Activation of Cdh1-dependent APC is required for G1 cell cycle arrest and DNA damage-induced G2 checkpoint in vertebrate cells (original) (raw)
Abstract
Anaphase-promoting complex (APC) is activated by two regulatory proteins, Cdc20 and Cdh1. In yeast and Drosophila, Cdh1-dependent APC (Cdh1–APC) activity targets mitotic cyclins from the end of mitosis to the G1 phase. To investigate the function of Cdh1 in vertebrate cells, we generated clones of chicken DT40 cells disrupted in their Cdh1 loci. Cdh1 was dispensable for viability and cell cycle progression. However, similarly to yeast and Drosophila, loss of Cdh1 induced unscheduled accumulation of mitotic cyclins in G1, resulting in abrogation of G1 arrest caused by treatment with rapamycin, an inducer of p27Kip1. Further more, we found that _Cdh1_–/– cells fail to maintain DNA damage-induced G2 arrest and that Cdh1–APC is activated by X-irradiation-induced DNA damage. Thus, activation of Cdh1–APC plays a crucial role in both cdk inhibitor-dependent G1 arrest and DNA damage-induced G2 arrest.
Keywords: Cdc27/DT40/mitotic cyclin/p27/rapamycin
Introduction
The ubiquitin–proteasome system for destruction of proteins plays a crucial role in the regulation of cell cycle progression (Kramer et al., 2000). Anaphase-promoting complex/cyclosome (APC) is a multisubunit complex that functions as a ubiquitin ligase specific for various cell cycle proteins, including cyclin B, cyclin A, mitotic kinases, inhibitors of anaphase, spindle-associated proteins and inhibitors of DNA replication (King et al., 1995, 1996; Sudakin et al., 1995; Cohen-Fix and Koshland, 1997; Juang et al., 1997). The activity of APC is tightly regulated to control cell cycle progression, being high from late mitosis until late in the G1 phase but low in S, G2 and early mitosis in yeast and mammalian cells.
Genetic and biochemical analyses in yeast, Drosophila and Xenopus have revealed that the function of the APC is regulated by two types of WD-40 repeat-containing protein, Cdc20/fizzy (fzy)/p55CDC and Hct1/srw1/fizzy-related (fzr)/Cdh1, in a substrate-specific manner. In metaphase, the Cdc20-activated APC (Cdc20–APC) ubiquitylates anaphase inhibitors, such as Pds1 in budding yeast (Visintin et al., 1997), Cut2 in fission yeast (Funabiki et al., 1996) and securin in Xenopus (Zou et al., 1999). Proteolysis of these anaphase inhibitors releases their binding partner Esp1 (in budding yeast), which in turn results in the cleavage of cohesin, allowing sister chromatid separation for transition from metaphase to anaphase (Uhlmann et al., 1999). It has also been reported that Cdc20 is essential not only for sister chromatid separation but also for proteolysis of mitotic cyclin clb2 in budding yeast (Lim et al., 1998; Yeong et al., 2000). These data suggest that Cdc20–APC is required for both initiation of anaphase and exit from mitosis. On the other hand, Hct1/srw1/fzr/Cdh1 is believed to maintain APC activity from the end of mitosis until the end of G1. In budding yeast, clb2 is highly stabilized in G1 phase in Hct1 mutants (Schwab et al., 1997; Visintin et al., 1997). Furthermore, in Drosophila, loss of fzr causes the unscheduled accumulation of mitotic cyclins in the G1 phase, following an extra division cycle in the epidermis (Sigrist and Lehner, 1997). These findings suggest that the Hct1/srw1/fzr/Cdh1-dependent APC activity targets mitotic cyclins for destruction from the end of mitosis to the G1 phase but is dispensable for metaphase–anaphase transition and exit from mitosis. However, it remains unclear whether this is also true in higher vertebrates.
Maintenance of genomic integrity after DNA damage depends on cell cycle checkpoints, which control a signaling system that produces changes in the activity of cyclin-dependent kinases (cdks), resulting in a delay in cell cycle progression. Arrest in G1 is considered to prevent aberrant replication of damaged DNA, and arrest in G2 allows cells to avoid segregation of defective chromosomes. G1 arrest after DNA damage is induced primarily by stabilization of p53 (Lakin and Jackson, 1999). However, it has been reported recently that the initial step in the DNA damage-induced G1 arrest is p53 independent and mediated by cyclin D1 proteolysis, which possibly is carried out by the APC (Agami and Bernards, 2000). This observation suggests the possibility that the APC is activated in response to DNA damage and contributes to the checkpoint activation.
In this study, we have investigated the role of Cdh1 in higher vertebrates by generating _Cdh1_–/– cells using the hyper-recombinogenic chicken B-cell line DT40. Pheno typic analysis of the mutant clones revealed that Cdh1 is dispensable for viability and cell cycle progression but plays a crucial role in down-regulation of mitotic cyclins in the G1 phase in vertebrate cells, similarly to yeast and Drosophila. Therefore, loss of Cdh1 induced unscheduled accumulation of mitotic cyclins in G1, resulting in abrogation of G1 arrest caused by rapamycin treatment, which is known to activate expression of p27Kip1. Furthermore, our results identify an unexpected role for Cdh1–APC in the G2 checkpoint activated by X-irradiation-induced DNA damage.
Results
Cdh1 targeting constructs and generation of Cdh1-deficient DT40 clones
Chicken Cdh1 cDNA was isolated by PCR using primers specific for human Cdh1 from the chicken cDNAs. Based on the sequences of chicken Cdh1 cDNA, ∼7 kb of the chicken Cdh1 locus was amplified by long-range PCR using genomic DNA extracted from DT40 cells as a template. Either the histidinol (his) or the blasticidin (bsr) resistance gene was inserted between sequences of 4 and 3 kb length (Figure 1A). Targeted integration of these constructs disrupts the reading frame of the Cdh1 gene at the first WD repeat. Targeted events were examined by PCR, Southern blotting analysis and RT–PCR (Figure 1B, C and D). We isolated two viable _Cdh1_–/– clones (clones 1 and 2), and the proliferative properties of _Cdh1_–/– clones, in comparison with wild-type cells, were monitored by using growth curves and cell cycle analysis. The growth curves of the _Cdh1_–/– clones were indistinguishable from that of wild-type cells, which divided approximately every 8 h (Figure 2A). Fluorescence-activated cell sorting (FACS) analysis showed that the cell cycle distributions were the same in both genotypes (Figure 2B). These data indicate that the Cdh1 gene is dispensable for viability and proliferation of DT40 cells. Low stringency Southern blot analysis using a Cdh1 probe did not detect any significant signal besides the Cdh1 gene (data not shown), suggesting that there is no additional _Cdh1_-related gene that may act redundantly, masking phenotypes of knockout cells.

Fig. 1. Generation of _Cdh1_–/– clones. (A) Schematic representation of a partial restriction map of the chicken Cdh1 locus, the two gene disruption constructs and the configuration of the targeted loci. Black boxes indicate the position of exons. The box designated ‘probe’ represents the region used for Southern blotting. Primer sites for PCR screening are indicated by arrowheads. Relevant _Bam_HI restriction enzyme sites are shown. (B) PCR analysis of genomic DNA from targeted cells of the genotypes indicated. Genomic DNAs from wild-type (+/+), heterozygous (+/–) and homozygous mutant (–/–) clones (clones 1 and 2) were amplified using three sets of primers (p2–p4, p1–p3 and p1–p5) depicted in (A). (C) Southern blot analysis of wild-type (+/+), heterozygous (+/–) and homozygous mutant (–/–) clones (clones 1 and 2). _Bam_HI-digested genomic DNA was hybridized with the labeled probe DNA depicted in (A). New 4 kb _Bam_HI fragments were detected with the probe following targeted integration of the knockout constructs. (D) Ethidium bromide-stained agarose gel showing RT–PCR products of the full-length Cdh1. mRNAs were isolated from wild-type (+/+), heterozygous (+/–) and homozygous mutant (–/–) clones. GAPDH cDNA was used as control amplification to ensure the successful completion of cDNA synthesis and the PCR.

