mRNA Instability Elements in the Human Papillomavirus Type 16 L2 Coding Region (original) (raw)
Abstract
Human papillomavirus capsid proteins L1 and L2 are detected only in terminally differentiated cells, indicating that expression of the L1 and L2 genes is blocked in dividing cells. The results presented here establish that the human papillomavirus type 16 L2 coding region contains _cis_-acting inhibitory sequences. When placed downstream of a reporter gene, the human papillomavirus type 16 L2 sequence reduced both mRNA and protein levels in an orientation-dependent manner. Deletion analysis revealed that the L2 sequence contains two _cis_-acting inhibitory RNA regions. We identified an inhibitory region in the 5′-most 845 nucleotides of L2 that acted by reducing cytoplasmic mRNA stability and a second, weaker inhibitory region in the 3′ end of L2. In contrast, human papillomavirus type 1 L1 and L2 genes did not encode strong inhibitory sequences. This result is consistent with observations of high virus production in human papillomavirus type 1-infected tissue, whereas only low levels of human papillomavirus type 16 virions are detectable in infected epithelium. The presence of inhibitory sequences in the L1 and L2 mRNAs may aid the virus in avoiding the host immunosurveillance and in establishing persistent infections.
Human papillomaviruses (HPVs) are nonenveloped DNA tumor viruses that can induce a variety of proliferative lesions upon infection of epithelial cells (28, 53, 70, 72). To date, more than 70 different HPV types have been identified (24). Each of these types infects either mucosal or cutaneous epithelium at distinct anatomical sites. Members of a subset of the HPV types are etiological agents of cancers, e.g., HPV-16, and are referred to as high-risk types, whereas certain HPV types are rarely or never found in cancers, e.g., HPV-1, and are referred to as low-risk types (28, 35, 53, 71). HPV-1 infects cutaneous epithelium at the plantar surface of the foot, whereas HPV-16 shows tropism for mucosal epithelial cells.
The production of HPV virions is strictly linked to the differentiation stage of the infected epithelial cell, and viral late-gene products, L1 and L2 (Fig. 1), are detected primarily in the terminally differentiated cells in the upper layers of the epithelium (11, 23, 32, 33, 53, 56, 61). One reason for the restriction of HPV late-gene expression to terminally differentiated cells and for the differences observed in the levels of expression of late-gene products from various HPV types may be the presence of negative regulatory elements in the HPV late mRNAs. Such elements were originally described by Kennedy et al. (29, 30), who reported the identification of an inhibitory sequence in the HPV-16 late 3′ untranslated region (UTR) (Fig. 1) which acted by reducing mRNA stability in vitro. Other investigators proposed that the activity of this negative regulatory element required the presence of a 5′ splice site-like sequence (21). In an attempt to produce HPV-16 L1 from eucaryotic expression plasmids encoding L1 cDNAs, we reasoned that deletion of the negative sequence in the late 3′ UTR would allow high L1 production. However, deletion of the late 3′ UTR from an HPV-16 L1 cDNA did not result in the production of detectable levels of L1 (58), indicating that the L1 coding region itself contained sequences that inhibit L1 production. These sequences acted in cis and inhibited the expression of a reporter gene to the extent of several hundredfold (58). Subsequent experiments demonstrated that inhibitory sequences were located primarily in the 5′ half of the L1 coding region and spanned several hundred nucleotides (58).
FIG. 1.

Genomic map of HPV-16. Numbers indicate nucleotide positions (51), the shaded box indicates the L2 coding region, and the white box indicates the L1 coding region. NCR, noncoding region, containing the late 3′ UTR; pA, HPV-16 late polyadenylation signal.
In the work described here, we investigated if HPV-16 L2 (Fig. 1) contains sequences that negatively affect L2 expression levels. The results presented here demonstrate that the HPV-16 L2 coding region contains _cis_-acting inhibitory RNA sequences that act by reducing mRNA and protein levels. A sequence in the 5′ end of HPV-16 L2 acted as an mRNA instability determinant, and a weaker, posttranscriptionally active sequence was found in the 3′ end of L2. We also show that the HPV-1 L1 and L2 coding regions do not contain strong inhibitory sequences. HPV-1 virions are easily detected in vivo, whereas HPV-16 virions are not. Therefore, the presence of inhibitory sequences in L1 and L2 correlates with the amounts of virus produced in vivo.
MATERIALS AND METHODS
Plasmid constructions.
The following plasmids have been described elsewhere: pE55 (59), pNLCATW (58), and pCS1X (58); pC16L1(A) and pC16L1(S) have been described as pCATL1A and pCATL1S (58), respectively, and pT7-16L2 has been described as pT7L2 (25).
pH16L2 was generated by excising a _Bss_HII-_Kpn_I fragment from pT7-16L2, followed by insertion into _Bss_HII- and _Kpn_I-digested pNLCATW (58), thereby replacing the chloramphenicol acetyltransferase (CAT) gene with the HPV-16 L2 open reading frame (ORF). To generate pCMV16L2, the HPV-16 L2 coding region was PCR amplified with oligonucleotides L2START (5′-CAGCGCGCCCTTAACAATGCGACACAAACG-3′) and L2STOP (5′-CAGTCGACCGTGGCCTCACTAGGCAGCC-3′) and inserted into _Hpa_I-digested, calf intestinal alkaline phosphatase (CIAP)-treated pLNCX (36). pC16L2(S) and pC16L2(A) were produced by subcloning, in the sense and antisense orientations, respectively, an HPV-16 L2 DNA fragment that had been PCR amplified from pT7-16L2 with oligonucleotides L2START and L2STOP into _Asp_718-digested, Klenow fragment-treated pNLCATW (58). pC16L2-Stop was constructed by annealing oligonucleotide GC-BAMHI (5′-CGCGCGGGGGGGGGGGGGGGGGGGGGGGGATCCCCCCCCCCCCCCCCCCCCCCCCG-3′), followed by subcloning into pC16L2(S) that had been digested with _Bss_HII and treated with CIAP. Digestion of pC16L2(S) with _Mlu_I and _Bss_HII, followed by T4 DNA polymerase treatment and religation, generated p16L2ΔC. pCL2D was generated by digestion of pC16L2(S) with _Sal_I and _Spe_I and religation. To generate pCL2C, pC16L2(S) was digested completely with _Sal_I and partially with _Spe_I and religated. A fragment that had been PCR amplified from pT7-16L2 with oligonucleotides L2START and L2M (5′-CGTCGACCTGGAGCTATATTAATAC-3′) was ligated into pBluescript that had been digested with _Eco_RV and treated with CIAP, generating pKSL2B. To generate pCL2B, a _Bss_HII-_Sal_I fragment from pKSL2B was ligated to pC16L2(S) that had been digested with _Mlu_I and _Sal_I. pCL2A was constructed by digestion of pC16L2(S) with _Spe_I, followed by religation. pCL2R1, pCL2R2, pCL2R4, pCL2R5, and pCL2R7 were constructed by insertion into _Asp_718-digested, Klenow fragment-treated pNLCATW (58) of HPV-16 L2 DNA fragments that had been PCR amplified with the following primer pairs: L2D (5′-CGTCGACGGAATTAATGAAGGAGCTTGG-3′) and L2B (5′-CACGCGTCAGTAACTAGTAGCACACCCA-3′), L2C (5′-CACGCGTAATATAGCTCCAGATCCTGAC-3′) and L2STOP, L2B and L2G (5′-CGTCGACGGATCAATAGTACTTAAA-3′), L2E (5′-CACGCGTCTATTGATCCTGCAGAAG-3′) and L2STOP, and L2C and L2D. To generate pL2HU(S) and pL2HU(A), HPV-16 L2 sequences were PCR amplified with oligonucleotides L2B and L2STOP and inserted in the sense and antisense orientations, respectively, into _Stu_I-digested pNLCATW (58).
pC1L1(S) and pC1L1(A) were generated by subcloning into _Asp_718-digested, Klenow fragment-treated, CIAP-treated pNLCATW (58), in the sense and antisense orientations, respectively, HPV-1 L1 sequences that had been PCR amplified from pHPV-1 (17) with oligonucleotides H1L1STOP (5′-GTTATATAGAATTCATACTAAGCC-3′) and H1L1START (5′-AGCGTCGACAAAGAGCTTATGT-3′). pC1L2(S) and pC1L2(A) were generated by subcloning into _Asp_718-digested, Klenow fragment-treated pNLCATW (58), in the sense and antisense orientations, respectively, HPV-1 L2 sequences that had been PCR amplified with H1L2S (5′-CGTCGACGTAACAAATGTATCGCCTACG-3′) and H1L2A (5′-AGAATTCCATTATACATAAGCTCTTTTACG-3′).
pHCMVtat was constructed by subcloning a _Sal_I-_Hpa_I human immunodeficiency virus type 1 (HIV-1) Tat-encoding fragment from pNL147 (48) into _Sal_I- and _Hpa_I-digested pCH16pA (58). pKSNLCAT was generated by ligation of a _Hin_dIII-_Eco_RI fragment from pNLCATW into pBluescript (Stratagene) that had been digested with _Hin_dIII and _Eco_RI.
Cells, transfections, and CAT ELISA.
For transfection of adherent cells (HLtat [48], 293, NIH 3T3, BHK-21, and CV-1), 3 × 105 cells were seeded per 60-mm-diameter plate 24 h prior to transfection. Plasmid pHCMVtat, producing HIV-1 Tat, was included in transfections. Transient transfections were carried out by the calcium phosphate coprecipitation technique (22) as described previously (60). For suspended cells (Jurkat and U937), 106 cells were transfected with Lipofectamine (Life Technologies) according to the manufacturer’s instructions. The cells were harvested 20 to 48 h posttransfection, and the amount of CAT protein was quantified in a CAT antigen capture enzyme-linked immunosorbent assay (ELISA; Boehringer GmbH). pCS1X (58) was included as an internal control in each transfection experiment, and secreted alkaline phosphatase (SEAP) activity was determined as previously described (58). In transfections executed for downstream RNA analysis, pHCMVtat was included as an internal control. When the vaccinia virus T7 RNA polymerase expression system (20) was used, cells were infected with 0.5 × 106 PFU of recombinant vaccinia virus vTF7-3 (20) expressing T7 RNA polymerase 1 to 2 h prior to transfection. Each experiment was repeated at least three times, and mean values of representative results are shown.
RNA preparation and primer extension.
Total cytoplasmic RNA was prepared from transfected HeLa or HLtat cells as previously described (60). For fractionation and preparation of nuclear RNA, the cells were lysed directly in culture dishes with Nonidet P-40 (NP-40) lysis buffer (10 mM Tris-Cl [pH 7.5], 150 mM NaCl, 1.5 mM MgCl2, 0.65% [NP-40 Sigma]). The nuclei were washed three times in NP-40 lysis buffer and then scraped off the culture dishes into 250 μl of NP-40 lysis buffer with a rubber policeman. Nuclei from two 60-mm-diameter culture dishes were pooled. Sodium dodecyl sulfate (SDS) was added to a final concentration of 0.2%, and nuclei were lysed on ice for 5 min, with repeated vortexing. Following freezing-thawing on dry ice and at 37°C, the samples were treated with DNase I for 10 min at 37°C. The samples were extracted twice with an equal volume of phenol-chloroform [at a ratio of 1:1; phenol was equilibrated in 1 M 3-(_N_-morpholino)propanesulfonic acid (MOPS; pH 6.0)], followed by extraction with chloroform and ethanol precipitation. Pellets were resuspended in water, RNA was quantified by spectrophotometry, and the quality of the RNA was checked on agarose gels.
