A Cholesterol-Lowering Gene Maps to Chromosome 13q (original) (raw)
Summary
A cholesterol-lowering gene has been postulated from familial hypercholesterolemia (FH) families having heterozygous persons with normal LDL levels and homozygous individuals with LDL levels similar to those in persons with heterozygous FH. We studied such a family with FH that also had members without FH and with lower-than-normal LDL levels. We performed linkage analyses and identified a locus at 13q, defined by markers D13S156 and D13S158. FASTLINK and GENEHUNTER yielded LOD scores >5 and >4, respectively, whereas an affected-sib-pair analysis gave a peak multipoint LOD score of 4.8, corresponding to a P value of 1.26×10-6. A multipoint quantitative-trait-locus (QTL) linkage analysis with maximum-likelihood binomial QTL verified this locus as a QTL for LDL levels. To test the relevance of this QTL in an independent normal population, we studied MZ and DZ twin subjects. An MZ-DZ comparison confirmed genetic variance with regard to lipid concentrations. We then performed an identity-by-descent linkage analysis on the DZ twins, with markers at the 13q locus. We found strong evidence for linkage at this locus with LDL (P<.0002), HDL (P<.004), total cholesterol (P<.0002), and body-mass index (P<.0001). These data provide support for the existence of a new gene influencing lipid concentrations in humans.
Introduction
Elevated serum LDL concentrations are a major cause of coronary atherosclerosis (Grundy 1997). Support for LDL's fundamental role derives from the discovery of the LDL receptor. Familial hypercholesterolemia (FH) is the most common (frequency 1/500) autosomal-dominant disease affecting lipid metabolism (Brown and Goldstein 1986); heterozygous affected persons have LDL levels twice normal levels and develop premature coronary disease, whereas homozygous individuals have sixfold-elevated LDL levels and often die of cardiovascular disease at age <20 years. Cholesterol-synthesis inhibitors have exerted a gratifying effect on the course of atherosclerosis (Gould et al. 1998). However, even more ingenious would be endogenous mechanisms that have a similar cholesterol-lowering effect. Hobbs et al. (1989) presented strong evidence supporting the notion that a “lipid-lowering” gene exists. They described a family with FH that featured affected members with lower-than-expected LDL concentrations. We recently identified a large Arab family with FH whose FH-affected family members often have normal LDL concentrations and do not manifest atherosclerosis. Thus far, 96 family members have been phenotyped and genotyped. The LDL-receptor mutation in this family has been identified elsewhere (Davis et al. 1986). The defect resides in the receptor's cytoplasmic tail and causes defective receptor internalization with almost-absent LDL-receptor activity. In the present investigation, we tested the hypothesis that a “cholesterol-lowering” gene exists in this family, by means of linkage analysis. We then studied a second, normal population and verified the relevance of our findings. We present evidence that such a gene indeed exists on chromosome 13q.
Methods
The FH family is Moslem and Arab and resides in Israel. Ninety-six members were examined after written informed consent was obtained. Venous blood was used for DNA extraction and automated serum lipid measurements. LDL-cholesterol levels were calculated by use of the Friedewald formula (Friedewald et al. 1972). Repeated blood sampling and determinations of lipoprotein concentrations were done to ensure validity of the measurements prior to initiation of cholesterol-lowering therapy. At the time of the determinations, the subjects ingested a diet typical for the region, which is relatively high in fat and dairy products. We have subsequently advised persons with LDL-receptor mutations to adopt a low-fat diet and have begun pharmacological treatment as clinically indicated.