Fig. 2. Proliferative characteristics of wild-type and _Cdh1_–/– DT40 cells. (A) Representative growth curves in wild-type cells (filled circles), _Cdh1_–/– clone 1 (filled triangles) and _Cdh1_–/– clone 2 (filled squares). Error bars show the standard error of the mean for three independent experiments. (B) Cell cycle distribution of wild-type DT40 cells and _Cdh1_–/– clones as measured by BrdU incorporation and DNA content by FACS analysis. Cells were pulse-labeled for 10 min before harvest, and subsequently stained with FITC–anti-BrdU to detect BrdU incorporation (vertical axis, log scale) and propidium iodine to detect total DNA (horizontal axis, linear scale). The upper gate identifies cells incorporating BrdU (S phase), the lower left gate identifies G1 cells and the lower right gate displays G2/M cells. Numbers show the percentages of cells falling in each gate.
Loss of Cdh1 results in unscheduled accumulation of mitotic cyclins in the G1 phase
In yeasts (budding and fission) and Drosophila, loss of hct1, ste9 and fzr, which are Cdh1 homologs, results in ectopic retention of mitotic cyclins in the G1 phase (Schwab et al., 1997; Sigrist and Lehner, 1997; Yamaguchi et al., 1997; Kitamura et al., 1998). We therefore investigated the protein levels of chicken mitotic cyclins, cyclin A and cyclin B, in wild-type and _Cdh1_–/– DT40 cells at different time points after release from nocodazole-induced metaphase block. FACS analysis showed that both genotypes were arrested in metaphase by nocodazole treatment and progress into G1 from M phase after release (Figure 3A). Immunoblot analysis revealed that the protein levels of both mitotic cyclins were significantly reduced as the G1 phase approached in wild-type cells. In contrast, in _Cdh1_–/– cells, the protein levels of mitotic cyclins failed to decline, resulting in ectopic accumulation of cyclin A and cyclin B in the G1 phase (Figure 3B), which is consistent with previous findings observed in yeast and Drosophila. These data suggest that Cdh1 takes part in the degradation of mitotic cyclins from M to G1 phase but is dispensable for exit from mitosis in higher vertebrates as well as in yeasts and Drosophila. It is noteworthy that mitotic cyclins are degraded to some extent in _Cdh1_–/– cells (Figure 3B), and this Cdh1-independent degradation of mitotic cyclins may be sufficient for cells to exit mitosis.
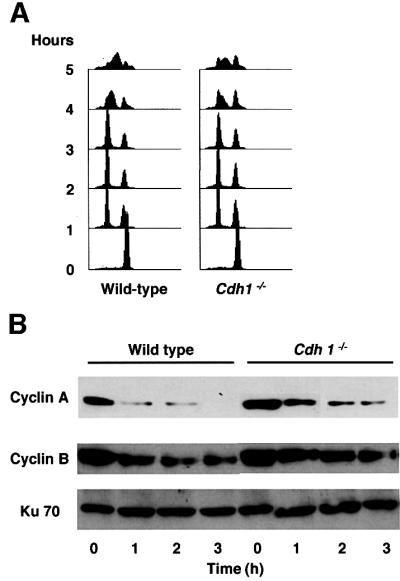
Fig. 3. Cdh1-deficient cells have unscheduled accumulation of mitotic cyclins in the G1 phase. (A) Cells, as indicated, were synchronized at metaphase by treatment with nocodazole for 10 h. At the indicated time points after release from nocodazole block, cells were harvested and analyzed by FACS. (B) The amounts of mitotic cyclins in wild-type and _Cdh1_–/– DT40 cells. Cells harvested in the experiment described in (A) were analyzed by immunoblotting with anti-cyclin A and B antibodies. The corresponding Ku70 levels are shown as a loading control.
Cdh1-deficient cells delay exit from mitosis
Although Cdh1 is dispensable for metaphase–anaphase transition and exit from mitosis, we examined whether loss of Cdh1 affects the duration of the mitotic phase. Wild-type or _Cdh1_–/– cells were synchronized at metaphase by treatment with nocodazole for 10 h. At different times after release from nocodazole block, the frequency of mitotic cells was determined by immunostaining of MPM-2 antigen, which is generally thought to represent mitotic phosphoproteins (Davis et al., 1983). At 45 min after release from metaphase block, ∼40–45% of _Cdh1_–/– cells remained in a mitotic state with positive MPM-2 staining, compared with only 20% MPM-2-positive cells in the wild type (Figure 4A). These observations suggest that Cdh1-deficient cells delay exit from mitosis. Furthermore, cytological examination of asynchronously growing cultures revealed that _Cdh1_–/– cells showed a significant increase in the number of cells in the mitotic phases, particularly in anaphase and telophase (Figure 4B), suggesting that the late mitotic phase is prolonged in _Cdh1_–/– cells. Taken together, Cdh1 is not essential for cell viability but plays a regulatory role in transition from late mitotic phase to G1.

Fig. 4. Exit from mitosis with delay in Cdh1-deficient cells. (A) After release from nocodazole treatment, as described in Figure 3A, cells were immunostained with MPM-2 antibody and analyzed by FACS. The graph displays the percentage of positively stained cells at the time indicated in wild-type cells (filled circles), Cdh1–/– clone 1 (filled triangles) and Cdh1–/– clone 2 (filled squares). The rate of positively stained cells was 100% at the time before release. Error bars show the standard error of the mean for three independent experiments. (B) The percentage of cells at mitotic phase (left panel) and at anaphase/telophase (right panel) in asynchronously growing wild-type DT40 cells and two Cdh1–/– clones. Chromatin is visualized by staining cells with aceto-orcein, with which by its strong staining mitotic condensed chromatin and chromatin segregation were easily identified. Columns and bars represent the mean and SD obtained from three independent experiments. Statistical differences were determined with Student’s _t_-test; *P <0.05.
Ectopic retention of mitotic cyclins abrogates G1 arrest in Cdh1–/– cells
During embryogenesis in Drosophila, fzr, a homolog of Cdh1, was shown to be required for removal of mitotic cyclin during G1 when proliferation of embryonic epidermal cell stops. In _fzr_-deficient embryos, epidermal cells progress through an extra division cycle without stopping in G1 after the terminal mitosis (mitosis 16). This defect is found to be caused by failure to degrade mitotic cyclins due to loss of fzr (Sigrist and Lehner, 1997). Since our _Cdh1_–/– DT40 cells appeared to have an unscheduled accumulation of mitotic cyclins in the G1 phase (Figure 3B), we attempted to investigate whether loss of Cdh1 function affects G1 arrest in higher vertebrates, as well as in Drosophila. During Drosophila embryogenesis, p27DAP, an inhibitor of cyclin E–cdk2, was shown to be expressed before mitosis 16 and to induce G1 arrest after completion of cycle 16 in epidermal cells (Lane et al., 1996). Unscheduled accumulation of mitotic cyclin by loss of fzr, therefore, appears to overcome the p27DAP-induced G1 arrest, resulting in progression through an extra division cycle (Du and Dyson, 1999). p27Kip1, a vertebrate homolog of p27DAP, inhibits the activity of cyclin E–cdk2, leading to a block of the G1–S transition in vertebrate cells (Polyak et al., 1994a). Accordingly, we tested the possibility that Cdh1-deficient cells abrogate G1 arrest induced by the expression of p27Kip1. (In the subsequent experiments, both _Cdh1_–/– clones behaved identically. Therefore, the results obtained from clone 1 are reported below.) We employed rapamycin, which is known to activate p27Kip1 expression (Nourse et al., 1994), to induce G1 arrest in DT40 cells. The p27Kip1 protein levels were up-regulated in both wild-type and _Cdh1_–/– DT40 cells after 72 h of rapamycin treatment (Figure 5A). FACS analysis revealed that rapamycin treatment for 72 h induced G1 arrest in wild-type cells (Figure 5B and C). In contrast, _Cdh1_–/– DT40 cells showed a significant decrease in the G1 fraction at 72 h of rapamycin treatment, and a corresponding increase in the number of cells with S phase DNA content (Figure 5B and C). While pRb was not phosphorylated in wild-type cells treated with rapamycin, _Cdh1_–/– cells dominantly expressed the phosphorylated form (Figure 5A). In addition, _Cdh1_–/– cells expressing the human Cdh1 transgene arrested in G1 in the presence of rapamycin, similarly to wild-type cells (Figure 5B). All these findings indicate that loss of Cdh1 results in abrogation of rapamycin-induced G1 arrest. Furthermore, _Cdh1_–/– cells treated with olomoucine, which is a specific cdk2 kinase inhibitor, were induced to undergo G1 arrest in the presence of rapamycin (Figure 5C). This result suggests that the abrogation of G1 arrest in Cdh1-deficient cells is caused by unscheduled activation of cdk2 kinase.