Primer extension was carried out by coprecipitating 50 μg of nuclear or cytoplasmic RNA with 105 cpm of [γ-32P]ATP-labelled oligonucleotides NL-PX2 (5′-GGGCACACACTACTTTGAG-3′), CMV-PX3 (5′-TGGATCGGTCCCGGTGTCTT-3′), and 47S-PX6 (5′-GCCAGAGCCCCGCGCGCATC-3′). Oligonucleotide NL-PX2 hybridizes to the 5′ end of mRNA transcribed from the HIV-1 long terminal repeat (LTR) promoter, oligonucleotide CMV-PX3 hybridizes to the 5′ end of mRNA transcribed from the human cytomegalovirus (CMV) immediate-early promoter, and oligonucleotide 47S-PX6 hybridizes to the 5′ end of the 47S rRNA precursor transcript. Annealing was performed by resuspending the RNA pellet in 8 μl of 1× RT buffer (50 mM Tris-Cl [pH 8.3], 75 mM KCl, 3 mM MgCl2, 0.5 mM each deoxynucleoside triphosphate [dNTP]), followed by incubation at 65°C for 1 min, at 37°C for 1 min, and on ice for 1 min. cDNA synthesis was carried out by adding 8 μl of 1× RT mixture (50 mM Tris-Cl [pH 8.3], 75 mM KCl, 3 mM MgCl2, 10 mM dithiothreitol, 0.5 mM each dNTP, 5 U of Molony murine leukemia virus reverse transcriptase [Life Technologies] per μl, 0.25 U of RNA Guard [Pharmacia] per μl), followed by incubation at 42°C for 60 min. The reaction was stopped by RNase treatment (1 μg of RNase A and 20 U of RNase T1 [Ambion]) at 37°C for 10 min, followed by ethanol precipitation. The pellet was resuspended in formamide loading dye (80% deionized formamide, 1 mM EDTA, 0.1% bromphenol blue, 0.1% xylene cyanol), and the suspension was boiled for 2 min. The reaction products were separated on 6% polyacrylamide–urea gels. Gels were analyzed by autoradiography, and RNA levels were quantified with a PhosphorImager (Molecular Dynamics). Each experiment was repeated at least three times, and representative results are shown.
Northern RNA blotting.
Northern RNA blotting was performed essentially as described previously (58). An antisense [α-32P]UTP-labelled riboprobe was generated from _Hin_dIII-linearized pKSNLCAT as described previously (68). The synthetic RNA was complementary to the 5′ ends of mRNAs produced from pNLCATW-derived plasmids and contained 180 nucleotides (nt) of the HIV-1 5′ LTR and 213 nt of the 5′ end of the CAT gene.
Extraction of poly(A)+ mRNA and reverse transcription (RT)-PCR.
Cytoplasmic poly(A)+ mRNA was isolated with Dynabeads Oligo (dT)25 (Dynal A. S.) as described previously (60). Briefly, transfected cells were lysed for 5 min on ice in NP-40 lysis buffer. Cytoplasmic and nuclear fractions were separated by centrifugation at 8,000 × g for 2 min. Supernatants were incubated with an equal volume of 2× binding buffer (20 mM Tris-HCl [pH 7.5], 1.0 M LiCl, 2 mM EDTA, 0.5% SDS) containing 400 μg of Dynabeads Oligo (dT)25. After three washes in washing buffer (10 mM Tris-HCl [pH 7.5], 0.15 M LiCl, 2 mM EDTA), poly(A)+ mRNAs were eluted from the beads with elution buffer (2 mM EDTA [pH 7.5]) at 65°C for 2 to 3 min and stored at −70°C until use.
RT-PCR was performed as previously described (60). Briefly, fourfold serially diluted cytoplasmic poly(A)+ mRNA was reverse transcribed at 42°C for 1 h in a total reaction volume of 30 μl with random hexamers. Five microliters of the cDNA product was PCR amplified in a 100-μl reaction volume with oligonucleotides CATS-2 (5′-CGTCTCAGCCAATCCCTGGGTG-3′) and CATA (5′-CTATTAGGCCCCGCCCTGCCACTC-3′) to detect cDNA of the CAT or CAT-HPV-16 L2 hybrid mRNA or EP (5′-AGGTGACGGTACAAGGGTCTCAGAAA-3′) and EW (5′-CCCACCATGTTCTTTCAAAGGC-3′) to detect cDNA of the equine infectious anemia virus (EIAV) gag mRNA produced from the internal control plasmid pE55 (59). PCR was performed in a total reaction volume of 100 μl for 25 cycles at 94°C for 1 min, 55°C for 1 min, and 72°C for 1 min, with a final extension at 72°C for 10 min. A 10-μl sample from each RT-PCR was analyzed by electrophoresis on 5% polyacrylamide gels.
Radioimmunoprecipitation.
Transfected cells were starved for 30 min in Met-free medium containing 0.5% fetal calf serum, followed by metabolic labelling for 1 h with 200 μCi of [35S]Met. The cells were washed and lysed in ice-cold RIPA buffer (25 mM Tris-HCl [pH 7.4], 75 mM NaCl, 0.5% Triton X-100, 0.5% sodium deoxycholate, 0.05% SDS). After three freezing-thawings, the cell extracts were centrifuged, and supernatants were collected, mixed with normal guinea pig serum, and incubated for 30 min at 4°C. Protein A-Sepharose (Pharmacia) beads were added, and incubation was continued for 1 h, followed by centrifugation and collection of supernatants. Normal guinea pig serum or guinea pig anti–HPV-16 L2 peptide antiserum (18) was added, and incubation was performed at 4°C for 16 h, followed by the addition of protein A-Sepharose beads and continued incubation for 3 h. The samples were heated, loaded onto 10% polyacrylamide–SDS gels (acrylamide/bisacrylamide ratio, 29:1) under reducing conditions, and electrophoresed at 180 V. The gels were dried and autoradiographed at −70°C.
RESULTS
The HPV-16 L2 protein can be efficiently produced in HeLa cells by use of the vaccinia virus T7 RNA polymerase-based expression system but not by use of eucaryotic expression plasmids.
We first attempted to express HPV-16 L2 (Fig. 1) from plasmids containing the HIV-1 LTR promoter (pH16L2) (Fig. 2A) or the CMV promoter (pCMV16L2) (Fig. 2A), which we have used previously for high expression of other virus genes, e.g., those encoding EIAV and HIV-1 proteins (48, 49, 59). However, the levels of L2 produced from these plasmids were undetectable (Fig. 2). In contrast, transfection of plasmid pT7-16L2, which contains the bacteriophage T7 promoter (Fig. 2A), into HeLa cells infected with a recombinant vaccinia virus producing T7 RNA polymerase (20) yielded high levels of L2 protein (Fig. 2). In the latter case, transcription of the plasmid occurs in the cytoplasm, while in the former case, nuclear factors are required. We do not know if the high L2 expression levels observed in the vaccinia virus T7 RNA polymerase expression system are a result of the bypassing of the nucleus, overall high transcription levels in this expression system, or interactions between vaccinia virus and the infected cell. We obtained similar results previously using the HPV-16 L1 gene (58). Our results indicated that the HPV-16 L2 coding region contains inhibitory sequences.
FIG. 2.

(A) Structures of the L2 expression plasmids. Shaded boxes indicate the HPV-16 L2 coding region, striped boxes represent HIV-1 LTRs, and triangles represent poly(A) signals (pA). Plasmid names are indicated on the left. T7, bacteriophage T7 RNA polymerase promoter; CMV, CMV immediate-early promoter. (B) Radioimmunoprecipitation of HPV-16 L2 from HeLa cells infected with recombinant vaccinia virus vTF7-3 (20) and transfected with pT7-16L2 or HLtat cells (48) transfected with pH16L2 or pCMV16L2. P, preimmune guinea pig serum; L, guinea pig anti–HPV-16 L2 peptide antiserum (18). Numbers indicate molecular masses in kilodaltons.
The HPV-16 L2 coding region contains _cis_-acting inhibitory sequences that act in an orientation-dependent manner.
To investigate if the HPV-16 L2 coding region contains sequences that inhibit gene expression, the entire HPV-16 L2 coding region was inserted, in sense and antisense orientations, downstream of the CAT reporter gene in plasmid pNLCATW (58), resulting in plasmids pC16L2(S) and pC16L2(A), respectively (Fig. 3). These plasmids were separately transfected in triplicate into HLtat cells (48) in the presence of the SEAP-producing plasmid pCS1X (58), included as an internal control for transfection efficiency. The standard deviation was less than 30% in all experiments shown, and all plasmids were analyzed in a minimum of three independent transfection experiments. Mean values of CAT levels produced after triplicate transfections revealed that pC16L2(S) produced 49-fold-lower levels of CAT than pNLCATW (58), whereas pC16L2(A) and pNLCATW (58) produced similar levels of CAT (Fig. 3). These results demonstrated that the HPV-16 L2 coding region contains _cis_-acting inhibitory sequences that acted in an orientation-dependent manner to reduce CAT production. For comparison, we analyzed the effect on CAT expression of the HPV-16 L1 coding region. The results demonstrated that pC16L1(S) produced 202-fold-lower CAT levels than pNLCATW (58) and that the presence of L1 in an antisense orientation had a lower inhibitory effect (Fig. 3), as we described previously (58). The HPV-16 L1 sequence had stronger inhibitory activity than the L2 sequence.
FIG. 3.

Schematic structures of CAT expression plasmids. The HIV-1 LTR (striped boxes), the CAT gene, and the HPV-1 or HPV-16 L1 and L2 sequences are indicated. Plasmids contained the L1 or L2 ORF from the first ATG to the translational stop codon. Plasmid names are shown on the left. Arrows indicate sense (→) and antisense (←) orientations. The depicted plasmids were transfected in triplicate into HLtat cells, and CAT levels were monitored in a CAT ELISA and normalized to SEAP levels produced from the internal control plasmid pCS1X (58). The mean CAT values obtained after triplicate transfections with pNLCATW (58) were divided by the mean CAT values obtained after triplicate transfections with the indicated plasmids to yield fold inhibition.
The HPV-1 L1 and L2 coding regions do not contain strong inhibitory sequences.
We next investigated if the presence of inhibitory sequences in the L1 and L2 coding regions is a general property of HPVs. The L1 and L2 coding sequences from HPV-1 were separately inserted in the sense and antisense orientations downstream of the CAT gene in pNLCATW (58), resulting in pC1L1(S), pC1L1(A), pC1L2(S), and pC1L2(A) (Fig. 3). These plasmids were transfected into HLtat cells (48), and the levels of CAT were determined. The results showed that HPV-1 L1 and L2 inhibited CAT expression 13- and 3-fold, respectively (Fig. 3), demonstrating that the HPV-1 L1 and L2 coding sequences encode only low inhibitory activity. The ratios between CAT levels produced from each plasmid containing HPV sequences in the sense orientation and CAT levels produced from the corresponding plasmid containing HPV sequences in the antisense orientation were 0.32 and 1.1 for the HPV-1 L1 [pC1L1(S)/pC1L1(A)] and L2 [pC1L2(S)/pC1L2(A)] plasmids, respectively, whereas the corresponding ratios for the HPV-16 L1 [pC16L1(S)/pC16L1(A)] and L2 [pC16L2(S)/pC16L2(A)] plasmids were 0.05 and 0.03, respectively. We concluded that the HPV-16 L1 and L2 coding regions contain strong inhibitory sequences located on the coding strand; the HPV-1 L2 sequence appeared to lack significant inhibitory activity, whereas the HPV-1 L1 sequence had weak inhibitory activity.
The inhibitory sequences in HPV-16 L2 are active in cells of different origins.