After we had determined the FH status (either heterozygous, homozygous, or not affected with FH) in all the family members, we examined the relationship between LDL cholesterol, age, gender, and body-mass index (BMI). We found no effects of gender and BMI in FH-heterozygous individuals; however, there was a modest age-related effect on LDL values in FH heterozygotes (see below). We also observed that the variation in LDL values increased with age. We then corrected the LDL-cholesterol values for age. The age-related effect on the variation was greater than the age-related effect on LDL values per se. We calculated the residuals of the LDL-cholesterol values by using standard linear regression, with age as an independent variable. This was done separately for FH-heterozygous affected and normal individuals in this family. We next corrected these residuals for age. This procedure allowed us to adjust the LDL values both for age and age-related variability. With these values we performed commingling analysis in the pedigree by using ILINK (Lathrop et al. 1984). These results allowed us to define FH-heterozygous persons with corrected LDL values ⩽150 mg/dl as “affected” by a putative cholesterol-lowering gene (for justification, see the Results section). Those persons having LDL values >150 mg/dl were defined as not affected by this gene. We did not have sufficient FH-homozygous subjects to correct their LDL values for age. Those homozygous persons with an LDL-cholesterol concentration ⩽500 mg/dl were classified as “affected” with the cholesterol-lowering gene, whereas those with LDL levels >500 mg/dl were classified as not affected. We picked these values of LDL concentrations because FH-heterozygous patients rarely have LDL concentrations >500 mg/dl and because <500 mg/dl in homozygous FH individuals are decidedly unusual. Moreover, such homozygous FH patients were asymptomatic in an earlier study (Sprecher et al. 1984). In persons carrying no FH mutation, there was a clear correlation between LDL values and age. LDL values were also higher in men than in women. Therefore, in this group the definition of “affected by the cholesterol-lowering gene” was more complex. We used the standardized linear-regression residuals of LDL versus age, done separately for men and women, to define persons as affected if LDL values were sharply (⩾1.5 SD) below the values predicted on the basis of their age and gender.
PCR-based mutation screening was performed in all 96 family members to categorize them as homozygous, heterozygous, or unaffected for FH. Genotyping was done with the ABI PRISM Genotyping System, including the Linkage Mapping Set, version 2 (LMS V2), PCR 9600 thermocyclers, the 877 Integrated Thermocycler, ABI DNA Sequencers, and GENESCAN and GENOTYPER software from PE Biosystems. The PCR primers contained in the LMS V2 amplify dinucleotide-repeat loci spaced at ∼10 cM. The markers are organized in a set of 28 panels, with 10–19 primer pairs per panel, whose products can be electrophoresed and detected in a single lane. The LMS V2 set includes forward primers that are labeled with 6-FAM, HEX, and NED (replacing TET) fluorescent dyes. These dyes can be distinguished by their different spectral properties. The LMS V2 reverse primers were redesigned to overcome the problem of nontemplated nucleotide adenylation, also known as “plus-A.” All markers were amplified under a common set of PCR conditions, by use of the True Allele PCR mix (PE Biosystems) containing Ampli_Taq_ Gold DNA Polymerase and a final MgCl2 concentration of 2.5 mM, with a 55°C annealing temperature. Electrophoresis and detection were done on an ABI 377 DNA Sequencer equipped with GENESCAN 2.1 software. Genotyping was performed with GENOTYPER 2.1 software. Genotypes were exported as a text file for subsequent linkage analysis.
We sought to determine whether any genes whose products are known to affect the interaction of LDL with its receptor cosegregate with the cholesterol-lowering phenotype. We looked for linkage between the cholesterol-lowering phenotype and the gene for the LDL receptor itself, as well as the genes for the receptor's two ligands, apo B-100 and apo E. Apo B gene mutations are known to cause low plasma-cholesterol levels (Collins et al. 1988). Apo B VNTR genotyping and apo E genotyping were done as described elsewhere (Hixon and Vernier 1990).
We selected 18 individuals for the first genotyping scan, to form a core pedigree. The selected group should contain a goodly number of closely related affected persons, which should minimize the danger of misspecifying the pedigree structure and should yield a maximum LOD score >3. The actual choice was made by visual inspection and by verification of the latter criterion by simulation analysis.
For total-genome scanning, we used FASTLINK (Cottingham et al. 1993), version 4.0, for the two-point analysis in the core pedigree. GENEHUNTER (Kruglyak et al. 1996) was used for multipoint analysis in split versions of both the core and extended pedigrees. GENEHUNTER is not able to accommodate the multiple loops in the extended pedigree unless the pedigree is split into subunits. We therefore also elected to perform an affected-sib-pair analysis by using a maximum-likelihood binomial (MLB) statistic as implemented in a modified version of GENEHUNTER (Abel and Müller-Myhsok 1998). The MLB statistic is a maximum-likelihood ratio–based sibship method especially geared to the analysis of sibships with multiple affected persons. The analysis is based on the estimation of a single parameter, α, which is related to p, the proportion of alleles shared by the sib pair, according to the relationship 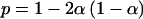 . The model used for parametric analysis was an autosomal-recessive–type model with an allele frequency of .01. We scrutinized all regions with a threshold p value of .1. Within the regions of interest, all 86 informative family members were genotyped. Because of computational restrictions, parametric multipoint linkage analysis within this highly consanguineous family was possible only by use of two flanking markers. When the results of the MLB analysis are considered, it should be noted that the LOD score obtained when this analysis is performed may be readily transformed into a χ2 variable, by multiplying it by
. The model used for parametric analysis was an autosomal-recessive–type model with an allele frequency of .01. We scrutinized all regions with a threshold p value of .1. Within the regions of interest, all 86 informative family members were genotyped. Because of computational restrictions, parametric multipoint linkage analysis within this highly consanguineous family was possible only by use of two flanking markers. When the results of the MLB analysis are considered, it should be noted that the LOD score obtained when this analysis is performed may be readily transformed into a χ2 variable, by multiplying it by 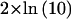 . This χ2 variable asymptotically follows a 50:50 distribution of 0 and 1 df.