Fig. 5. Abrogation of rapamycin-induced G1 arrest in _Cdh1_–/– cells. (A) Expression of p27Kip1, pRb, cyclin A and cyclin B in wild-type and _Cdh1_–/– DT40 cells treated with 100 nM rapamycin for 36 or 72 h. The corresponding Ku70 levels are shown as a loading control. ‘AS’ indicates extracts from asynchronously proliferating cells. (B) Wild type, _Cdh1_–/– cells and _Cdh1_–/– cells expressing the human Cdh1 transgene were treated with 100 nM rapamycin for 72 h. Cells were harvested and analyzed by FACS. (C) G1 arrest was induced in rapamycin-treated _Cdh1_–/– cells by treatment with olomoucine, a specific inhibitor for cdk2. Wild-type and _Cdh1_–/– cells were treated with 100 nM rapamycin for 48 h (upper panels). Thereafter, DMSO (control) or olomoucine (100 mM) was added to the culture medium and incubated for another 24 h. Cells were harvested and analyzed by FACS. Cells were pulse-labeled for 10 min before harvest, and subsequently stained with FITC–anti-BrdU to detect BrdU incorporation and propidium iodide to detect total DNA. (D) Cell extracts of the wild-type and _Cdh1_–/– DT40 cells were harvested after rapamycin treatment for 72 h and immunoprecipitated with anti-p27Kip1 antibody or anti-cyclin E antibody. Cyclin A and cyclin E co-immunoprecipitated with p27Kip1 were detected by immunoblotting with their specific antibodies. Immunoprecipitates with anti-cyclin E antibody were assayed for histone H1 kinase activity.
The expression of p27Kip1 induces G1 arrest by inhibiting the activity of the cyclin E–cdk2 complex, which is essential for initiation of DNA replication (Polyak et al., 1994b). It has also been shown that p27Kip1 interacts with the cyclin A–cdk2 complex and inhibits its kinase activity (Blain et al., 1997). Therefore, it can be postulated that p27Kip1 is sequestered by the cyclin A–cdk2 complex ectopically accumulated during the G1 phase in _Cdh1_–/– cells, resulting in activation of the cyclin E–cdk2 complex and leading to abrogation of the rapamycin-induced G1 arrest. To investigate this, cyclin E and cyclin A immunoprecipitated with p27Kip1 were examined in wild-type and _Cdh1_–/– cells that were treated with rapamycin for 72 h. While a large amount of cyclin E was associated with p27Kip1 in wild-type cells, cyclin E was reduced in p27Kip1 immunoprecipitates in _Cdh1_–/– cells (Figure 5D). Conversely, association between cyclin A and p27Kip1 was markedly increased in _Cdh1_–/– cells. Furthermore, the cyclin E-associated kinase activity was significantly higher in the lysate derived from _Cdh1_–/– cells than in that from wild-type cells (Figure 5D). These findings suggest that the cyclin E–cdk2 complex dissociated from p27Kip1, which is sequestered by ectopically accumulated cyclin A–cdk2 complex, leads to abrogation of G1 arrest in _Cdh1_–/– cells.
Loss of Cdh1 causes premature entry into mitosis after DNA damage
In the normal cell cycle, cyclin A and cyclin B begin to accumulate from early S phase and G2 phase, respectively. We tested whether loss of Cdh1 affected levels of mitotic cyclins in S and G2 and cell cycle progression from S to mitotic entry. Wild-type or _Cdh1_–/– DT40 cells were synchronized in early S phase by an aphidicolin block. At different times after release from the S block, the protein levels of cyclin A and cyclin B and the mitotic index were determined. Immunoblot analyses showed that the expression of mitotic cyclins from S through G2 and early M phases was comparable in wild-type and _Cdh1_–/– cells (data not shown). Furthermore, the rate of mitotic entry of _Cdh1_–/– cells after release from S block was similar to that of wild-type cells (Figure 6A). These data suggest that Cdh1 deficiency does not impair transition from S phase to mitotic entry in normal cell cycle progression.


Fig. 6. _Cdh1_-deficient cells are unable to maintain G2 arrest after X-irradiation. (A) Cells were synchronized at early S phase by an aphidicolin block. Mitotic indices of wild-type cells (filled circles) and _Cdh1_–/– cells (filled triangles) were determined by counting cells with mitotic spindles at the indicated times after the release from aphidicolin block. Mitotic spindles were determined by staining cells with an anti-α-tubulin antibody. (B) Cells were treated with 10 Gy of X radiation (upper panel) or 5 J/m2 of UV radiation (lower panel) 1 h after the release from aphidicolin block. Mitotic indices of wild-type cells (filled circles) and _Cdh1_–/– cells (filled triangles) were determined by the same method as described in (A) at the indicated times. (C) Wild-type cells and _Cdh1_–/– cells were synchronized at early S phase and exposed to 10 Gy of X radiation 1 h after the release from the block. At the indicated time points after the release, cells were harvested and analyzed by FACS.
Although our original goal in this work was to understand the involvement of Cdh1 in normal cell cycle progression, our results identify a new role for Cdh1 in cell cycle checkpoints responsive to damaged DNA. Cells released from early S phase block were exposed to 10 Gy of X radiation at mid-S phase, and the cell cycle progression of wild-type and _Cdh1_–/– cells after irradiation was assessed by counting the rate of mitotic entry and by FACS analysis of DNA. Morphological examination revealed that both genotypes entered mitosis within 4 h after release from S block without irradiation (Figure 6A). Following treatment with 10 Gy of X radiation, the vast majority of wild-type cells appeared to arrest in the G2 phase until 6 h after irradiation, suggesting that wild-type cells have the damaged DNA-induced G2 checkpoint function (Figure 6B, upper panel). However, substantially more _Cdh1_–/– cells entered mitosis relative to wild-type cells at 5 h after irradiation, indicative of a defect in the G2 checkpoint. Moreover, in FACS analysis, _Cdh1_–/– cells showed a relative decrease in the G2 fraction as early as 8 h after irradiation and a corresponding increase in the number of cells having sub-G1 DNA content, which is characteristic of dead cells (Figure 6C). These findings suggest that the massive cell death of Cdh1-deficient cells after irradiation was due to premature entry into mitosis with DNA damage and, thereby, that Cdh1 plays a crucial role in the damaged DNA-induced G2 checkpoint.
DNA strand damage caused by UV-irradiation also activates the G2 checkpoint, but molecular mechanisms involved in regulating G2 delay after UV-irradiation are different from those after X-irradiation (Bulavin et al., 2001). In contrast to X-irradiation, UV-irradiation induced a G2 delay in both genotypes (Figure 6B, lower panel), suggesting that Cdh1 deficiency has no effect on the UV-induced G2 checkpoint.
