B, Nalbantoglu J, Karpati G. Transient immunosuppression by FK506 permits a sustained high- level dystrophin expression after adenovirus-mediated dystrophin minigene transfer to skeletal muscles of adult dystrophic (mdx) mice. Gene Ther. 1996;3:706–716. [PubMed] [Google Scholar]
- 24.Mittereder N, March K L, Trapnell B C. Evaluation of the concentration and bioactivity of adenovirus vectors for gene therapy. J Virol. 1996;70:7498–7509. doi: 10.1128/jvi.70.11.7498-7509.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nevins J R. Mechanism of activation of early viral transcription by the adenovirus E1a gene product. Cell. 1981;26:213–220. doi: 10.1016/0092-8674(81)90304-4. [DOI] [PubMed] [Google Scholar]
- 26.Nevins J R, Imperiale M J, Kao H T, Strickland S, Feldman L T. Detection of an adenovirus E1a-like activity in mammalian cells. Curr Top Microbiol Immunol. 1984;113:15–19. doi: 10.1007/978-3-642-69860-6_3. [DOI] [PubMed] [Google Scholar]
- 27.Niwa M, Rose S D, Berget S M. In vitro polyadenylation is stimulated by the presence of an upstream intron. Genes Dev. 1990;4:1552–1559. doi: 10.1101/gad.4.9.1552. [DOI] [PubMed] [Google Scholar]
- 28.Ohashi T, Watabe K, Uehara K, Sly W S, Vogler C, Eto Y. Adenovirus-mediated gene transfer and expression of human beta- glucuronidase gene in the liver, spleen, and central nervous system in mucopolysaccharidosis type VII mice. Proc Natl Acad Sci USA. 1997;94:1287–1292. doi: 10.1073/pnas.94.4.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parks R J, Chen L, Anton M, Sankar U, Rudnicki M A, Graham F L. A helper-dependent adenovirus vector system—removal of helper virus by cre-mediated excision of the viral packaging signal. Proc Natl Acad Sci USA. 1996;93:13565–13570. doi: 10.1073/pnas.93.24.13565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ragot T, Vincent N, Chafey P, Vigne E, Gilgenkrantz H, Couton D, Cartaud J, Briand P, Kaplan J C, Perricaudet M, et al. Efficient adenovirus-mediated transfer of a human minidystrophin gene to skeletal muscle of mdx mice. Nature. 1993;361:647–650. doi: 10.1038/361647a0. [DOI] [PubMed] [Google Scholar]
- 31.Sawchuk S J, Boivin G P, Duwel L E, Ball W, Bove F, Trapnell B, Hirsch R. Anti-T cell receptor monoclonal antibody prolongs transgene expression following adenovirus-mediated in vivo gene transfer to mouse synovium. Hum Gene Ther. 1996;7:499–506. doi: 10.1089/hum.1996.7.4-499. [DOI] [PubMed] [Google Scholar]
- 32.Smith T A G, White B D, Gardner J M, Kaleko M, McClelland A. Transient immunosuppression permits successful repetitive intravenous administration of an adenovirus vector. Gene Ther. 1996;3:496–502. [PubMed] [Google Scholar]
- 33.Stratford-Perricaudet L D, Makeh I, Perricaudet M, Briand P. Widespread long-term gene transfer to mouse skeletal muscles and heart. J Clin Invest. 1992;90:626–630. doi: 10.1172/JCI115902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tripathy S K, Black H B, Goldwasser E, Leiden J M. Immune responses to transgene-encoded proteins limit the stability of gene expression after injection of replication-defective adenovirus vectors. Nat Med. 1996;2:545–550. doi: 10.1038/nm0596-545. [DOI] [PubMed] [Google Scholar]
- 35.Vincent N, Ragot T, Gilgenkrantz H, Couton D, Chafey P, Gregoire A, Briand P, Kaplan J C, Kahn A, Perricaudet M. Long-term correction of mouse dystrophic degeneration by adenovirus-mediated transfer of a minidystrophin gene. Nat Genet. 1993;5:130–134. doi: 10.1038/ng1093-130. [DOI] [PubMed] [Google Scholar]
- 36.Wolff J A, Ludtke J J, Acsadi G, Williams P, Jani A. Long-term persistence of plasmid DNA and foreign gene expression in mouse muscle. Hum Mol Genet. 1992;1:363–369. doi: 10.1093/hmg/1.6.363. [DOI] [PubMed] [Google Scholar]
- 37.Xiao X A, Li J A, Samulski R J. Efficient long-term gene transfer into muscle tissue of immunocompetent mice by adeno-associated virus vector. J Virol. 1996;70:8098–8108. doi: 10.1128/jvi.70.11.8098-8108.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang Y, Ertl H C, Wilson J M. MHC class I-restricted cytotoxic T lymphocytes to viral antigens destroy hepatocytes in mice infected with E1-deleted recombinant adenoviruses. Immunity. 1994;1:433–442. doi: 10.1016/1074-7613(94)90074-4. [DOI] [PubMed] [Google Scholar]
- 39.Yang Y, Greenough K, Wilson J M. Transient immune blockade prevents formation of neutralizing antibody to recombinant adenovirus and allows repeated gene transfer to mouse liver. Gene Ther. 1996;3:412–420. [PubMed] [Google Scholar]
- 40.Yang Y, Li Q, Ertl H C, Wilson J M. Cellular and humoral immune responses to viral antigens create barriers to lung-directed gene therapy with recombinant adenoviruses. J Virol. 1995;69:2004–2015. doi: 10.1128/jvi.69.4.2004-2015.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang Y, Nunes F A, Berencsi K, Furth E E, Gonczol E, Wilson J M. Cellular immunity to viral antigens limits E1-deleted adenoviruses for gene therapy. Proc Natl Acad Sci USA. 1994;91:4407–4411. doi: 10.1073/pnas.91.10.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang Y, Nunes F A, Berencsi K, Gonczol E, Engelhardt J F, Wilson J M. Inactivation of E2a in recombinant adenoviruses improves the prospect for gene therapy in cystic fibrosis. Nat Genet. 1994;7:362–369. doi: 10.1038/ng0794-362. [DOI] [PubMed] [Google Scholar]
- 43.Yang Y, Su Q, Wilson J M. Role of viral antigens in destructive cellular immune responses to adenovirus vector-transduced cells in mouse lungs. J Virol. 1996;70:7209–7212. doi: 10.1128/jvi.70.10.7209-7212.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang Y, Wilson J M. Clearance of adenovirus-infected hepatocytes by MHC class I-restricted CD4+ CTLs in vivo. J Immunol. 1995;155:2564–2570. [PubMed] [Google Scholar]
- 45.Yang Y P, Xiang Z Q, Ertl H C J, Wilson J M. Upregulation of class I major histocompatibility complex antigens by interferon gamma is necessary for T-cell-mediated elimination of recombinant adenovirus-infected hepatocytes in vivo. Proc Natl Acad Sci USA. 1995;92:7257–7261. doi: 10.1073/pnas.92.16.7257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou H S, Oneal W, Morral N, Beaudet A L. Development of a complementing cell line and a system for construction of adenovirus vectors with E1 and E2a deleted. J Virol. 1996;70:7030–7038. doi: 10.1128/jvi.70.10.7030-7038.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]