. This χ2 variable asymptotically follows a 50:50 distribution of 0 and 1 df.
We also subjected our total genome–scan data to a test for linkage in terms of a quantitative-trait locus (QTL), using the novel QTL approach as implemented in MLBQTL (Alcais and Abel 1999). We chose this analysis because, contrary to the classically used Haseman-Elston approach, the MLBQTL does not necessarily assume a normal distribution of the trait or derived measures. For the analysis, we considered the phenotypic information from FH-pedigree members, who, after correction for gender, status at the FH locus, age, and the known covariate BMI, were ⩾1 SD above or below the mean. Thus, we applied a combined discordant and concordant sib-pair QTL analysis.
Finally, to test the hypothesis that the cholesterol-modifying gene is present in the general population, we performed studies in 122 pairs of MZ and 100 pairs of DZ twins and in available parents of the DZ twins. The subjects were all healthy, normotensive white individuals recruited from various parts of Germany. The protocol was approved by Humboldt University's committee on the protection of human subjects, and written informed consent was obtained from all participants. Persons with histories of familial lipid disorders were excluded. Details of our twin analysis have been published elsewhere (Knoblauch et al. 1997). Blood was obtained for total cholesterol, HDL, and triglycerides, and LDL was calculated by use of the Friedewald equation (Friedewald et al. 1972). Blood was also obtained for the determination of zygosity and other molecular-genetic studies. Microsatellite markers D13S1306, D13S170, D13S1241, D13S265, D13S159, and D13S158, spanning the “cholesterol-lowering gene” locus on chromosome 13, were analyzed.
For the twin linkage analysis, DZ pairs and their parents were included as described elsewhere (Busjahn et al. 1999). Analysis was done by use of a variance-component approach (Eaves et al. 1996). Phenotypic variance was decomposed into variance due to genetic background (A), variance due to the QTL effect (Q), and environmental variance (E): _Var_=_A_2+_Q_2+_E_2. For the three possible identical by descent (IBD) states (sharing zero, one, or two alleles), covariance of a sib pair was then defined by Cov _IBD_0=0.5_A_2, Cov _IBD_1=0.5_A_2+0.5_Q_2, and Cov _IBD_2=0.5_A_2+_Q_2. To improve estimates of total variance and genetic background, MZ twins were included in the analysis, with the covariance defined as Cov _MZ_=_A_2+_Q_2.
To test for a QTL effect, the difference in model fit for models with and without a QTL effect was calculated as a χ2 statistic. For each sib pair and each locus, the proportion of alleles IBD, based on parental genotypes and independent allele-frequency estimates, was calculated by use of a multipoint approach, as implemented in MAPMAKER/SIBS (Kruglyak and Lander 1995). The higher power of the variance-covariance–based analysis, compared with the squared trait differences–based approach by the Haseman-Elston method, has been shown in a recent simulation study (Fulker and Cherny 1996). Because we used a candidate-gene approach, we accepted P<.01 to test for significant linkage, in accordance with the criteria defined by Lander and Kruglyak (1995).
Heritability was estimated by structural equation modeling (Neale and Cardon 1992, p. 496), by use of the MX program developed by Neale (1997). The variability of any given phenotype within a population can be decomposed into genetic influences (A), environmental influences shared by the twins within a family (C), and effects of random environment (E): _Var_=_A_2+_C_2+_E_2. For MZ and DZ, the covariance of their phenotype is given by Cov _MZ_=_A_2+_C_2 and Cov _DZ_=0.5_A_2+_C_2. Genetic, as well as environmental, effects were estimated by the best-fitting model as selected by the χ2 value. Statistical analysis was conducted by use of the SPSS program. Adjustment of phenotypic values for sex and age was done by multiple linear regression, with the unstandardized residuals as the corrected phenotypes. In case of significant deviations from a normal distribution, the appropriate transformations were applied prior to analysis.