DNA damage induces activation of Cdh1–APC
The activity of APC is largely dependent on its interaction with Cdc20 or Cdh1 (Kramer et al., 2000). The phosphorylation of Cdh1 is known to prevent binding to and activation of the APC. Hct1, a budding yeast homolog of Cdh1, was shown to be inactivated through phosphorylation by maturation-promoting factor (MPF) and activated through dephosphorylation by Cdc14 from late mitosis to the end of the G1 phase (Visintin et al., 1998; Jaspersen et al., 1999). Moreover, it has been suggested that interaction of Cdh1 and APC (active Cdh1–APC) is maintained until late in the G1 phase and diminished by the phosphorylation of Cdh1 with cdk2 from S phase in mammalian cells (Lukas et al., 1999; Kramer et al., 2000). These previous observations indicate that Cdh1–APC activity in normal cells is down-regulated by the dissociation of Cdh1 from the APC during S phase through to mitotic entry in the normal cell cycle. Based on our findings obtained using _Cdh1_–/– DT40 cells, we investigated whether APC is activated in the G2 phase through interaction with Cdh1 in response to DNA damage. _Cdh1_–/– cells expressing the human Cdh1 transgene were synchronized in early S phase by treatment with aphidicolin. The cells released from early S phase block were exposed to X-irradiation, and interaction between Cdh1 and APC was investigated. In the cells not treated with X-irradiation, Cdh1 had little association with Cdc27, a component of the APC complex, during G2 phase (Figure 7A), which is consistent with findings reported previously (Kramer et al., 2000). In contrast, when cells were exposed to X-irradiation at 1 h after release from S block, Cdh1 was induced to interact with Cdc27 in the G2 phase (Figure 7A). Interaction between Cdh1 and the APC complex induced by DNA damage was observed not only in DT40 cells but also in both rodent fibroblast (Rat1 cell) and a human cancer cell line (HeLa cell) (Figure 7B).

Fig. 7. Activation of the Cdh1–APC complex after X-irradiation-induced DNA damage. (A) _Cdh1_–/– cells expressing HA-tagged human Cdh1 were synchronized in early S phase by aphidicolin treatment and treated with 10 Gy of X radiation. The cell extracts harvested at 4 h after the release were immunoprecipitated with antibodies against HA. Cdc27, a component of the APC complex contained in total cell lysates was determined by immunoblotting (left panel). The relative amount of Cdc27, precipitated with the HA-human Cdh1, was determined by immunoblotting (right panel). (B) HeLa cells were synchronized at the beginning of S phase by the double thymidine block and treated with 10 Gy of X radiation 3 h after the release from S block. Rat1 fibroblasts were synchronized at early S phase by aphidicolin treatment and treated with 10 Gy of X radiation 1 h after the release from aphidicolin block. The cell extracts harvested at 8 h (HeLa) or 4 h (Rat1) after the release were immunoprecipitated with an antibody against Cdc27. Cdc27 and Cdh1 co-immunoprecipitated with Cdc27 antibody were detected by immunoblotting with their specific antibodies. (C) In vitro ubiquitination assay. HeLa cells synchronized at G2 phase were treated with or without X-irradiation as described in (B). APC immunoprecipitated with anti-Cdc27 antibody from the cell lysates was subjected to the in vitro ubiquitylation assay as described in Materials and methods. The reaction was terminated at the times indicated. His-Cdc20 (arrow) was used as a substrate. ‘Ub-Cdc20’ indicates ubiquitylated Cdc20. (D) Ubiquitylation of Cdh1-associated complex after X-irradiation. HeLa cells transfected with ubiquitin and HA-Cdh1 were treated with 10 Gy of X radiation 3 h after the release from S block and immunoprecipitated with anti-HA antibody. Ubiquitylated proteins precipitated with HA-tagged Cdh1 were detected by immunoblotting with an anti-ubiquitin antibody. (E) The amounts of mitotic cyclins in wild-type and _Cdh1_–/– DT40 cells. Cells were synchronized by aphidicolin treatment for 10 h, irradiated with 10 Gy 1 h after the release and analyzed by immunoblotting with anti-cyclin A and B antibodies. The time indicated was that after the release. The corresponding Ku70 levels are shown as a loading control.
Having determined the complex formed by APC and Cdh1 after DNA damage, we asked whether the Cdh1– APC complex becomes catalytically active in response to DNA damage. First, we performed an in vitro ubiquitylation assay using human Cdc20 as substrate. Human Cdc20 contains a KEN box, which was shown to be the Cdh1-specific targeting motif (Pfleger and Kirschner, 2000) and, thereby, is considered to be a specific substrate for Cdh1–APC. APC immunoprecipitated with anti-Cdc27 antibody from cells in normal G2 phase showed no activity to ubiquitylate human Cdc20 (Figure 7C, lanes 5–8), which is consistent with results reported previously (Kramer et al., 2000). In contrast, APC purified from cells arrested in the G2 phase after X-irradiation ubiquitylated human Cdc20 in a time-dependent manner (Figure 7C, lane 1–4), suggesting that Cdh1–APC becomes catalytically active by DNA damage induced by X-irradiation. Furthermore, we attempted to investigate the in vivo ubiquitylation of the Cdh1–APC complex after X-irradiation in the G2 phase. HeLa cells stably expressing human ubiquitin were established and used to examine the ubiquitylation of the Cdh1-associated complex. Ubiquitylation of the complex immunoprecipitaed with hemagglutinin (HA)-tagged Cdh1 was very low in cells in the G2 phase without DNA damage (Figure 7D, left lane). However, Cdh1-associated complex in X-irradiated cells in the G2 phase showed significantly increased ubiquitylation (Figure 7D, right lane). All these findings suggest that Cdh1–APC is activated at the X-irradiation-induced G2 checkpoint.
Our findings indicate the possibility that activation of Cdh1–APC by DNA damage may induce degradation of regulators for G2–M transition, resulting in G2 arrest. In HeLa and other cells, it is known that the expression of cyclin B is down-regulated in S and G2 phase in response to DNA damage (Muschel et al., 1993). To investigate whether abrogation of the damaged DNA-induced G2 checkpoint in _Cdh1_–/– cells is due to an inability to degrade mitotic cyclins, the protein levels of mitotic cyclins after X-irradiation were examined. Immunoblot analyses showed that the levels of mitotic cyclins did not change significantly but remained similarly stable in both wild-type and _Cdh1_–/– cells (Figure 7E). These data suggest that the degradation of mitotic cyclins mediated by the Cdh1–APC complex plays little role in the G2 checkpoint after DNA damage.
Discussion
Role of Cdh1 in the mitotic phase
In the present study, we have generated _Cdh1_-deficient cells using DT40 chicken B lymphocytes. _Cdh1_–/– DT40 cells appeared to be exiting mitosis and proliferating normally despite the fact that expression of cyclin A and cyclin B in these cells in M and G1 phases was significantly elevated. These findings suggest that Cdh1 plays a major role in proteolysis of mitotic cyclins from the M to G1 phases but is dispensable for viability and proliferation of higher vertebrate cells, which is similar to observations in yeast and Drosophila (Schwab et al., 1997; Sigrist and Lehner, 1997). In higher vertebrate cells, cyclin B destruction has been believed to be required for exit from mitosis. However, recent observation has clearly demonstrated that mammalian cells overexpressing cyclin B1 are capable of exiting mitosis (Jin et al., 1998), which appears consistent with our result.