We transfected pC16L2(A) or pC16L2(S) (Fig. 2A) into cell lines of different origins (Table 1) to test if the inhibitory activity of the L2 sequences was restricted to epithelial cells. Table 1 shows a 14- to 126-fold difference in CAT production between pC16L2(A) and pC16L2(S) in the cell lines, indicating that the inhibitory sequences were functional in different cell types and in cells from different species. However, the inhibitory element in HPV-16 L2 acted most efficiently in human epithelial cells. The results indicated that the regulatory mechanism involving the HPV-16 L2 sequences is evolutionarily conserved and is not cell type specific.
TABLE 1.
The negative element in HPV-16 L2 is active in cells of different origins
| Cell line | Origin | Fold inhibition |
|---|---|---|
| HeLa | Human epithelioid | 126 |
| 293 | Human epithelial morphology | 14 |
| Jurkat | Human T cell | 50 |
| U937 | Human monocyte | 14 |
| CV-1 | Monkey fibroblast morphology | 39 |
| BHK-21 | Hamster fibroblast morphology | 55 |
| NIH 3T3 | Mouse fibroblast | 68 |
The HPV-16 L2 coding region contains _cis_-acting sequences that reduce cytoplasmic and nuclear mRNA levels.
To further investigate the inhibitory effect of the HPV-16 L2 coding sequence, plasmids pC16L2(S), pC16L2(A), and pNLCATW were separately transfected into HLtat cells in the presence of the internal control plasmid pHCMVtat (see Materials and Methods), and cytoplasmic mRNA levels were monitored by primer extension. The results revealed that pNLCATW and pC16L2(A) produced similar mRNA and protein levels, whereas pC16L2(S) produced significantly lower mRNA and protein levels than pC16L2(A) and pNLCATW (Fig. 4A), respectively. The differences were 20- to 13-fold at the mRNA level and 133- to 147-fold at the protein level. Quantitation of mRNA and protein levels in cells transfected with serially diluted pNLCATW verified that the analysis was performed in the linear range of the assays (Fig. 4B). In addition, we analyzed cytoplasmic poly(A)+ mRNA by RT-PCR with fourfold serially diluted mRNA. The results demonstrated that the mRNA levels produced from pC16L2(S) were approximately 30- to 60-fold lower than those produced from pNLCATW (Fig. 4C). The levels of internal control EIAV gag mRNA did not vary significantly between the two transfections. The CAT protein levels produced from pC16L2(S) were 140-fold lower than those produced from pNLCATW in the same transfection experiment (Fig. 4C).
FIG. 4.
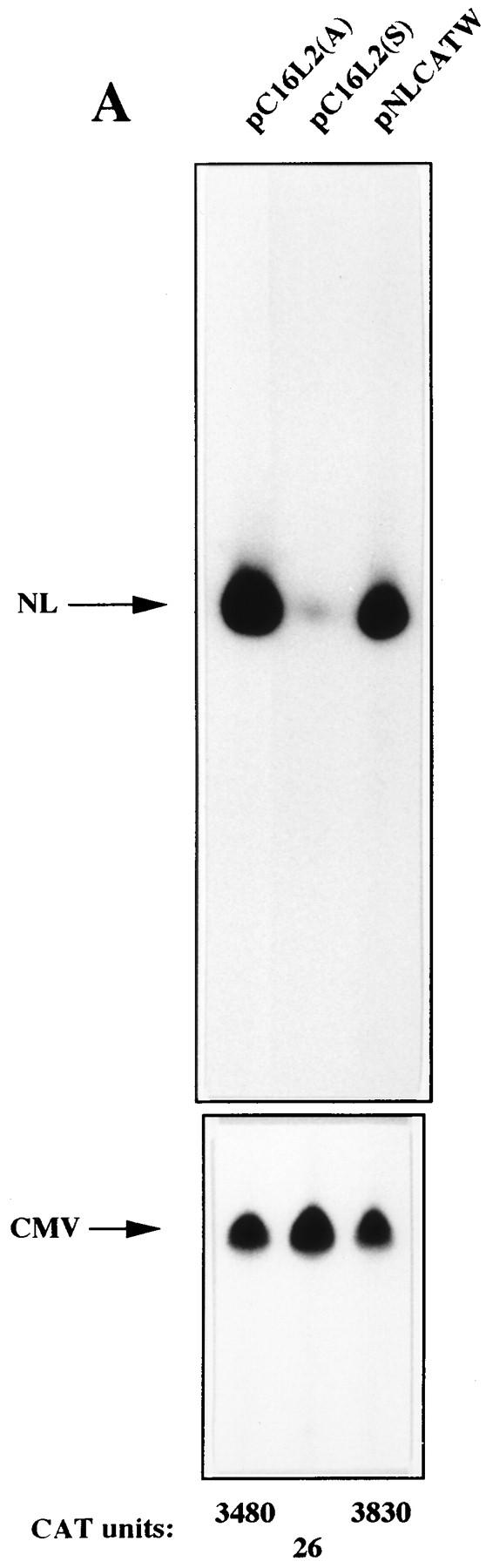
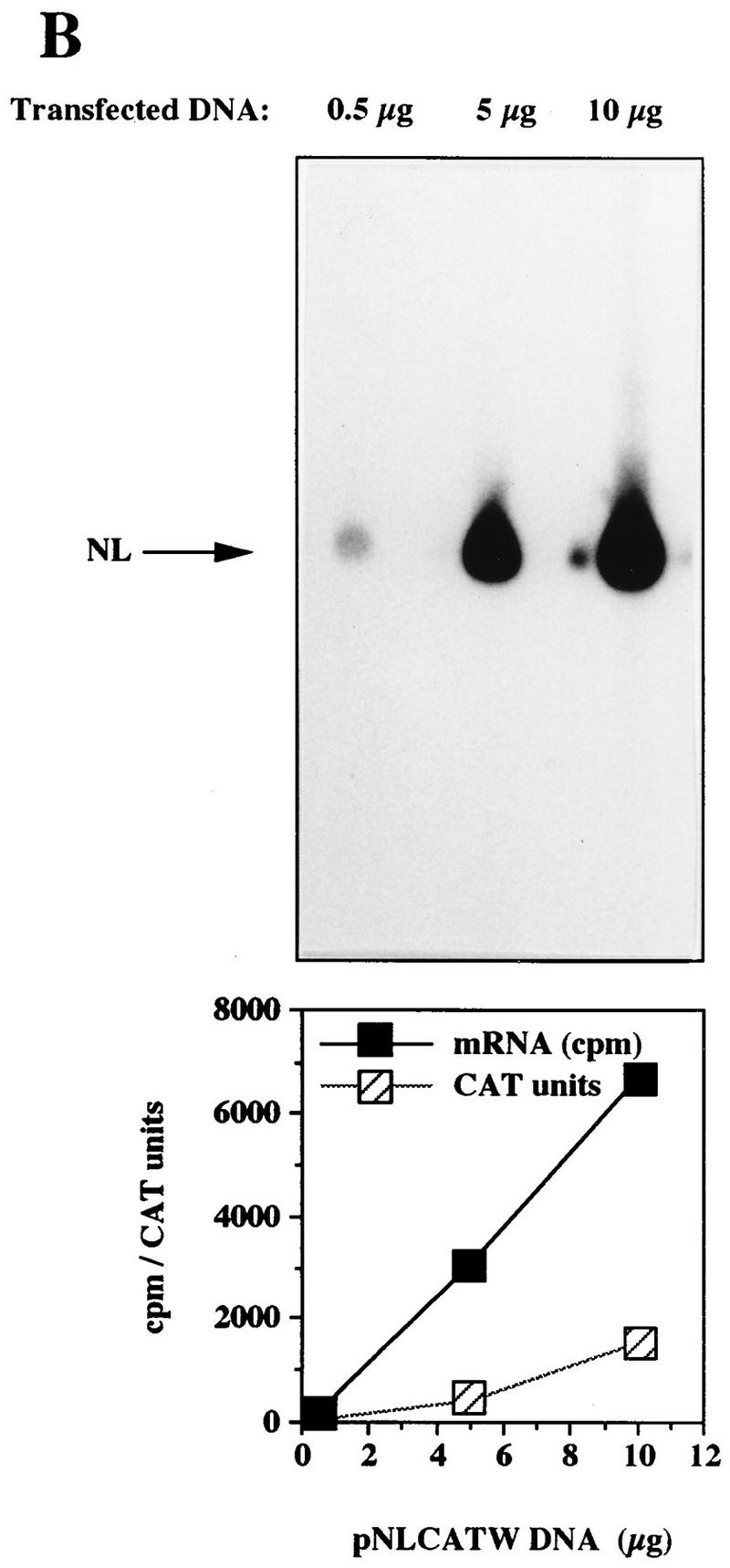

The HPV-16 L2 inhibitory sequences reduce cytoplasmic mRNA levels. (A) Cytoplasmic mRNA levels produced from the indicated plasmids were detected by primer extension. NL, extension products of mRNAs derived from the HIV-1 LTR promoter; CMV, extension products of the internal control mRNA derived from the CMV immediate-early promoter in pHCMVtat; CAT units, CAT protein levels produced from the indicated plasmids. (B) Serially diluted pNLCATW plasmid DNA was transfected into HLtat cells. Cytoplasmic mRNA levels (counts per minute [cpm]) were quantified by use of a PhosphorImager, and CAT protein levels (CAT units) were quantified by a CAT antigen capture ELISA. NL, primer extension products of mRNAs derived from the HIV-1 LTR promoter in pNLCATW. (C) RT-PCR of fourfold serially diluted cytoplasmic poly(A)+ mRNAs extracted from HLtat cells transfected with plasmid pNLCATW or pC16L2(S) as indicated. The upper panel shows PCR amplification with oligonucleotide primers specific for cDNA generated from the CAT mRNAs produced from pNLCATW and pC16L2(S), and the lower panel shows PCR amplification with oligonucleotide primers specific for cDNA generated from the EIAV gag mRNAs produced from the internal control plasmid pE55 (59). RT-, amplification from poly(A)+ mRNA in the absence of reverse transcriptase.
Next, cytoplasmic and nuclear RNAs were extracted and analyzed by primer extension. The mRNA levels produced from pC16L2(S) were lower than those produced from pNLCATW in both cellular compartments (Fig. 5A). The results in Fig. 5B show that the mRNA levels produced from pC16L2(S) were 13-fold lower in the cytoplasm and 6-fold lower in the nucleus than those produced from pNLCATW. Analysis of the subcellular distribution of the pC16L2(S) and pNLCATW mRNAs showed that 25 and 41% were cytoplasmic, respectively (Fig. 5). pHCMVtat mRNA was evenly distributed in the cytoplasmic and nuclear compartments (Fig. 5A; 46 and 40% of the pHCMVtat mRNAs were cytoplasmic in the two transfections). The 47S rRNA precursor was found only in the nuclear fractions (Fig. 5A), as expected. CAT protein levels produced from pC16L2(S) in the same transfection experiment were 73-fold lower than those produced from pNLCATW (Fig. 5B). It was observed in all transfection experiments that the inhibitory effect was greater at the protein level than at the mRNA level. These results indicated that the L2 sequence acted by reducing mRNA levels in the nuclear and cytoplasmic compartments and suggested an additional inhibitory effect on mRNA utilization.
FIG. 5.