Results
In the FH-heterozygous family members, we found a modest, albeit significant (P<.04), effect of age on LDL values, allowing us to correct for the effects of age. Figure 1 shows the frequency distribution of corrected LDL-cholesterol values in FH-heterozygous subjects. Commingling analysis using ILINK gave significant evidence (P<.03) against a unimodal distribution of the corrected LDL values. The mean LDL value of the lowest genotypic distribution in the model was 138 mg/dl; the SD was 29 mg/dl. The other means were close together, at 198 and 203 mg/dl, respectively. This finding was interpreted as indicating a recessive trait, which was in accordance with a segregation analysis that used LOKI (Heath 1997). The classification threshold (150 mg/dl) was calculated as the 5th percentile in the group of persons heterozygous for the putative cholesterol-lowering gene. This approach gave a 95% certainty of correct classification for heterozygous individuals. We classified FH-heterozygous persons with values ⩽150 mg/dl as affected by a cholesterol-lowering trait. The extended pedigree is shown in figure 2. FH-heterozygous individuals are indicated, as well as the uncorrected LDL-cholesterol values. Persons affected by the cholesterol-lowering gene are also indicated.
Figure 1.

Frequency distribution of the subjects, in terms of their corrected LDL-cholesterol concentrations. Commingling analysis by use of ILINK allowed us to establish phenotypic criteria in terms of being “affected” by a putative cholesterol-lowering gene (LDL cholesterol <150 mg/dl).
Figure 2.

Extended pedigree showing all 96 individuals from whom lipid measurements were obtained. The subject's pedigree identification number is given, as well as the actual LDL-cholesterol concentration (mg/dl). FH-heterozygous individuals are half-blackened, whereas homozygous persons are fully blackened. Gray symbols represent those individuals selected for the core pedigree. Individuals with FH bearing the cholesterol-lowering gene are denoted by an asterisk (*). Their LDL-cholesterol levels are decidedly lower than would be expected for patients with FH.
We first looked for linkage between the cholesterol-lowering–gene locus and the gene for the LDL receptor itself, as well as the genes for the receptor's two ligands, apo B-100 and apo E. No significant evidence for linkage was found with either of these candidates. We next constructed a core pedigree consisting of 29 individuals composed of normal and FH-heterozygous individuals only. A subset of 18 individuals from this core pedigree (table 1) was genotyped for 300 microsatellite markers. Markers on eight chromosomal regions (two regions each on chromosomes 1 and 12 and one region each on chromosomes 6, 13, 16, and 18) were candidates (P<.1) for linkage to the low-cholesterol phenotype.
Table 1.
Demographic Information on Individuals in the Core Pedigree
| Apo Genotype | |||||
|---|---|---|---|---|---|
| Individual | Agea (Sex) | LDL (age and FH adjusted)b (mg/dl) | FH | ApoB | ApoEc |
| IV:1 | 62 years (F) | 341 (238) | 1 | 1,2 | 3,3 |
| IV:2 | 63 years (M) | 111* (126*) | 1 | 1,3 | 3,3 |
| IV:4 | 57 years (F) | 71* (104*) | 1 | 3,3 | 3,3 |
| IV:5 | 54 years (M) | 135* (137*) | 1 | 1,3 | 3,4 |
| IV:6 | 55 years (M) | 190 (167) | 1 | 1,3 | 3,4 |
| IV:7 | 55 years (F) | 223 (185) | 1 | 1,4 | 3,3 |