Our results show that mitotic cyclins accumulate significantly but are partially degraded at the end of mitosis in _Cdh1_–/– cells, suggesting that not only Cdh1 but also another molecule, such as Cdc20, is involved in the proteolysis of mitotic cyclins in the mitotic phase. In budding yeast, Hct1/Cdh1 and Cdc20 have been shown to target some common molecules, and the main difference between them is the time during the cell cycle when each is active (Yeong et al., 2000). Furthermore, Cdc20–APC can only be activated under conditions in which Hct1/Cdh1–APC is inactive, and this dependency helps to ensure that the events of mitosis occur in the proper sequence (Lim et al., 1998; Visintin et al., 1998; Shirayama et al., 1999; Yeong et al., 2000). Therefore, collectively, these previous data suggest that Cdc20 and Hct1/Cdh1 target distinct, but overlapping sets of proteins for degradation in crucial mitotic events, such as separation of sister chromatids and exit from mitosis. It was found recently that in early Xenopus embryos, Cdh1 is not expressed (Lorca et al., 1998), indicating that Cdh1 is not required for cyclin oscillations in early embryonic cell cycles. Similarly, Cdh1 is not expressed in early fly embryos, where Cdc20 is required for cyclin destruction (Sigrist and Lehner, 1997). Additionally, vertebrate Cdc20–APC and Cdh1–APC both ubiquitylate mitotic cyclins in vitro (Fang et al., 1998). Thus, it is reasonable to speculate that Cdc20 promotes not only the initiation of anaphase but also the partial destruction of mitotic cyclins, which is sufficient for exit from mitosis, in _Cdh1_–/– DT40 cells. Our observations suggest that proteolysis of mitotic cyclins is regulated by two steps, mediated by Cdc20 and Cdh1, in higher vertebrate cells.
Although Cdh1 is dispensable for viability of DT40 cells, the length of time between anaphase and the end of mitosis was found to be prolonged in _Cdh1_–/– DT40 cells. This result suggests that Cdh1–APC regulates the progression of late mitosis despite the fact that it is not essential for exiting mitosis.
Involvement of Cdh1 in G1–S transition
In cultured human cells, ectopic overexpression of cyclin A can drive premature entry into S phase and causes a decrease in the percentage of cells in G1 (Resnitzky et al., 1995). These results demonstrate that the expression of cyclin A is rate limiting for the G1–S transition. However, our observations showed that _Cdh1_–/– DT40 cells have a normal G1 phase and do not enter S phase prematurely despite the high expression of both cyclin A and cyclin B. These findings suggest that the accumulation of mitotic cyclins due to loss of Cdh1 does not affect the duration of the G1 phase in normal exponential conditions. This discrepancy may be explained by a difference in the expression levels of cyclin A. The level of cyclin A induced by the exogenous expression system in the study of Resnitzky et al. (1995) appears to be much higher than physiological levels. Thus, it is possible that the levels of mitotic cyclins accumulated at G1 phase in Cdh1-deficient cells are not enough to induce premature entry into S phase.
In _fzr_-deficient Drosophila embryos, epidermal cells progress through an extra division cycle without stopping in G1 after terminal mitosis. This defect is believed to be caused by failure to decrease mitotic cyclins (Sigrist and Lehner, 1997). Furthermore, _srw1_-mutant fission yeasts also do not arrest at G1 under conditions of nitrogen starvation (Yamaguchi et al., 1997). Similarly, we found that _Cdh1_–/– DT40 cells abrogate the G1 arrest induced by rapamycin, which is an activator of p27Kip1, following entry into S phase. p27Kip1 is known to induce G1 arrest by inhibiting the activity of the cyclin E–cdk2 complex. p27Kip1 has also been shown to interact with cyclin A–cdk2. Since the induction of p27Kip1 expression by rapamycin was similar in both wild-type and _Cdh1_–/– DT40 cells (Figure 5A), one mechanistic explanation as to how inactivation of Cdh1 can cause an abrogation of G1 cell cycle arrest is by sequestration of p27Kip1 by accumulated cyclin A–cdk2 complex to activate cyclin E–cdk2. Four lines of evidence presented here support this hypothesis. First, inhibition of cdk2 kinase activity by treatment with olomoucine led _Cdh1_–/– cells to arrest at the G1 phase in the presence of rapamycin (Figure 5C). Secondly, the amounts of cyclin E associated with p27Kip1 were decreased in _Cdh1_–/– cells. Thirdly, p27Kip1 interacted predominantly with ectopically accumulated cyclin A in _Cdh1_–/– cells. Finally, the level of cyclin E-associated kinase activity was dramatically elevated in _Cdh1_–/– cells (Figure 5D). In fission yeast, cdk1 activated by B-type cyclin can promote G1–S progression in the absence of G1 cyclin (Fisher and Nurse, 1996). Therefore, the possibility cannot be ruled out that cdk1 activated with ectopically accumulated cyclin B may promote G1–S transition in rapamycin-treated _Cdh1_–/– cells. It is also possible that high levels of cyclin A promote transition of _Cdh1_–/– cells from G1 to S phase. In any case, our findings suggest that activation of Cdh1 plays a crucial role in establishment of G1 arrest by cooperating with inhibitors of G1 cyclin-dependent kinases.
Activation of Cdh1–APC in the DNA damage-induced G2 checkpoint
Recent reports have shown that Cdh1–APC is inactivated from S phase until the mid-mitotic phase through phosphorylation by cyclin A–cdk2 or cyclin B–cdk1 in mammalian cells (Lukas et al., 1999). These data indicate that Cdh1–APC is not required for progression of the cell cycle from S phase to mitotic entry, which is consistent with our finding that Cdh1 deficiency does not affect the cell cycle progression from S phase to the mitotic phase in physiological conditions. However, we found that _Cdh1_–/– cells failed to maintain X-irradiation-induced arrest in the G2 phase of the cell cycle. This finding strongly suggests that the activation of Cdh1–APC plays a role in DNA damage-induced G2 checkpoint function. In fact, we demonstrate here that X-irradiation induces interaction of Cdh1 with APC in the G2 phase (Figure 7A and B) and consequently activates the Cdh1–APC (Figure 7C and D). These findings indicate that Cdh1–APC is activated at G2 by X-irradiation-induced DNA damage.
How can Cdh1–APC induce G2 arrest in response to DNA damage? One obvious possibility is that Cdh1–APC targets regulators for G2–M transition. However, our findings showed that the levels of mitotic cyclins in _Cdh1_–/– cells after DNA damage were similar to those in wild-type cells (Figure 7E). These data imply that cyclin A and cyclin B cannot be substrates for Cdh1–APC when it is activated irregularly by DNA damage at G2. Recent reports have demonstrated that Cdc20 strictly recognizes a substrate with a well-defined D-box, whereas Cdh1–APC recognizes both D-box and non-D-box substrates (Pfleger and Kirschner, 2000) for destruction. Therefore, the regulation of the interaction between Cdh1–APC and those diverse substrates may be dependent on the cell cycle phases and events. It seems likely that critical molecules for promoting the G2–M transition, besides mitotic cyclins, are potential substrates of activated Cdh1– APC in the DNA damage checkpoint and that the degradation of those substrates by the activated Cdh1– APC induces G2 arrest.
We demonstrate that UV radiation induced G2 delay not only in wild-type cells but also in _Cdh1_–/– cells (Figure 6B), suggesting that Cdh1 deficiency has no effect on the UV-induced G2 checkpoint. Recently, it has been reported that molecular mechanisms regulating UV-induced G2 delay are distinct from those induced by X radiation (Bulavin et al., 2001). Therefore, this result shows that Cdh1 is involved in the X-irradiation-induced G2 checkpoint pathway but not in the UV-induced G2 checkpoint pathway.