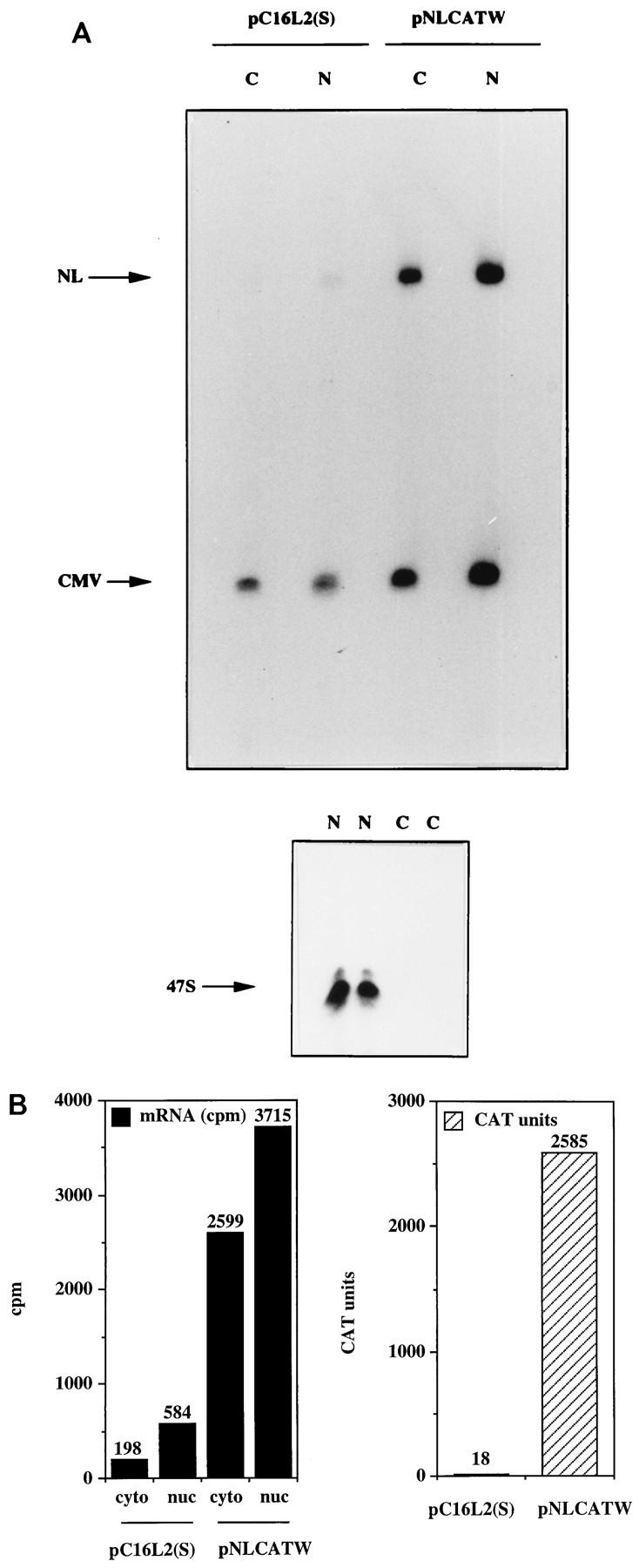
The HPV-16 L2 inhibitory sequences reduce mRNA levels in both cytoplasmic and nuclear compartments. (A) Cytoplasmic (C) and nuclear (N) mRNA levels produced from the indicated plasmids were detected by primer extension. NL, extension products of mRNAs derived from the HIV-1 LTR promoter; CMV, extension products of the internal control mRNA derived from the CMV immediate-early promoter in pHCMVtat; 47S, extension product of the 47S precursor rRNA. (B) Histogram showing cytoplasmic (cyto) and nuclear (nuc) mRNA levels (counts per minute [cpm]) quantified by use of a PhosphorImager and CAT protein levels (CAT units) produced from the indicated plasmids.
HPV-16 L2 contains cytoplasmic mRNA instability determinants.
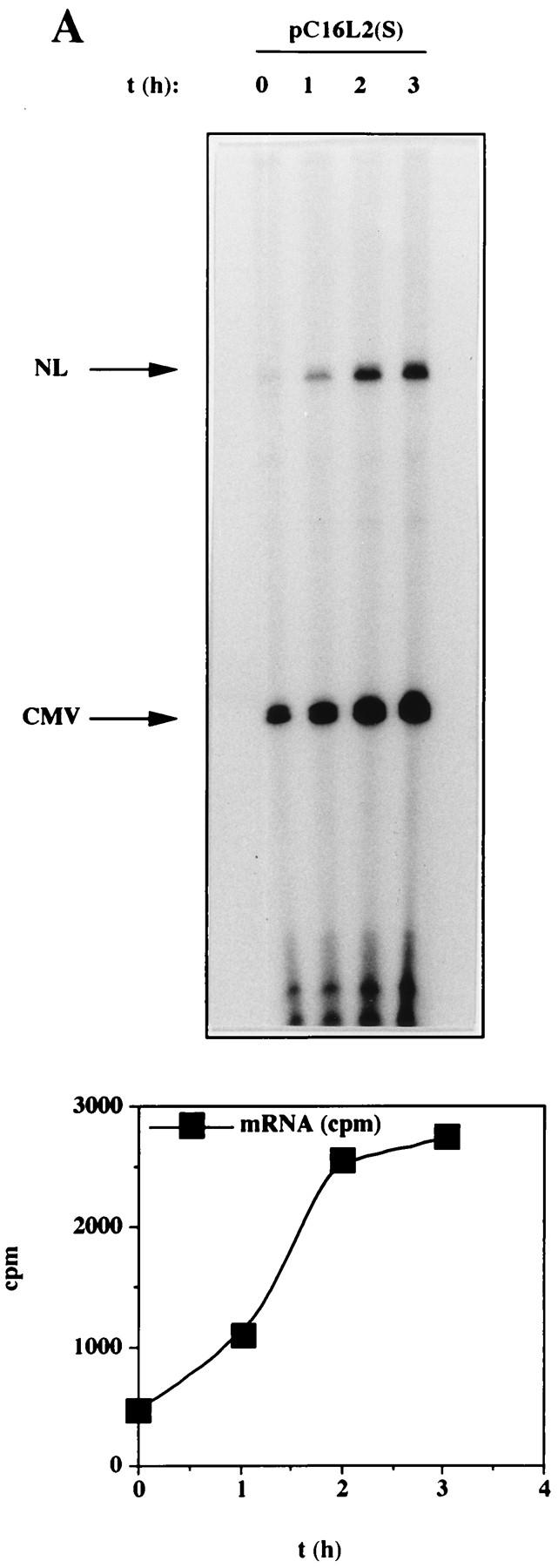
To investigate if the decreased levels of L2-containing mRNAs could be explained by a reduced mRNA half-life, HLtat cells were transfected with pC16L2(S) or pNLCATW and treated with actinomycin D for 0, 30, and 60 min. Cytoplasmic RNA was extracted, and mRNA levels were quantified by primer extension. Figure 6A shows that the mRNAs produced from pC16L2(S) were less stable than those produced from pNLCATW, a result which explains, at least in part, the reduced steady-state levels of L2-containing mRNAs. The cytoplasmic half-life of pC16L2(S) mRNAs was 61 min, whereas the pNLCATW-derived mRNAs had a half-life of 161 min (Fig. 6B). The mRNA half-life was reduced approximately threefold when the L2 sequence was present on the mRNA. The pC16L2(S)-derived mRNAs were more stable in the nucleus than in the cytoplasm (data not shown), and we therefore focused on cytoplasmic L2 mRNA. The stability of several cellular mRNAs is affected by translation inhibitors (reviewed in reference 41). To test if CAT-L2 mRNA levels could be induced by translation inhibitors, we treated pC16L2(S)-transfected cells with cycloheximide for 0, 1, 2, and 3 h. Levels of cytoplasmic mRNAs produced from pC16L2(S) continuously increased during the cycloheximide treatment time course (Fig. 7A), indicating that the effect on cytoplasmic mRNA stability was dependent on protein synthesis. There was an approximate fivefold induction after 3 h of cycloheximide treatment (Fig. 7A), while the difference in steady-state mRNA levels between pC16L2(S) and pNLCATW was 10- to 30-fold, indicating that inhibition of translation did not entirely prevent premature cytoplasmic degradation of CAT-L2 mRNAs. To test if specific inhibition of translation of the mRNA produced by pC16L2(S) resulted in increased cytoplasmic mRNA levels, a stable GC-rich hairpin loop that blocks translation of the mRNA was inserted at the 5′ end of pC16L2(S), generating pC16L2-Stop (Fig. 7B). This plasmid did not produce detectable levels of CAT protein. The mRNA levels produced from pC16L2-Stop were twofold lower than those produced from pC16L2(S) (Fig. 7C). The reason for this may be that the introduction of a stable RNA structure may have effects on the mRNA other than inhibiting translation. These results indicated that the reduction of the cytoplasmic mRNA levels by the L2 sequence was not dependent on translation of the L2-CAT mRNAs. The results presented here also demonstrate that HPV-16 L2 contains a rapid mRNA degradation determinant and suggest that a labile factor targets HPV-16 L2 mRNAs for premature degradation in the cytoplasm.
FIG. 6.
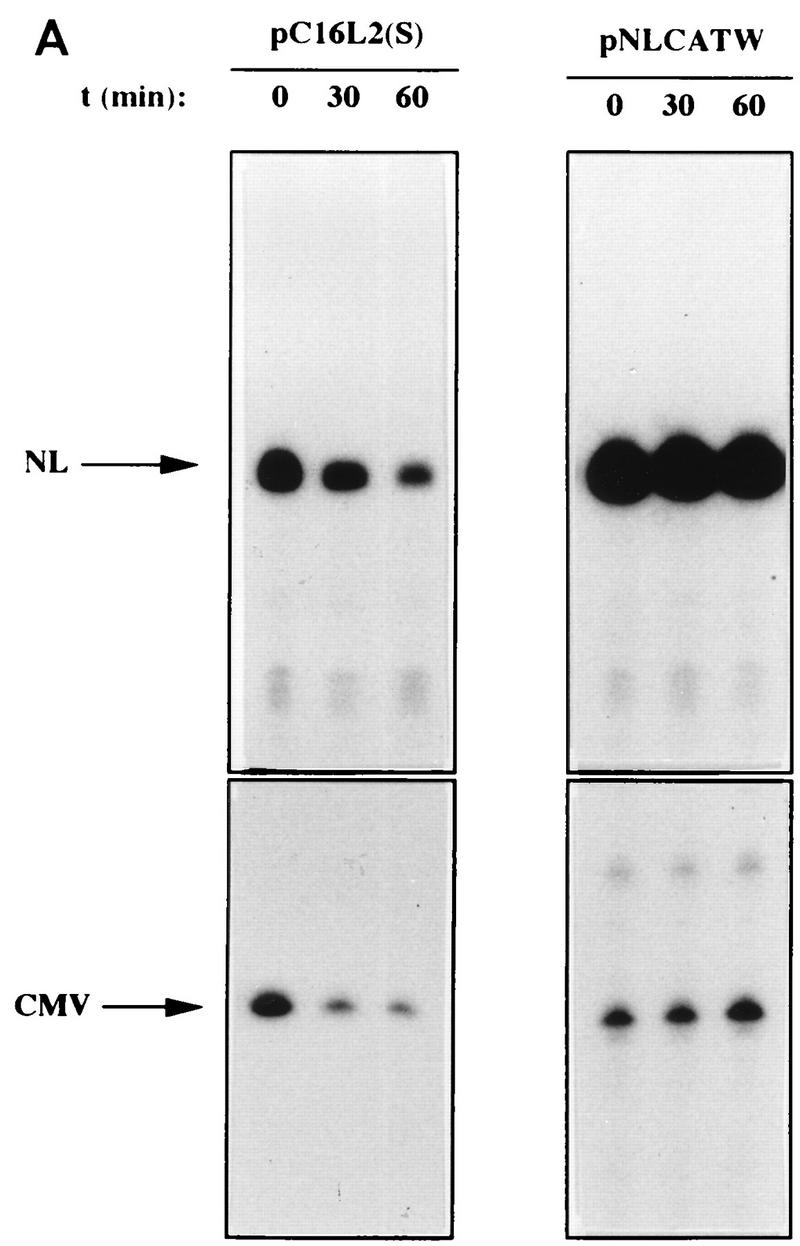

The HPV-16 L2 inhibitory sequences reduce cytoplasmic mRNA stability. (A) Cytoplasmic mRNA levels produced from the indicated plasmids were detected by primer extension at different times [t (min)] after the addition of 10 μg of actinomycin D (Sigma) per ml to the media. NL, extension products of mRNAs derived from the HIV-1 LTR promoter; CMV, extension products of the internal control mRNA derived from the CMV immediate-early promoter in pHCMVtat. (B) Quantitation of mRNA levels at the indicated times by use of a PhosphorImager after normalization to the internal control mRNA levels. lg, log. A representative experiment is shown.