| IV:8 | 60 years (F) | 221 (181) | 1 | 1,2 | 3,3 |
| IV:9 | 65 years (M) | 159 (ND) | 0 | 3,3 | 3,3 |
| V:1 | 42 years (F) | 140* (140*) | 1 | 2,3 | 3,3 |
| V:3 | 35 years (F) | 165 (159) | 1 | 2,3 | ND |
| V:5 | … (F) | 207 (…) | 1 | 1,1 | 3,4 |
| V:6 | 24 years (F) | 136* (136*) | 1 | 1,1 | 3,3 |
| V:7 | 32 years (M) | 211 (197) | 1 | 1,1 | 3,4 |
| V:8 | 32 years (F) | 187 (178) | 1 | 2,3 | 3,3 |
| VI:1 | 3 years (F) | 142* (143*) | 1 | 2,2 | 3,3 |
| VI:2 | 7 years (M) | 111* (85*) | 1 | 1,3 | 3,3 |
| VI:3 | 5 years (M) | 126* (110*) | 1 | 1,3 | 3,3 |
| VI:4 | 3 years (M) | 115* (80*) | 1 | 1,2 | 3,4 |
Eighty-six individuals from the 96-member pedigree were then used to test for linkage with additional markers from the eight chromosomal regions. Ten persons were not included because they were not informative for the linkage analysis. In the core pedigree, a maximum pairwise LOD score of 3.69 was obtained with marker D13S129, at a recombination fraction (θ) of .01. Also in the core pedigree, marker D13S254 yielded a maximum pairwise LOD score of 3.32 at θ=.05 (table 2). The maximum two-point LOD score in the entire pedigree was 5.22 with marker D13S129 at θ=.05. Two additional markers exceeding the critical threshold of 3.0 were D13S265, with a LOD score of 4.06 at θ=.10, and D13S1241, with a LOD of 3.01 at θ=.10. We also analyzed the data set as an “affecteds-only” analysis (table 3). Multipoint linkage analysis in the extended pedigree was hampered by the complex consanguinity and by possible additional, unrecognized interconnections. Nevertheless, a three-point analysis using FASTLINK gave a LOD score of 5.67 at a location close to D13S129. GENEHUNTER yielded a maximum multipoint LOD score of 4.50 at θ=.0, at the neighboring marker D13S1300, which flanks D13S129. To circumvent potential limitations of FASTLINK and GENEHUNTER, we performed the affected-sib-pair analysis with the MLB statistic. Significant positive LOD scores were obtained exclusively for markers from the same region on chromosome 13, as shown in figure 3. The maximum multipoint LOD score (when the MLB statistic was used) was 4.82 (corresponding to a nominal P value of 1.26×10-6) at D13S1241. This region demonstrated complete sharing among affected sibs. The critical region on the basis of the “_z_max-2” method for this analysis, was defined by two polymorphic markers, D13S156 and D13S158, which are ∼37 cM apart. We next verified our findings by showing independently that the 13q locus is a QTL for LDL concentrations in this pedigree. QTL analysis using MLBQTL gave a maximum LOD of 4.23, with the peak located at D13S794 and with the “_z_max-2” support interval also extending from D13S156 to D13S158. No other significant peaks (LOD score >3) in the genome were identified by use of the QTL analysis. One region on chromosome 6 (around D6S257) showed a LOD score of 1.95. Haplotype data show the cosegregation of the 13q markers with the phenotype, as shown in figure 4. The gray-shaded zone represents the area of complete allele sharing within all affected sib pairs. Nevertheless, there was no common region of homozygosity in all affected pedigree members. Two persons unaffected by the cholesterol-lowering trait have the same haplotypes as their affected siblings. These two individuals may represent incomplete penetrance.
Table 2.