Involvement of Cdh1–APC in cancers
The maintenance of genomic integrity after DNA damage depends on the coordinated action of the DNA repair and checkpoint systems. The failure of a checkpoint leads to genomic instability and predisposition to cancer (Lengauer et al., 1998). Since our findings suggest that activation of Cdh1–APC is required for both cdk inhibitor-dependent G1 arrest and DNA damage-induced G2 arrest, it is conceivable that abnormal regulation of Cdh1 activation may involve failure in normal cell cycle progression, leading to genetic and chromosomal instabilities, which result in development of malignant tumors. Investigation of this possibility is currently underway using various cancer cells.
Our findings also have potential relevance for the treatment of cancer. Abrogation of the DNA damage-induced G2 checkpoint sensitizes cells to genotoxic stress. This suggests that a specific inhibitor of DNA damage-induced Cdh1 activation could make radiotherapy and/or chemotherapy more effective.
Materials and methods
Cell culture
DT40 cells were cultured in RPMI1640 medium supplemented with 10–5 M β-mercaptoethanol, penicillin/streptomycin, 10% fetal calf serum and 1% chicken serum (Sigma) at 37°C. Rat1 and HeLa cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM)-F12 supplemented with penicillin/streptomycin and 10% fetal calf serum.
Plasmid constructs
Chicken Cdh1 cDNA was amplified by RT–PCR using mRNA extracted from DT40 cells with primers for human Cdh1. Using this cDNA, ∼9 kb of the chicken genomic cdh1 locus was isolated from DT40 genomic DNA using long-range PCR. Chicken Cdh1 disruption constructs, _Cdh1_-Bsr (blasticidin) and _Cdh1_-His (histidinol), were made by replacing ∼1 kb of the genomic sequence of Cdh1 with Bsr and His selection marker cassettes (Figure 1A).
Gene targeting
For gene targeting of the chicken Cdh1 locus, DT40 cells (107) were suspended in 0.5 ml of phosphate-buffered saline (PBS) containing 30 µg of linearized targeting vector for each transfection and electroporated with a Gene Pulser apparatus (BTX) at 210 V and 475 µF. Following electroporation, cells were transferred into 20 ml of fresh medium and incubated for 24 h. Cells were then resuspended in 80 ml of medium containing 30 µg/ml blasticidin-S (Calbiochem) or 1 mg/ml histidinol (Sigma) and divided into four 96-well plates. After 7–10 days, drug-resistant colonies were transferred to 24-well plates. Genomic DNA was extracted from each expanded clone, and successful targeted recombination was identified by Southern blot and PCR analyses. To generate _Cdh1_–/– cells expressing the human Cdh1 transgene, we transfected cells with the pUHG plasmid containing human Cdh1 and selected stable transformants with 2.5 mg/ml hygromycin (Calbiochem).
Genomic PCR and RT–PCR
To evaluate the targeted allele, genomic DNA from DT40 cells was amplified using the three sets of primers (p2–p4, p1–p3 and p1–p5) illustrated in Figure 1(A). Genomic DNA was prepared using InstaGene Matrix (Bio-Rad). mRNA was isolated using a Micro-FastTrack 2.0 kit (Invitrogen) and reverse-transcribed using the Superscript cDNA synthesis system as described by the suppliers (Gibco-BRL). Full-length chicken Cdh1 cDNA was amplified by PCR with 35 cycles of 94°C (1 min), 60°C (1 min) and 72°C (1 min). Primers for the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were used along with the primers for the amplification of Cdh1 cDNA.
Antibodies
Proteins were detected by anti-chicken cyclin A antibody (Transduction Laboratories), anti-chicken cyclin B2 antibody (Gallant and Nigg, 1992), anti-cyclin E antibody (Santa Cruz), anti-Rb antibody (Transduction Laboratories), anti-ubiquitin antibody (Sigma), anti-chicken Ku70 antibody (Takata et al., 1998) and anti-Cdh1 antibody (Wang et al., 2000).
Cell cycle analysis
To analyze the intracellular DNA content, cells were fixed in 70% methanol at –20°C for several hours. Cells were centrifuged at 2000 r.p.m. and resuspended in PBS containing RNase A (Sigma) at 0.1 mg/ml. Samples were incubated at 37°C for 15 min, and propidium iodide (Sigma) was added to a final concentration of 25 µg/ml. Samples were processed by a FACScan (Becton Dickinson) using Cell Quest software (Becton Dickinson). For determination of the bromodeoxyuridine (BrdU) incorporation, cells were labeled for 10 min with 20 µM BrdU (Amersham). They were harvested and fixed in 70% methanol at –20°C for several hours. Then, cells were stained with an anti-BrdU antibody as described previously (Sonoda et al., 1998).
Cell synchronization
In DT40 cells, to synchronize cells at the G1, early S phase or metaphase, cells were cultured in the presence of 100 nM rapamycin (Calbiochem) for 72 h, 1 µg/ml aphidicolin (Wako) for 10 h or 0.5 µg/ml nocodazole (Sigma) for 10 h, respectively. Rat1 cells were synchronized at early S phase by 1 µg/ml aphidicolin treatment for 12 h. HeLa cells were synchronized at the beginning of S phase by means of a double thymidine/aphidicolin block. Briefly, HeLa cells were incubated with 2.0 mM thymidine (Sigma) for 24 h, released for 8 h and then treated with 1 µg/ml aphidicolin for a further 16 h. Cells were exposed to 10 Gy of X-rays at 1 h (DT40 and Rat1) or 3 h (HeLa) after release from S block.
Detection of cells at mitosis
Cells with mitotic condensed chromatin were visualized by staining with aceto-orcein (Merck) in 60% acetic acid (Hirota et al., 2000). Cells at anaphase and telophase are easily identified by aceto-orcein staining. To avoid the morphological confusion between mitotic cells and apoptotic cells in the X- and UV-irradiation experiments, mitotic indices were determined by counting cells with mitotic spindles. To detect the mitotic spindles, DT40 cells were fixed with 100% methanol for 10 min, washed with PBS twice and incubated in a blocking buffer (1% bovine serum albumin in PBS) for 1 h at room temperature. The cells were incubated with the monoclonal antibody against α-tubulin (Sigma) for 20 h at 4°C, washed with PBS and incubated with fluorescein isothiocyanate (FITC)-conjugated anti-mouse IgG (Biosource) for 4 h at room temperature.
The proportion of cells in the mitotic phase was also determined by FACS analysis using anti-MPM-2 antibody (Upstate Biotechnology). Briefly, cells were harvested and fixed in 70% methanol at –20°C for several hours. Fixed cells were stained for 1 h at room temperature with anti-MPM-2 antibody, washed three times in PBS and stained with FITC-conjugated anti-mouse IgG (Biosource). Stained cells were analyzed with FACScan using Cell Quest software.
Immunoprecipitation
Cells were washed with PBS and used immediately, or were frozen in liquid nitrogen and stored at –80°C. The cells were then lysed in 0.5% Triton X-100 lysis buffer (20 mM Tris–HCl pH 7.7, 100 mM KCl, 50 mM sucrose, 0.1 mM CaCl2, 1 mM MgCl2, 0.5% Triton X-100, 20 mM β-glycerophosphate, 1 mM Na3VO4 and a cocktail of protease inhibitors). The extracts were centrifuged at 14 000 r.p.m. for 15 min at 4°C, and the supernatants were used for immunoprecipitation. Aliquots of supernatants were incubated for 2 h at 4°C with a monoclonal anti-p27_Kip1_ antibody (Transduction Laboratories), anti-Cdc27 antibody (Santa Cruz) and anti-HA antibody (Boehringer Mannheim), respectively. Cells were incubated for another 2 h after addition of protein G plus protein A–agarose beads (Oncogene Science). The washed immunoprecipitants and the supernatant were collected for western blotting.