FIG. 7.

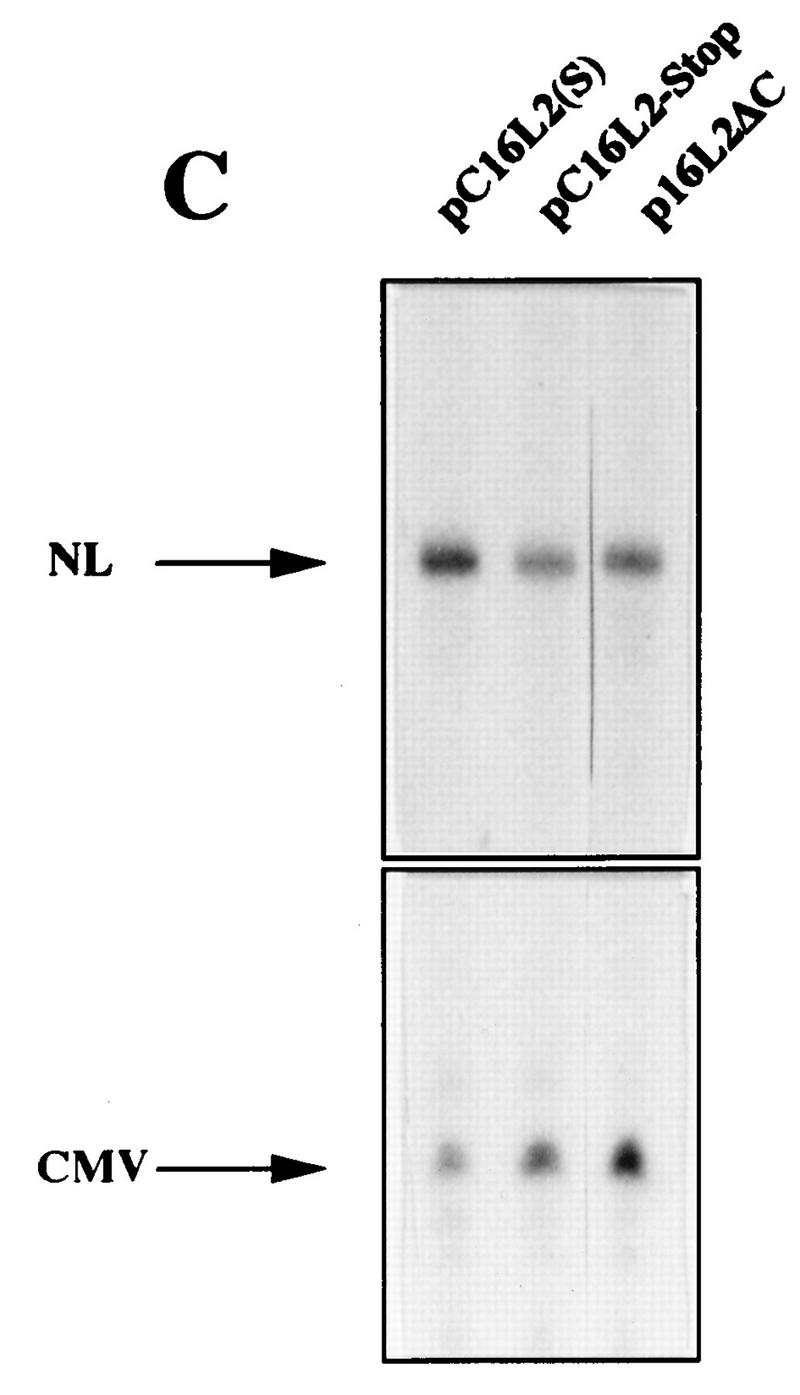
(A) The effect on cytoplasmic mRNA levels of the HPV-16 L2 inhibitory sequences could be reduced by translation inhibitors. Cytoplasmic mRNA levels produced from the indicated plasmids were detected by primer extension at different times [t (h)]) after the addition of 10 μg of cycloheximide (Sigma) per ml to the cell culture media. NL, extension products of mRNAs derived from the HIV-1 LTR promoter; CMV, extension products of the internal control mRNA derived from the CMV immediate-early promoter in pHCMVtat. The graph shows quantitation of the mRNA levels at the indicated times by use of a PhosphorImager after normalization to the internal control mRNA levels. A representative experiment is shown. (B) Translation of the CAT-L2 mRNA specifically is not required for the reduction of cytoplasmic mRNA levels. Plasmid structures are shown. Shaded boxes represent the HPV-16 L2 coding region, numbers indicate nucleotide positions (51), striped boxes indicate HIV-1 LTRs, black boxes indicate the CAT gene, and a GC-rich hairpin is shown as a stem-loop structure. Plasmid names are indicated on the left. (C) Cytoplasmic mRNA levels produced from the indicated plasmids were detected by primer extension. NL, extension products of mRNAs derived from the HIV-1 LTR promoter; CMV, extension products of the internal control mRNA derived from the CMV immediate-early promoter in pHCMVtat.
We wished to confirm that the low cytoplasmic mRNA levels produced from the CAT-L2 hybrid plasmids were a result of the presence of L2 and were not caused by the combination of CAT and L2 sequences. We therefore compared the steady-state cytoplasmic mRNA levels produced from p16L2ΔC (Fig. 7B), designed to express the L2 gene from the HIV-1 LTR promoter in the absence of the CAT gene, with those produced from pC16L2(S). The two plasmids produced similar low cytoplasmic mRNA levels (Fig. 7C), and L2 protein could not be detected in cells transfected with p16L2ΔC (data not shown). In conclusion, the presence of the HPV-16 L2 sequence in the mRNA resulted in decreased mRNA levels, caused by rapid mRNA degradation.
The cytoplasmic mRNA instability sequence is contained in the 5′-most 845 nt of HPV-16 L2.
To map the negative sequences in HPV-16 L2, we introduced deletions in the L2 sequence contained in pC16L2(S), resulting in plasmids pCL2D, pCL2C, pCL2B, and pCL2A (Fig. 8A). The L2 sequences present in pCL2A, pCL2B, and pCL2C inhibited CAT expression when compared to pNLCATW but less so than did the entire L2 sequence present in pC16L2(S) (Fig. 8A). pCL2D produced CAT levels similar to those produced from pNLCATW (Fig. 8A), demonstrating that the inhibitory sequences had been destroyed. Therefore, sequences in the 5′ and 3′ ends of the L2 coding region contributed to the inhibitory activity. Alternatively, inhibitory sequences were located in the 5′ end and in the middle portion of the L2 coding region.
FIG. 8.

The 5′ end of the L2 sequence contains a cytoplasmic mRNA instability determinant. (A) Schematic structures of CAT expression plasmids. Striped boxes indicate HIV-1 LTRs, black boxes represent the CAT gene, shaded boxes represent the HPV-16 L2 coding region, and arrows indicate antisense (←) and sense (→) orientations. Numbers refer to nucleotide positions in the HPV-16 genomic clone (51). Plasmid names are shown on the left. The depicted plasmids were transfected in triplicate into HLtat cells, and CAT levels were monitored in a CAT antigen capture ELISA and normalized to SEAP levels produced from the internal control plasmid pCS1X (58). The mean CAT values obtained after triplicate transfections with pNLCATW (58) were divided by the mean CAT values obtained after triplicate transfections with the indicated plasmids to yield fold inhibition (for plasmids from top to bottom, fold inhibition was 1, 1.2, 88, 1, 9, 14, and 56). (B) Histogram showing the cytoplasmic mRNA levels quantified by use of a PhosphorImager (counts per minute [cpm]) and CAT protein levels (CAT units) produced from plasmids pNLCATW, pC16L2(S), pCL2D, pCL2C, pCL2B, and pCL2A. (sets of bars from left to right). (C) Serially diluted pCL2B plasmid DNA was transfected as indicated. Cytoplasmic mRNA levels (cpm) were quantified by use of a PhosphorImager. NL, primer extension products of mRNAs derived from the HIV-1 LTR promoter; CMV, extension products of the internal control mRNA derived from the CMV immediate-early promoter in pHCMVtat. (D) Cytoplasmic mRNA levels produced from the indicated plasmids were detected by primer extension at different times [t (min)] after the addition of 10 μg of actinomycin D (Sigma) per ml to the media. NL, extension products of mRNAs derived from the HIV-1 LTR promoter; CMV, extension products of the internal control mRNA derived from the CMV immediate-early promoter in pHCMVtat. The graphs show quantitation of the mRNA levels at the indicated times by use of a PhosphorImager after normalization to the internal control mRNA levels. lg, log. A representative experiment is shown.
Next, the cytoplasmic mRNA levels produced from pNLCATW and the HPV-16 L2-containing plasmids pC16L2(S), pCL2D, pCL2C, pCL2B, and pCL2A were assayed and quantified by primer extension. pCL2D produced mRNA and CAT protein levels similar to those produced from pNLCATW, demonstrating that the mRNA instability determinant had been destroyed by the deletion. pCL2C was partially inhibitory, indicating that L2 sequences between nt 4228 and nt 4865 were required for the function of the mRNA instability determinant. However, these sequences did not encompass the entire mRNA instability determinant. pCL2B produced mRNA levels comparable to those produced from pC16L2(S), indicating that the first 845 nt contained the complete mRNA instability sequence. To confirm this result, mRNA levels produced from serially diluted pCL2B were determined and compared to mRNA levels produced from pNLCATW and pC16L2(S) (Fig. 8C). The results verified that pCL2B produced low levels of mRNA similar to those produced from pC16L2(S) (Fig. 8C) and that pCL2B and pC16L2(S) produced lower mRNA levels than did pNLCATW (Fig. 8C). Actinomycin D time course experiments with pCL2B- and pNLCATW-transfected cells, followed by analysis of the cytoplasmic mRNA levels produced from these plasmids, showed that the cytoplasmic half-life of pCL2B mRNAs was threefold lower than that of pNLCATW mRNAs (Fig. 8D). These results mapped the HPV-16 L2 mRNA instability sequence to the first 845 nt of the L2 ORF. Interestingly, pCL2A produced higher mRNA levels than did pC16L2(S), but these were lower than those produced from pNLCATW, indicating that the mRNA instability determinant was affected but not destroyed. However, at the protein level, the inhibitory activity of the L2 sequence in pCL2A was stronger than that in pCL2B (Fig. 8B), suggesting the existence of additional inhibitory sequences in the 3′ end of L2.
Identification of inhibitory sequences in the 3′ end of L2.