Two-Point LOD Scores in the Core Pedigree
| LOD Score at θ = | |||||||
|---|---|---|---|---|---|---|---|
| Marker | 0 | .01 | .05 | .1 | .2 | .3 | .4 |
| D13S175 | −∞ | −.48 | .32 | .56 | .55 | .36 | .17 |
| D13S217 | −∞ | −5.82 | −2.75 | −1.46 | −.42 | −.07 | .02 |
| D13S171 | −∞ | −4.15 | −1.72 | −.74 | −.05 | .1 | .09 |
| D13S263 | −∞ | −7.04 | −2.95 | −1.38 | −.19 | .15 | .16 |
| D13S153 | −∞ | −7.06 | −2.92 | −1.35 | −.18 | .14 | .15 |
| D13S156 | −∞ | −2.2 | −.37 | .35 | .74 | .58 | .27 |
| D13S1306 | .55 | .67 | .85 | .84 | .62 | .34 | .12 |
| D13S789 | −∞ | −2.84 | −1.18 | −.4 | .16 | .25 | .16 |
| D13S170 | −∞ | 1.94 | 2.29 | 2.14 | 1.53 | .83 | .26 |
| D13S271 | −∞ | .13 | 1.17 | 1.38 | 1.19 | .77 | .33 |
| D13S265 | −∞ | .67 | 1.56 | 1.71 | 1.44 | .92 | .39 |
| D13S794 | −∞ | −1 | .68 | 1.09 | 1.03 | .62 | .21 |
| D13S795 | −∞ | −1.93 | −.54 | 0 | .28 | .22 | .1 |
| D13S1300 | .54 | .58 | .88 | 1 | .81 | .46 | .16 |
| D13S129 | 3.67 | 3.69 | 3.56 | 3.2 | 2.25 | 1.21 | .39 |
| D13S125 | −.29 | −.09 | .4 | .58 | .52 | .29 | .09 |
| D13S254 | −∞ | 3.06 | 3.32 | 3.06 | 2.23 | 1.29 | .49 |
| D13S154 | −∞ | −1.2 | .49 | 1 | 1 | .56 | .14 |
| D13S1241 | −∞ | 1.67 | 2.25 | 2.22 | 1.67 | .92 | .29 |
| D13S786 | −∞ | −2.54 | −.41 | .33 | .67 | .54 | .26 |
| D13S159 | −∞ | −1.82 | .19 | .81 | .95 | .67 | .3 |
| D13S158 | −∞ | −2.16 | −.22 | .43 | .63 | .39 | .12 |
| D13S173 | −∞ | −1.97 | −.65 | −.18 | .08 | .09 | .05 |
| D13S285 | −∞ | −5.04 | −2.39 | −1.22 | −.31 | −.03 | .02 |
Table 3.
Two-Point LOD Scores in the Whole Pedigree (Affecteds Only)
| LOD Score at θ = | |||||||
|---|---|---|---|---|---|---|---|
| Marker | 0 | .01 | .05 | .1 | .2 | .3 | .4 |
| D13S175 | −.41 | −.35 | −.17 | −.06 | .02 | .03 | .01 |
| D13S217 | −1.68 | −1.54 | −1.12 | −.78 | −.39 | −.18 | −.07 |
| D13S171 | −1.64 | −1.51 | −1.11 | −.79 | −.41 | −.2 | −.07 |
| D13S263 | −1.63 | −1.42 | −.89 | −.51 | −.15 | −.01 | .03 |
| D13S153 | −2.84 | −2.52 | −1.67 | −1.03 | −.37 | −.08 | .01 |
| D13S156 | −.12 | .03 | .32 | .42 | .36 | .2 | .07 |
| D13S1306 | .9 | .89 | .82 | .71 | .46 | .23 | .07 |
| D13S789 | .69 | .67 | .6 | .5 | .33 | .18 | .07 |
| D13S170 | 1.63 | 1.63 | 1.51 | 1.29 | .78 | .36 | .09 |
| D13S271 | 1.59 | 1.54 | 1.34 | 1.11 | .68 | .34 | .11 |
| D13S265 | 2.37 | 2.3 | 2 | 1.65 | 1.04 | .55 | .19 |
| D13S794 | 1.34 | 1.3 | 1.15 | .96 | .58 | .27 | .07 |
| D13S795 | .58 | .57 | .53 | .47 | .31 | .15 | .04 |
| D13S1300 | 1.78 | 1.72 | 1.45 | 1.14 | .62 | .26 | .06 |
| D13S129 | 1.96 | 1.9 | 1.67 | 1.36 | .8 | .38 | .11 |
| D13S125 | .68 | .66 | .59 | .51 | .35 | .2 | .08 |
| D13S254 | 1.48 | 1.49 | 1.42 | 1.25 | .83 | .44 | .15 |
| D13S154 | .72 | .71 | .65 | .54 | .31 | .12 | .01 |
| D13S1241 | .02 | .14 | .38 | .45 | .33 | .15 | .02 |
| D13S786 | .71 | .77 | .86 | .82 | .6 | .33 | .12 |
| D13S159 | .44 | .56 | .78 | .81 | .64 | .37 | .14 |
| D13S158 | −1.15 | −1 | −.63 | −.39 | −.18 | −.09 | −.05 |
| D13S173 | .46 | .44 | .38 | .31 | .19 | .09 | .02 |
| D13S285 | −.5 | −.43 | −.25 | −.11 | −.01 | −.01 | −−.02 |
Figure 3.