Histone H1 kinase assay
Cells were washed with PBS twice and suspended in 120 µl of lysis buffer (20 mM Tris–HCl pH 7.5, 10 mM EDTA, 100 mM NaCl, 1% Triton X-100, 1 mM NaF, 1 mM β-glycerophosphate and a cocktail of protease inhibitors). A 100 µl aliquot of lysates and 5 µl of an anti-cyclin E antibody were mixed and incubated for 1 h at 4°C. Then, a 20 µl aliquot of protein G plus protein A–agarose beads was added. After gentle agitation for 30 min at 4°C, beads were washed four times in lysis buffer, the second wash containing 1 M NaCl, and twice in kinase buffer [25 mM MOPS pH 7.2, 15 mM MgCl2, 5 mM EDTA, 1 mM dithiothreitol (DTT), 60 mM β-glycerophosphate, 0.1 mM sodium orthovanadate and a cocktail of protease inhibitors]. Pelleted beads were mixed with an equal volume of a reaction mix containing Triton X-100 (1%), [γ-32P]ATP and histone H1 (1 mg/ml) and incubated for 15 min at 30°C. The reaction was chilled and stopped by addition of a sample buffer. The samples were loaded on an SDS–polyacrylamide gel and visualized by autoradiography.
In vitro ubiquitination assay
The N-terminal half of human Cdc20 cDNA encoding amino acid residues 1–250 was inserted into pET23d and expressed in NovaBlue (DE3) (Novagene). The His-tagged recombinant Cdc20 protein was purified and biotinylated (Pierce). The in vitro ubiquitination assay was carried out as described previously (Kotani et al., 1998, 1999). In brief, synchronized HeLa cells were treated with or without X-irradiation at the G2 phase, and the cell extracts harvested 8 h after the release were immunoprecipitated with anti-Cdc27 antibody. Immunoprecipitants were incubated with 2 µg of His-Cdc20 in 30 µl of 5 mM Tris pH 7.6, 0.5 mM MgCl2, 2 mM ATP, 2 mM DTT, 2 mM creatinine phosphate, 1 µg/ml creatine, phosphokinase, 0.2 mg/ml bovine ubiquitin (Sigma), 40 µg/ml mouse recombinant E1 and 50 µg/ml human recombinant hE2-C. After the reaction, His-Cdc20 was purified using ProBond resin, subjected to SDS–PAGE and transferred onto a nitrocellulose membrane followed by detection with streptavidin–horseradish peroxidase.
In vivo ubiquitylation of Cdh1-associated complex
HeLa cells were transfected with myc-tagged ubiquitin expression plasmid (pcDNA3/myc-Ub), and neomycin-resistant clones were isolated and cultured in the presence of 1 mg/ml geneticin (Gibco-BRL). To synchronize the myc-ubiquitin-expressing HeLa cells at early S phase, asynchronously growing cells were treated with 2 mM thymidine for 16 h. Cells were then released from the block by changing to complete growth medium containing 24 mM each of thymidine and deoxycytidine. At 6 h after the release, cells were transfected with Cdh1/pCGN-HA using FuGENE 6 (Roche) reagent. After 2 h, thymidine was added to the medium to a final concentration of 2 mM and cells were cultured for another 16 h. Before harvest at 8 h after the release, 10 mM MG132 (Calbiochem) was added to the medium and incubated for 1 h. An immunoprecipitation analysis with anti-HA antibody was performed as described above. The samples were subjected to SDS–PAGE followed by immunoblot analysis using anti-ubiquitin antibody (Sigma).
Acknowledgments
Acknowledgements
We thank Dr A.Venkitaraman for helpful discussions and critical reading of the manuscript, Dr Q.Hu for providing the pCGN plasmids, Drs E.Nigg and J.Braun for providing the anti-cyclin B2 and Cdh1 antibodies, respectively, Dr J.Moon for editorial assistance, Drs K.Yonezawa, K.Todokoro, T.Hirota, T.Morisaki and T.Marumoto for helpful discussions, and T.Arino and Y.Fukushima for secretarial assistance. This work was supported by a grant for cancer research from the Ministry of Education, Science and Culture of Japan (H.S.).
References
- Agami R. and Bernards,R. (2000) Distinct initiation and maintenance mechanisms cooperate to induce G1 cell cycle arrest in response to DNA damage. Cell, 102, 55–66. [DOI] [PubMed] [Google Scholar]
- Blain S.W., Montalvo,E. and Massague,J. (1997) Differential interaction of the cyclin-dependent kinase (Cdk) inhibitor p27Kip1 with cyclin A–Cdk2 and cyclin D2–Cdk4. J. Biol. Chem., 272, 25863–25872. [DOI] [PubMed] [Google Scholar]
- Bulavin D.V., Higashimoto,Y., Popoff,I.J., Gaarde,W.A., Basrur,V., Potapova,O., Apella,E. and Fornace,A.J. (2001) Initiation of a G2/M checkpoint after ultraviolet radiation requires p38 kinase. Nature, 411, 102–107. [DOI] [PubMed] [Google Scholar]
- Cohen-Fix O. and Koshland,D. (1997) The metaphase-to-anaphase transition: avoiding a mid-life crisis. Curr. Opin. Cell Biol., 9, 800–806. [DOI] [PubMed] [Google Scholar]
- Davis F.M., Tsao,T.Y., Fowler,S.K. and Rao,P.N. (1983) Monoclonal antibodies to mitotic cells. Proc. Natl Acad. Sci. USA, 80, 2926–2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du W. and Dyson,N. (1999) The role of RBF in the introduction of G1 regulation during Drosophila embryogenesis. EMBO J., 18, 916–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang G., Yu,H. and Kirschner,M.W. (1998) Direct binding of CDC20 protein family members activates the anaphase-promoting complex in mitosis and G1. Mol. Cell, 2, 163–171. [DOI] [PubMed] [Google Scholar]
- Fisher D.L. and Nurse,P. (1996) A single fission yeast mitotic cyclin B p34cdc2 kinase promotes both S-phase and mitosis in the absence of G1 cyclins. EMBO J., 15, 850–860. [PMC free article] [PubMed] [Google Scholar]
- Funabiki H., Yamano,H., Kumada,K., Nagao,K., Hunt,T. and Yanagida,M. (1996) Cut2 proteolysis required for sister-chromatid separation in fission yeast. Nature, 381, 438–441. [DOI] [PubMed] [Google Scholar]
- Gallant P. and Nigg,E.A. (1992) Cyclin B2 undergoes cell cycle-dependent nuclear translocation and, when expressed as a non-destructible mutant, causes mitotic arrest in HeLa cells. J. Cell Biol., 117, 213–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota T. et al. (2000) Zyxin, a regulator of actin filament assembly, targets the mitotic apparatus by interacting with h-warts/LATS1 tumor supressor. J. Cell Biol., 149, 1073–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaspersen S.L., Charles,J.F. and Morgan,D.O. (1999) Inhibitory phosphorylation of the APC regulator Hct1 is controlled by the kinase Cdc28 and the phosphatase Cdc14. Curr. Biol., 9, 227–236. [DOI] [PubMed] [Google Scholar]
- Jin P., Hardy,S. and Morgan,D.O. (1998) Nuclear localization of cyclin B1 controls mitotic entry after DNA damage. J. Cell Biol., 141, 875–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juang Y.L., Huang,J., Peters,J.M., McLaughlin,M.E., Tai,C.Y. and Pellman,D. (1997) APC-mediated proteolysis of Ase1 and the morphogenesis of the mitotic spindle. Science, 275, 1311–1314. [DOI] [PubMed] [Google Scholar]