To confirm the presence of inhibitory sequences in the 3′ end of L2, sequences spanning various parts of the 3′-most 800 nt of HPV-16 L2 (nt 4865 to nt 5665) were inserted downstream of CAT in pNLCATW. Analysis of the CAT levels produced from pCL2R1 and pCL2R2 (Fig. 9A) revealed that the L2 sequences present in these two plasmids inhibited CAT production to similar extents (Fig. 9A), suggesting that an inhibitory region was located in the middle of these two overlapping sequences. The borders of this region should be nt 5060 and nt 5520. However, the presence of this fragment downstream of CAT, as in pCL2R7, did not result in strong inhibition (Fig. 9A), indicating that additional sequences in the 5′ and 3′ ends were required for efficient inhibition. This suggestion was consistent with larger deletions of 5′ and 3′ sequences, as in pCL2R5 and pCL2R4, respectively, resulting in a loss of inhibition (Fig. 9A). We concluded that an inhibitory region was located downstream of nt 4860 in the L2 coding sequence and that efficient inhibition required the presence of 500 to 600 nt of the L2 3′ end.
FIG. 9.
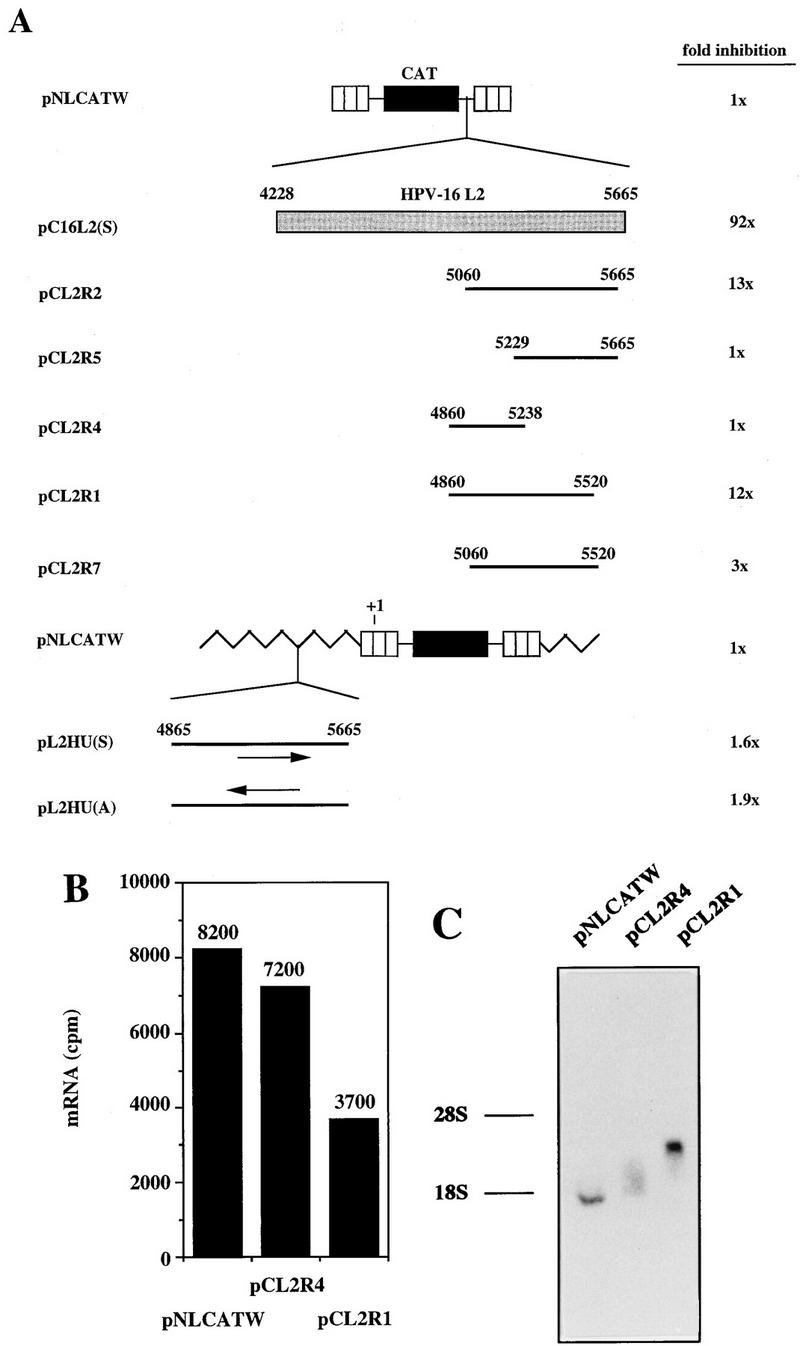
The 3′ end of L2 contains weak inhibitory sequences. (A) Schematic structures of CAT expression plasmids. Striped boxes indicate HIV-1 LTRs, black boxes represent the CAT gene, shaded boxes represent the HPV-16 L2 coding region, and thick black lines indicate subfragments of the L2 coding region. Numbers refer to nucleotide positions in the HPV-16 genomic clone (51). Arrows indicate sense (→) and antisense (←) orientations. Jagged line, vector sequence; +1, start of transcription in the HIV-1 LTR promoter. Plasmid names are shown on the left. The depicted plasmids were transfected in triplicate into HLtat cells, and CAT levels were monitored in a CAT antigen capture ELISA and normalized to SEAP levels produced from the internal control plasmid pCS1X (58). The mean CAT values obtained after triplicate transfections with pNLCATW (58) were divided by the mean CAT values obtained after triplicate transfections with the indicated plasmids to yield fold inhibition. (B) Cytoplasmic mRNA levels produced from the indicated plasmids were detected by primer extension. The histogram shows the mRNA levels quantified by use of a PhosphorImager (counts per minute [cpm]). (C) Northern RNA blotting of cytoplasmic RNA extracted from cells transfected with the indicated plasmids. The positions of the 28S and 18S rRNA species are shown.
Next we monitored the cytoplasmic mRNA levels produced from plasmids pNLCATW, pCL2R1, and pCL2R4 (Fig. 9A). Primer extension revealed that pCL2R1 produced approximately twofold-lower cytoplasmic mRNA levels than did pCL2R4 and pNLCATW (Fig. 9B). The mRNAs produced from these plasmids were of the expected sizes when analyzed by Northern RNA blotting (Fig. 9C). These results indicated that the 12-fold inhibitory effect conferred by the L2 sequence in pCL2R1 (Fig. 9A) was at the posttranscriptional level. The presence of the 3′-most 800 nt of the L2 sequence upstream of the transcribed region, as in plasmids pL2HU(S) and pL2HU(A) (Fig. 9A), had no effect on CAT levels (Fig. 9A), further indicating that inhibition occurred posttranscriptionally. We concluded that weaker inhibitory sequences were present in the 3′-most 800 nt of L2. These inhibitory sequences acted independently of those in the 5′ end of L2.
DISCUSSION
Here we show that the HPV-16 L2 coding region contains _cis_-acting inhibitory sequences that act in an orientation-dependent manner to reduce cytoplasmic and nuclear mRNA levels. The low cytoplasmic mRNA steady-state levels were a result of the reduced stability of mRNAs containing L2. Mapping experiments demonstrated that the 5′ end of the L2 coding region contained an mRNA destabilization sequence and that the 3′ end of L2 contained a weaker inhibitory sequence that acted posttranscriptionally to reduce protein levels. Furthermore, we showed that strong inhibitory sequences in the L1 and L2 coding regions are found in HPV-16 but not in HPV-1, suggesting that late-gene expression is differently regulated by these two HPV types.
Regulation of mRNA stability is a common mechanism of posttranscriptional gene regulation in eucaryotes (reviewed in references 2, 3, 9, and 40 to 42). Mammalian c-fos, c-myc, and β-tubulin mRNAs have been shown to encode mRNA instability determinants in their coding regions (4, 6, 27, 45, 54, 55, 62–64, 66, 67). Interestingly, c-fos and c-myc mRNAs also contain mRNA instability determinants in their 3′ UTRs (9, 41), a property that they share with late HPV-16 L1 and L2 mRNAs (21, 29, 30, 58). The mRNA instability determinants in the c-fos, c-myc, and β-tubulin coding regions are located in the middle, 3′, and 5′ regions and span approximately 320 (54, 55), 320 (63), and 40 (8, 13–15, 38) nt, respectively. The HPV-16 L2 coding region mRNA destabilizing determinant is contained within the first 845 nt (Fig. 8). Mapping of the 5′ end of the L2 sequence will show if the size of the functional element is smaller. The L2 coding region does not contain AUUUA or UUUUU motifs, which are commonly found in AU-rich mRNA instability elements (9, 41), but does have a 60% A+U content, similar to that of an mRNA instability sequence located in the HIV-1 gag ORF (50). The human insulinlike growth factor II mRNA contains an RNA cleavage-promoting sequence in the 3′ UTR, consisting of two elements located approximately 2,000 nt apart (44). These RNA elements hybridize to form the functionally active stem-loop structure (44). It remains to be investigated if it is the RNA primary or secondary structure that is the major determinant of the activity of the HPV-16 L2 mRNA instability determinant.
It was previously concluded that translation of the c-myc and c-fos mRNAs specifically was required for deadenylation and rapid degradation (27, 45, 64). The function of the negative element in the β-tubulin coding region is dependent on the production of the first amino acids of the β-tubulin protein (66, 67). Furthermore, the yeast MATα1 mRNA is rapidly degraded as a result of the presence in the mRNA coding region of rare codons (7, 26). Similarly, we showed that treatment of cells with the translation elongation inhibitor cycloheximide rendered the CAT-L2 hybrid mRNAs more stable in the cytoplasm (Fig. 7A). However, the function of the mRNA instability sequence in the CAT-L2 hybrid mRNAs was not dependent on translation of these mRNAs specifically (Fig. 7B and C), suggesting that a labile factor targets the L2 mRNA for rapid degradation. This suggestion is not without precedent, since premature degradation of a c-fos 3′ UTR-containing mRNA was shown to be inhibited by cycloheximide but not by insertion into the mRNA of sequences that blocked the translation of c-fos (31). This idea is consistent with our observations of reduced CAT-L2 mRNA levels in both the cytoplasmic and the nuclear compartments (Fig. 5), suggesting that mRNA destabilization occurs in the nucleus and the cytoplasm. Alternatively, inhibition of translation by cycloheximide affects the stability of the mRNA only in the cytoplasm. The c-fos coding region instability determinant interacts with cellular factors (10), and the c-myc mRNA instability determinant binds a 70-kDa protein which protects against degradation of the c-myc mRNA (39). A recent report indicates that the HPV-16 L2 protein interacts nonspecifically with nucleic acids (69). However, production of the HPV-16 L2 protein is most likely not required for the inhibitory activity of the L2 mRNA. It remains to be investigated by what mechanism L2-containing mRNAs are degraded in the cytoplasm and the nucleus and if cellular proteins bind to them. We cannot exclude the possibility that L2-containing mRNAs are retained in the nucleus, where they are prematurely degraded.
HPVs infect dividing cells in the basal cell layer of the stratified epithelium. As the infected cell moves toward the upper cell layers and differentiates, HPV late-gene expression is activated (11, 23, 28, 32, 33, 56). This differentiation-dependent HPV late-gene expression pattern has been observed for several different HPV types, illustrating that the expression of late genes is suppressed in the lower cell layers, while in the upper strata, with differentiated cells, this block is relieved. Interestingly, the expression of c-fos in squamous cell epithelium also is differentiation dependent, with higher c-fos mRNA and protein levels in the upper strata (19). Therefore, the inhibitory activity of the c-fos and HPV-16 L2 coding region determinants must be relieved as the cell differentiates. Interestingly, c-myc production decreases as myoblasts differentiate. A recent report indicated that this effect is attributable to the c-myc coding region mRNA instability determinant (65), demonstrating an inverse correlation between the inhibitory activity of the RNA sequence and cell differentiation for c-fos and HPV-16 late mRNAs. It remains to be investigated if the inhibitory sequences in HPV-16 L1 and L2 are as active in terminally differentiated cells as in dividing cells.
An attempt to classify HPVs into different groups based on the amount of virus present in benign lesions has been made and has suggested that HPVs can be divided into three groups: productive, weakly productive, and nonproductive (57). It is of interest to note that HPV-1 categorically falls into the productive group, characterized by the detection of moderate to large amounts of virus in lesions, whereas HPV-16 belongs to the weakly productive group, where only minute amounts of virus can be detected. Since we have identified strong negative sequences in HPV-16 L1 and L2 (58; this study) but not in HPV-1 L1 and L2, it is reasonable to speculate that the negative sequences present in the coding regions may explain the different amounts of virions present in lesions caused by HPV-1 and HPV-16. In contrast, both HPV-1 and HPV-16 show strict cell differentiation-dependent expression of late genes. This property correlates with the presence of a negative element in the 3′ UTRs of the late mRNAs of both HPV types (29, 30, 58, 60), suggesting that the block in late-gene expression caused by the 3′ UTR element is alleviated as the cell differentiates. It would be of interest to determine the effect of cell differentiation on the activity of the 3′ UTR and coding region inhibitory sequences.
The regulation of viral late-gene expression caused by the presence of negative regulatory sequences on the mRNAs coding for structural proteins appears to be a common strategy used by many viruses. Although such inhibitory RNA sequences have not yet been characterized in detail, several reports on negative sequences on viral late mRNAs have been presented. For example, HIV-1 contains inhibitory sequences in the Gag, Pol, and Env coding regions (16, 34, 37, 46, 47, 50) and human T-cell leukemia virus type 1 contains inhibitory sequences in the 5′ UTR (52) and in the Pol and Env coding regions (43). An mRNA instability sequence previously identified in the HIV-1 gag ORF was shown to reduce mRNA levels in both cytoplasmic and nuclear compartments (50), similar to the data presented here for HPV-16 L2 (Fig. 5). The HIV-1 gag mRNA inhibitory element interacted specifically with poly(A)-binding protein 1 (1), but in preliminary experiments we did not detect binding to the L2 sequences of proteins with an affinity for poly(A).
The presence of multiple inhibitory sequences on viral late mRNAs encoding structural proteins appears to be a common property of HIV-1, HTLV-1, and HPV-16. For example, HIV-1 contains inhibitory sequences in the Env coding region (12, 37) that would be present on the Env-producing and Gag-Pol-producing mRNAs as well as inhibitory sequences in the Gag and Pol coding regions (16, 34, 47, 50). A similar arrangement is observed for HPV-16, where the HPV-16 L1 coding region contains inhibitory elements (58) that would be present on both the L1 mRNAs and the L2-L1 mRNAs (5). In addition to these negative elements, the L2-L1 mRNAs would contain the L2 coding region inhibitory sequences described here. Therefore, the inhibitory sequences in HPV-16 L2 would allow independent regulation of L1 and L2 production. Perhaps the presence of inhibitory sequences in the L2 coding region allows a balanced production of L1 and L2 and reflects a requirement for the production of a certain ratio between L1 and L2 molecules to generate correctly assembled virions. Alternatively, perhaps the posttranscriptional processing and regulation of expression of a polycistronic mRNA are different from those of a monocistronic mRNA and therefore require more complex _cis_-acting signals. In conclusion, the presence of negative elements on viral late mRNAs allows the virus to regulate late-gene expression and virus production. This ability may be of critical importance for the virus in avoiding host immune system surveillance and in establishing persistent infections. Subgenomic virus expression plasmids encoding late genes lacking negative sequences may be valuable tools for the development of DNA vaccines.
ACKNOWLEDGMENTS
We thank M. Yaniv, F. Thierry, H. zur Hausen, G. N. Pavlakis, and B. K. Felber for plasmids, J. Dillner for guinea pig anti-HPV-16 L2 peptide antiserum, S. Leonov for help with electrotransfer equipment, and S. Izadi for participating in some experiments.
Part of this work was performed at the Department of Medical Immunology and Microbiology, Biomedical Center, Uppsala University. This work was supported by the Swedish Cancer Society, the Swedish Medical Research Council, the Swedish Society for Medical Research, the Swedish Society of Medicine, Anders Otto Swärds Stiftelse, Stiftelsen Lars Hiertas Minne, Jeanssonska Stiftelserna, Magnus Bergvalls Stiftelse, and Åke Wibergs Stiftelse.
REFERENCES
- 1.Afonia E, Neumann M, Pavlakis G N. Preferential binding of poly(A)-binding protein 1 to an inhibitory RNA element in the human immunodeficiency virus type 1 gag mRNA. J Biol Chem. 1997;272:2307–2311. doi: 10.1074/jbc.272.4.2307. [DOI] [PubMed] [Google Scholar]
- 2.Belasco J G, Brawerman G, editors. Control of messenger RNA stability. San Diego, Calif: Academic Press, Inc.; 1993. [Google Scholar]
- 3.Bellman C A, Parker R. Degradation of mRNA in eucaryotes. Cell. 1995;81:179–183. doi: 10.1016/0092-8674(95)90326-7. [DOI] [PubMed] [Google Scholar]
- 4.Bernstein P L, Herrick D J, Prokipcak R D, Ross J. Control of c-myc mRNA half-life in vitro by a protein capable of binding to a coding region stability determinant. Genes Dev. 1992;6:642–654. doi: 10.1101/gad.6.4.642. [DOI] [PubMed] [Google Scholar]
- 5.Billakanti S R, Calef C E, Farmer A D, Halpern A L, Myers G L. Human papillomaviruses: a compilation and analysis of nucleic acid and amino acid sequences. Los Alamos, N.Mex: Theoretical Biology and Biophysics, Los Alamos National Laboratory; 1996. [Google Scholar]
- 6.Brewer G, Ross J. Poly(A) shortening and degradation of the 3′ A+U-rich sequences of human c-myc mRNA in a cell-free system. Mol Cell Biol. 1988;8:1697–1708. doi: 10.1128/mcb.8.4.1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caponigro G, Parker R. mRNA turnover in yeast promoted by the MATalpha1 instability element. Nucleic Acids Res. 1996;24:4304–4312. doi: 10.1093/nar/24.21.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Caron J M, Jones A L, Rall L B, Kirshner M W. Autoregulation of tubulin synthesis in enucleated cells. Nature (London) 1985;317:645–651. doi: 10.1038/317648a0. [DOI] [PubMed] [Google Scholar]
- 9.Chen C-Y A, Shyu A-B. AU-rich elements: characterization and importance in mRNA degradation. Trends Biochem Sci. 1995;20:465–470. doi: 10.1016/s0968-0004(00)89102-1. [DOI] [PubMed] [Google Scholar]
- 10.Chen C-Y A, You Y, Shyu A-B. Two cellular proteins bind specifically to a purine-rich sequence necessary for the destabilization function of a c-fos protein-coding region determinant of mRNA instability. Mol Cell Biol. 1992;12:5748–5757. doi: 10.1128/mcb.12.12.5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chow L T, Broker T R. Papillomavirus DNA replication. Intervirology. 1994;37:150–158. doi: 10.1159/000150373. [DOI] [PubMed] [Google Scholar]
- 12.Churchill M J, Moore J L, Rosenberg M, Brighty D W. The rev-responsive element negatively regulates human immunodeficiency virus type 1 mRNA expression in primate cells. J Virol. 1996;70:5786–5790. doi: 10.1128/jvi.70.9.5786-5790.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cleveland D W, Havercroft J C. Is apparent autoregulatory control of tubulin synthesis nontranscriptionally regulated? J Biol Chem. 1983;97:919–924. doi: 10.1083/jcb.97.3.919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cleveland D W, Lopata M A, Sherline P, Kirschner M W. Unpolymerized tubulin modulates the level of tubulin mRNAs. Cell. 1981;25:537–546. doi: 10.1016/0092-8674(81)90072-6. [DOI] [PubMed] [Google Scholar]
- 15.Cleveland D W, Pittenger M F, Feramisco J R. Elevation of tubulin levels by microinjection suppresses new tubulin synthesis. Nature (London) 1983;305:738–740. doi: 10.1038/305738a0. [DOI] [PubMed] [Google Scholar]
- 16.Cochrane A W, Jones K S, Beidas S, Dillon P J, Skalka A M, Rosen C. Identification and characterization of intragenic sequences which repress human immunodeficiency virus structural gene expression. J Virol. 1991;65:5305–5313. doi: 10.1128/jvi.65.10.5305-5313.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Danos O, Katinka M, Yaniv M. Human papillomavirus 1a complete DNA sequence: a novel type of genome organization among papovaviridae. EMBO J. 1982;1:231–236. doi: 10.1002/j.1460-2075.1982.tb01152.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dillner L, Heino P, Moreno-Lopez J, Dillner J. Antigenic and immunogenic epitopes shared by human papillomavirus type 16 and bovine, canine, and avian papillomaviruses. J Virol. 1991;65:6862–6871. doi: 10.1128/jvi.65.12.6862-6871.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fisher C, Byers M R, Iadarola M J, Powers E A. Patterns of epithelial expression of Fos protein suggest important role in the transition from viable to cornified cell during keratinization. Development. 1991;111:253–258. doi: 10.1242/dev.111.2.253. [DOI] [PubMed] [Google Scholar]
- 20.Fuerst T R, Niles E G, Studier F W, Moss B. Eukaryotic transient-expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc Natl Acad Sci USA. 1986;83:8122–8126. doi: 10.1073/pnas.83.21.8122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Furth P A, Choe W-T, Rex J H, Byrne J C, Baker C C. Sequences homologous to 5′ splice sites are required for the inhibitory activity of papillomavirus late 3′ untranslated regions. Mol Cell Biol. 1995;14:5278–5289. doi: 10.1128/mcb.14.8.5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Graham F J, van der Eb A J. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology. 1973;52:456–460. doi: 10.1016/0042-6822(73)90341-3. [DOI] [PubMed] [Google Scholar]
- 23.Hagensee M E, Galloway D A. Growing human papillomaviruses and virus-like particles in the laboratory. In: Lacey C, editor. Papillomavirus reviews: current research on papillomaviruses. Leeds, England: Leeds University Press; 1996. pp. 85–92. [Google Scholar]
- 24.Halpern A, Farmer A, Calef C. A survey of papillomavirus variants. In: Myers G, editor. Human papillomaviruses, 1996. Los Alamos, N.Mex: Theoretical Biology and Biophysics, Los Alamos National Laboratory; 1996. pp. i43–i185. [Google Scholar]
- 25.Heino P, Dillner J, Schwartz S. Human papillomavirus type 16 capsid proteins produced from recombinant Semliki Forest virus assemble into virus like particles. Virology. 1995;214:349–359. doi: 10.1006/viro.1995.0044. [DOI] [PubMed] [Google Scholar]
- 26.Hennigan A N, Jacobson A. Functional mapping of the translation-dependent instability element of yeast MATα1 mRNA. Mol Cell Biol. 1996;16:3833–3843. doi: 10.1128/mcb.16.7.3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Herrick D J, Ross J. The half-life of c-myc mRNA in growing and serum-stimulated cells: influence of the coding and 3′ untranslated regions and role of ribosome translocation. Mol Cell Biol. 1994;14:2119–2128. doi: 10.1128/mcb.14.3.2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Howley P M. Papillomavirinae: the viruses and their replication. In: Fields B N, Knipe D M, Howley P M, editors. Fields’ virology. 3rd ed. Vol. 2. Philadelphia, Pa: Lippincott-Raven Publishers; 1996. pp. 2045–2076. [Google Scholar]