- King R.W., Peters,J.M., Tugendreich,S., Rolfe,M., Hieter,P. and Kirschner,M.W. (1995) A 20S complex containing CDC27 and CDC16 catalyzes the mitosis-specific conjugation of ubiquitin to cyclin B. Cell, 81, 279–288. [DOI] [PubMed] [Google Scholar]
- King R.W., Deshaies,R.J., Peters,J.M. and Kirschner,M.W. (1996) How proteolysis drives the cell cycle. Science, 274, 1652–1659. [DOI] [PubMed] [Google Scholar]
- Kitamura K., Maekawa,H. and Shimoda,C. (1998) Fission yeast Ste9, a homolog of Hct1/Cdh1 and Fizzy-related, is a novel negative regulator of cell cycle progression during G1-phase. Mol. Biol. Cell, 9, 1065–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotani S., Tugendreich,S., Fujii,M., Jorgensen,P.M., Watanabe,N., Hoog,C., Hieter,P. and Todokoro,K. (1998) PKA and MPF-activated polo-like kinase regulate anaphase-promoting complex activity and mitosis progression. Mol. Cell, 1, 371–380. [DOI] [PubMed] [Google Scholar]
- Kotani S., Tanaka,H., Yasuda,H. and Todokoro,K. (1999) Regulation of APC activity by phosphorylation and regulatory factors. J. Cell Biol. 146, 791–800. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Kramer E.R., Scheuringer,N., Podtelejnikov,A.V., Mann,M. and Peters,J.M. (2000) Mitotic regulation of the APC activator proteins CDC20 and CDH1. Mol. Biol. Cell, 11, 1555–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakin N.D. and Jackson,S.P. (1999) Regulation of p53 in response to DNA damage. Oncogene, 18, 7644–7655. [DOI] [PubMed] [Google Scholar]
- Lane M.E., Sauer,K., Wallace,K., Jan,Y.N., Lehner,C.F. and Vaessin,H. (1996) Dacapo, a cyclin-dependent kinase inhibitor, stops cell proliferation during Drosophila development. Cell, 87, 1225–1235. [DOI] [PubMed] [Google Scholar]
- Lengauer C., Kinzler,K.W. and Vogelstein,B. (1998) Genetic instabilities in human cancers. Nature, 396, 643–649. [DOI] [PubMed] [Google Scholar]
- Lim H.H., Goh,P.Y. and Surana,U. (1998) Cdc20 is essential for the cyclosome-mediated proteolysis of both Pds1 and Clb2 during M phase in budding yeast. Curr. Biol., 8, 231–234. [DOI] [PubMed] [Google Scholar]
- Lorca T., Castro,A., Martinez,A.M., Vigneron,S., Morin,N., Sigrist,S., Lehner,C., Doree,M. and Labbe,J.C. (1998) Fizzy is required for activation of the APC/cyclosome in Xenopus egg extracts. EMBO J., 17, 3565–3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukas C., Sorensen,C.S., Kramer,E., Santoni-Rugiu,E., Lindeneg,C., Peters,J.M., Bartek,J. and Lukas,J. (1999) Accumulation of cyclin B1 requires E2F and cyclin-A-dependent rearrangement of the anaphase-promoting complex. Nature, 401, 815–818. [DOI] [PubMed] [Google Scholar]
- Muschel R.J., Zhang,H.B. and McKenna,W.G. (1993) Differential effect of ionizing radiation on the expression of cyclin A and cyclin B in HeLa cells. Cancer Res., 53, 1128–1135. [PubMed] [Google Scholar]
- Nourse J., Firpo,E., Flanagan,W.M., Coats,S., Polyak,K., Lee,M.H., Massague,J., Crabtree,G.R. and Roberts,J.M. (1994) Interleukin-2-mediated elimination of the p27Kip1 cyclin-dependent kinase inhibitor prevented by rapamycin. Nature, 372, 570–573. [DOI] [PubMed] [Google Scholar]
- Pfleger C.M. and Kirschner,M.W. (2000) The KEN box: an APC recognition signal distinct from the D box targeted by Cdh1. Genes Dev., 14, 655–65. [PMC free article] [PubMed] [Google Scholar]
- Polyak K., Lee,M.H., Erdjument-Bromage,H., Koff,A., Roberts,J.M., Tempst,P. and Massague,J. (1994a) Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell, 78, 59–66. [DOI] [PubMed] [Google Scholar]
- Polyak K., Kato,J.Y., Solomon,M.J., Sherr,C.J., Masague,J., Roberts,J.M and Koff,A. (1994b) p27Kip1, a cyclin–Cdk inhibitor, links transforming growth factor-β and contact inhibition to cell cycle arrest. Genes Dev., 8, 9–22. [DOI] [PubMed] [Google Scholar]
- Resnitzky D., Hengst,L. and Reed,S.I. (1995) Cyclin A-associated kinase activity is rate limiting for entrance into S phase and is negatively regulated in G1 by p27Kip1. Mol. Cell. Biol., 15, 4347–4352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab M., Lutum,A.S. and Seufert,W. (1997) Yeast Hct1 is a regulator of Clb2 cyclin proteolysis. Cell, 90, 683–693. [DOI] [PubMed] [Google Scholar]
- Shirayama M., Toth,A., Galova,M. and Nasmyth,K. (1999) APC(Cdc20) promotes exit from mitosis by destroying the anaphase inhibitor Pds1 and cyclin Clb5. Nature, 402, 203–207. [DOI] [PubMed] [Google Scholar]
- Sigrist S.J. and Lehner,C.F. (1997) Drosophila fizzy-related down-regulates mitotic cyclins and is required for cell proliferation arrest and entry into endocycles. Cell, 90, 671–681. [DOI] [PubMed] [Google Scholar]
- Sonoda E., Sasaki,S., Buerstedde,J.M., Bezzubova,O., Shinohara,A., Ogawa,H., Takata,M., Yamaguchi-Iwai,Y. and Takeda,S. (1998) Rad51-deficient vertebrate cells accumulate chromosomal breaks prior to cell death. EMBO J., 17, 598–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudakin V., Ganoth,D., Dahan,A., Heller,H., Hershko,J., Luca,F.C., Ruderman,J.V. and Hershko,A. (1995) The cyclosome, a large complex containing cyclin-selective ubiquitin ligase activity, targets cyclins for destruction at the end of mitosis. Mol. Biol. Cell, 6, 185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata M. et al. (1998) Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J., 17, 5497–5508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlmann F., Lottspeich,F. and Nasmyth,K. (1999) Sister-chromatid separation at anaphase onset is promoted by cleavage of the cohesin subunit Scc1. Nature, 400, 37–42. [DOI] [PubMed] [Google Scholar]
- Visintin R., Prinz,S. and Amon,A. (1997) CDC20 and CDH1: a family of substrate-specific activators of APC-dependent proteolysis. Science, 278, 460–463. [DOI] [PubMed] [Google Scholar]
- Visintin R., Craig,K., Hwang,E.S., Prinz,S., Tyers,M. and Amon,A. (1998) The phosphatase Cdc14 triggers mitotic exit by reversal of Cdk-dependent phosphorylation. Mol. Cell, 2, 709–718. [DOI] [PubMed] [Google Scholar]
- Wang C.X., Fisk,B.C., Wadehra,M., Su,H. and Braun,J. (2000) Overexpression of murine fizzy-related (fzr) increases natural killer cell-mediated cell death and suppresses tumor growth. Blood, 96, 259–263. [PubMed] [Google Scholar]
- Yamaguchi S., Murakami,H. and Okayama,H. (1997) A WD repeat protein controls the cell cycle and differentiation by negatively regulating Cdc2/B-type cyclin complexes. Mol. Biol. Cell, 8, 2475–2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeong F.M., Lim,H.H., Padmashree,C.G. and Surana,U. (2000) Exit from mitosis in budding yeast: biphasic inactivation of the Cdc28–Clb2 mitotic kinase and the role of Cdc20. Mol. Cell, 5, 501–511. [DOI] [PubMed] [Google Scholar]
- Zou H., McGarry,T.J., Bernal,T. and Kirschner,M.W. (1999) Identification of a vertebrate sister-chromatid separation inhibitor involved in transformation and tumorigenesis. Science, 285, 418–422. [DOI] [PubMed] [Google Scholar]