- 29.Kennedy I M, Haddow J K, Clements J B. Analysis of human papillomavirus type 16 late mRNA 3′ processing signals in vitro and in vivo. J Virol. 1990;64:1825–1829. doi: 10.1128/jvi.64.4.1825-1829.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kennedy I M, Haddow J K, Clements J B. A negative regulatory element in the human papillomavirus type 16 genome acts at the level of late mRNA stability. J Virol. 1991;65:2093–2097. doi: 10.1128/jvi.65.4.2093-2097.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koeller D M, Horowitz J A, Casey J L, Klausner R D, Harford J B. Translation and the stability of mRNAs encoding the transferrin receptor and c-fos. Proc Natl Acad Sci USA. 1991;88:7778–7782. doi: 10.1073/pnas.88.17.7778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Laimins L A. The biology of human papillomaviruses: from warts to cancer. Infect Agents Dis. 1993;2:74–86. [PubMed] [Google Scholar]
- 33.Lambert P F. Papillomavirus DNA replication. J Virol. 1991;65:3417–3420. doi: 10.1128/jvi.65.7.3417-3420.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maldarelli F, Martin M A, Strebel K. Identification of posttranscriptionally active inhibitory sequences in human immunodeficiency virus type 1 RNA: novel level of gene regulation. J Virol. 1991;65:5732–5743. doi: 10.1128/jvi.65.11.5732-5743.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meyers C, Laimins L A. In vitro model systems for the study of HPV induced neoplasias. In: Lacey C, editor. Papillomavirus reviews: current research on papillomaviruses. Leeds, England: Leeds University Press; 1996. pp. 79–83. [Google Scholar]
- 36.Miller A D, Rosman G J. Improved retroviral vectors for gene transfer and expression. BioTechniques. 1989;7:980–990. [PMC free article] [PubMed] [Google Scholar]
- 37.Nasioulas G, Zolotukhin A S, Tabernero C, Solomin L, Cunningham C P, Pavlakis G N, Felber B K. Elements distinct from the human immunodeficiency virus type 1 splice sites are responsible for the Rev dependence of env mRNA. J Virol. 1994;68:2986–2993. doi: 10.1128/jvi.68.5.2986-2993.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pittenger M F, Cleveland D W. Retention of autoregulatory control of tubulin synthesis in cytoplasts; demonstration of a cytoplasmic mechanism that regulates the level of tubulin expression. J Cell Biol. 1985;101:1941–1952. doi: 10.1083/jcb.101.5.1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prokipcak R D, Herrick D J, Ross J. Purification and properties of a protein that binds to the C-terminal coding region of human c-myc mRNA. J Biol Chem. 1994;269:9261–9269. [PubMed] [Google Scholar]
- 40.Ross J. Control of messenger RNA stability in higher eucaryotes. Trends Biochem Sci. 1996;12:171–176. doi: 10.1016/0168-9525(96)10016-0. [DOI] [PubMed] [Google Scholar]
- 41.Ross J. mRNA stability in mammalian cells. Microbiol Rev. 1995;59:15–95. doi: 10.1128/mr.59.3.423-450.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sachs A B. Messenger RNA degradation in eucaryotes. Cell. 1993;74:413–421. doi: 10.1016/0092-8674(93)80043-e. [DOI] [PubMed] [Google Scholar]
- 43.Saiga A, Orita S, Minoura-Tada N, Maeda M, Aono Y, Asakawa M, Nakahara K, Kubota R, Osame M, Igarashi H. cis-Acting inhibitory elements within the pol-env region of human T-cell leukemia virus type 1 possibly involved in viral persistence. J Virol. 1997;71:4485–4494. doi: 10.1128/jvi.71.6.4485-4494.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scheper W, Meinsma D, Holthuizen P E, Sussenbach J S. Long-range RNA interaction of two sequence elements required for endonucleolytic cleavage of human insulin-like growth factor II mRNAs. Mol Cell Biol. 1995;15:235–245. doi: 10.1128/mcb.15.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schiavi S C, Wellington C L, Shyu A-B, Chen C-Y A, Greenberg M E, Belasco J G. Multiple elements in the c-fos protein-coding region facilitate mRNA deadenylation and decay by a mechanism coupled to translation. J Biol Chem. 1994;269:3441–3448. [PubMed] [Google Scholar]
- 46.Schneider R, Campbell M, Nasioulas G, Felber B K, Pavlakis G N. Inactivation of human immunodeficiency virus type 1 inhibitory elements allows Rev-independent expression of Gag and Gag/protease and particle formation. J Virol. 1997;71:4892–4903. doi: 10.1128/jvi.71.7.4892-4903.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schwartz S, Campbell M, Nasioulas G, Harrison J, Felber B K, Pavlakis G N. Mutational inactivation of an inhibitory sequence in human immunodeficiency virus type 1 results in Rev-independent gag expression. J Virol. 1992;66:7176–7182. doi: 10.1128/jvi.66.12.7176-7182.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schwartz S, Felber B K, Benko D M, Fenyö E M, Pavlakis G N. Cloning and functional analysis of multiply spliced mRNA species of human immunodeficiency virus type 1. J Virol. 1990;64:2519–2529. doi: 10.1128/jvi.64.6.2519-2529.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schwartz S, Felber B K, Fenyö E M, Pavlakis G N. Env and Vpu proteins of human immunodeficiency virus type 1 are produced from multiple bicistronic mRNAs. J Virol. 1990;64:5448–5456. doi: 10.1128/jvi.64.11.5448-5456.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schwartz S, Felber B K, Pavlakis G N. Distinct RNA sequences in the gag region of human immunodeficiency virus type 1 decrease RNA stability and inhibit expression in the absence of Rev protein. J Virol. 1992;66:150–159. doi: 10.1128/jvi.66.1.150-159.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Seedorf K, Krämer G, Dürst M, Suhai S, Röwekamp W G. Human papillomavirus type 16 DNA sequence. Virology. 1985;145:181–185. doi: 10.1016/0042-6822(85)90214-4. [DOI] [PubMed] [Google Scholar]
- 52.Seiki M, Hikikoshi A, Yoshida M. The U5 sequence is a cis-acting repressive element for genomic RNA expression of human T cell leukemia virus type 1. Virology. 1990;186:81–86. doi: 10.1016/0042-6822(90)90232-g. [DOI] [PubMed] [Google Scholar]
- 53.Shah K V, Howley P M. Papillomaviruses. In: Fields B N, Knipe D M, Howley P M, editors. Fields’ virology. 3rd ed. Vol. 2. Philadelphia, Pa: Lippincott-Raven Publishers; 1996. pp. 2077–2109. [Google Scholar]
- 54.Shyu A-B, Belasco J G, Greenberg M E. Two distinct destabilizing elements in the c-fos message trigger deadenylation as a first step in rapid mRNA decay. Genes Dev. 1991;5:221–231. doi: 10.1101/gad.5.2.221. [DOI] [PubMed] [Google Scholar]
- 55.Shyu A-B, Greenberg M E, Belasco J G. The c-fos transcript is targeted for rapid decay by two distinct mRNA degradation pathways. Genes Dev. 1989;3:60–72. doi: 10.1101/gad.3.1.60. [DOI] [PubMed] [Google Scholar]
- 56.Stanley M A. Replication of human papillomaviruses in cell culture. Antivir Res. 1994;24:1–15. doi: 10.1016/0166-3542(94)90047-7. [DOI] [PubMed] [Google Scholar]
- 57.Taichman L B, LaPorta R F. The expression of papillomaviruses in epithelial cells. In: Salzman N P, Howley P M, editors. The papovaviridae. New York, N.Y: Plenum Press; 1987. pp. 109–139. [Google Scholar]
- 58.Tan W, Felber B K, Zolotukhin A S, Pavlakis G N, Schwartz S. Efficient expression of human papillomavirus type 16 L1 protein in epithelial cells by using rev and the rev-responsive element of human immunodeficiency virus or the cis-acting transactivation element of simian retrovirus type 1. J Virol. 1995;69:5607–5620. doi: 10.1128/jvi.69.9.5607-5620.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tan W, Schalling M, Zhao C, Luukkonen M, Nilsson M, Fenyö E M, Pavlakis G N, Schwartz S. Inhibitory activity of the equine infectious anemia virus major 5′ splice site in the absence of Rev. J Virol. 1996;70:3645–3658. doi: 10.1128/jvi.70.6.3645-3658.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tan W, Schwartz S. The Rev protein of human immunodeficiency virus type 1 counteracts the effect of an AU-rich negative element in the human papillomavirus type 1 late 3′ untranslated region. J Virol. 1995;69:2932–2945. doi: 10.1128/jvi.69.5.2932-2945.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Turek L P. The structure, function and regulation of papillomaviral genes in infection and cervical cancer. Adv Virus Res. 1994;44:305–356. doi: 10.1016/s0065-3527(08)60332-2. [DOI] [PubMed] [Google Scholar]
- 62.Wellington C L, Greenberg M E, Belasco J G. The destabilizing elements in the coding region of c-fos mRNA are recognized as RNA. Mol Cell Biol. 1993;13:5034–5042. doi: 10.1128/mcb.13.8.5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wisdom R, Lee W. The protein-coding region of c-myc mRNA contains a sequence that specifies rapid mRNA turnover and induction by protein synthesis inhibitors. Genes Dev. 1991;5:232–243. doi: 10.1101/gad.5.2.232. [DOI] [PubMed] [Google Scholar]
- 64.Wisdom R, Lee W. Translation of c-myc mRNA is required for its posttranscriptional regulation during myogenesis. J Biol Chem. 1990;265:19015–19021. [PubMed] [Google Scholar]
- 65.Yeilding N M, Rehman M T, Lee W M F. Identification of sequences in c-myc mRNA that regulate its steady-state levels. Mol Cell Biol. 1996;16:3511–3522. doi: 10.1128/mcb.16.7.3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yen T J, Gay D A, Pachter J S, Cleveland D W. Autoregulated changes in stability of polyribosome-bound β-tubulin mRNA are specified by the first 13 translated nucleotides. Mol Cell Biol. 1988;8:1224–1235. doi: 10.1128/mcb.8.3.1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yen T J, Machlin P S, Cleveland D W. Autoregulated instability of β-tubulin mRNAs by recognition of the nascent amino terminus of β-tubulin. Nature (London) 1988;334:580–585. doi: 10.1038/334580a0. [DOI] [PubMed] [Google Scholar]
- 68.Zhao C, Tan W, Sokolowski M, Schwartz S. Identification of nuclear and cytoplasmic factors that interact specifically with an AU-rich, cis-acting inhibitory sequence in the 3′ untranslated region of human papillomavirus type 1 late mRNAs. J Virol. 1996;70:3659–3667. doi: 10.1128/jvi.70.6.3659-3667.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhou J, Sun X Y, Louis K, Frazer I H. Interaction of human papillomavirus (HPV) type 16 capsid proteins with HPV DNA requires an intact L2 N-terminal sequence. J Virol. 1994;68:619–625. doi: 10.1128/jvi.68.2.619-625.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.zur Hausen H. Papillomavirus infections—a major cause of human cancers. Biochim Biophys Acta. 1996;1288:55–78. doi: 10.1016/0304-419x(96)00020-0. [DOI] [PubMed] [Google Scholar]
- 71.zur Hausen H. Viruses in human cancers. Science. 1991;254:1167–1173. doi: 10.1126/science.1659743. [DOI] [PubMed] [Google Scholar]
- 72.zur Hausen H, de Villiers E-M. Human papillomaviruses. Annu Rev Microbiol. 1994;48:427–447. doi: 10.1146/annurev.mi.48.100194.002235. [DOI] [PubMed] [Google Scholar]