Results of linkage analysis using MLB and MLBQTL in the FH pedigree, together with linkage results for LDL in the DZ twins (P values have been transformed into LOD scores). In the twins, the peak level of significance was .0002, right on marker D13S1241.
Figure 4.

Nuclear pedigrees selected for sib-pair analysis, shown in terms of the cholesterol-lowering trait. Unblackened and blackened symbols represent unaffected and affected individuals, respectively; gray symbols represent unknown status. Haplotypes from the candidate region on chromosome 13 are shown below the individuals. Identical haplotypes are marked in the same color. The smallest area of complete sharing among all sibs is indicated by the horizontal gray bar.
To test the relevance of our findings, we performed a twin analysis in German subjects (table 4). Females were twice as common as males. The subjects were young adults of normal height, weight, and BMI. Total cholesterol, HDL, LDL, and triglyceride values were all within normal limits. Table 4 also shows the results of the heritability analysis. A major genetic effect was demonstrated for all lipid parameters, although strong environmental effects were also shown. Table 5 shows the results of the linkage analysis. Significant linkage was shown for HDL, LDL, total-cholesterol concentrations, and BMI. Figure 3 gives the results of the variance-component multipoint linkage analysis for LDL. The peak level of significance was .0002, right on marker D13S1241. For this locus, the lower boundary-effect estimates, on a 95% confidence interval, for LDL cholesterol and BMI were ∼26% and ∼22%, respectively.
Table 4.
Clinical Data and Serum Lipid Values for the German Twin Sample[Note]
| Variable | MZ Twins | DZ Twins | GeneticEffect | P |
|---|---|---|---|---|
| n (pairs) | 122 | 100 | ||
| Age (years) | 34 ± 15 | 34 ± 13 | ||
| Sex (male/female) | 80/164 | 60/140 | ||
| Height (cm) | 169 ± 9 | 170 ± 9 | ||
| Weight (kg) | 67 ± 13 | 71 ± 14 | ||
| BMI (kg/m2) | 23 ± 4 | 24 ± 4 | .97 | .01 |
| Total cholesterol (mg/dl) | 183 ± 39 | 193 ± 42 | .64 | .01 |
| HDL cholesterol (mg/dl) | 51 ± 14 | 57 ± 17 | .59 | .01 |
| LDL cholesterol (mg/dl) | 115 ± 34 | 115 ± 32 | .66 | .01 |
| Triglycerides (mg/dl) | 87 ± 67 | 100 ± 64 | .72 | .01 |
Table 5.
Results of Linkage Analysis
| Model | χ2 Model Difference (P)a |
|---|---|
| Total cholesterol | 13.63 (.0002) |
| HDL cholesterol | 8.20 (.004) |
| LDL cholesterol | 13.60 (.0002) |
| Triglycerides | 6.39 (.011) |
| BMI | 14.26 (.0001) |
Discussion
The important finding in our study is that we were able to identify significant linkage between a locus on chromosome 13q and a putative cholesterol-lowering gene in a family with FH that had members with unexpectedly low LDL concentrations. To solidify our results, we used more than one linkage analysis, including parametric linkage analysis, multipoint linkage analysis, an affected-sib-pair method, and a QTL analysis. At first glance, there appears to be a discrepancy between the parametric analysis and the affected-sib-pair analysis. The peak LOD score from MLB and QTLMLB analysis is located near D13S786. This marker and flanking markers yielded a LOD score of −∞ in the two-point linkage analysis. This result may be due to uncertainties in the genetic models used in the analysis. More specifically, these problems may reside in the penetrance matrix. None of these recombination events was present in an affected individual. This fact is shown by the table of LOD scores from the affecteds-only analysis in the entire pedigree (table 3). We observed a series of 10 markers (D13S1306–D13S254) showing positive LOD scores, with θ=0 in every case.
We interpreted the lack of a common homozygosity region in this family as being due to a high frequency of this modifier gene in the general population. A rough estimate suggests a population allele frequency of .10, which is also the value used for the linkage analysis. Furthermore, the family that we studied is from an isolated but stable population. The expected region of homozygosity may be detectable only at the intragenic level, as has been described by others (Aksentijevich et al. 1999). Aksentijevich et al. (1999) performed haplotype studies of familial Mediterranean fever in the Ashkenazi Jewish population. There were two completely different haplotypes (for six markers) in that study, both carrying the sequence alteration V726A in the same family. Similarly, Aksentijevich et al. (1999) point out that the mutation E148Q may extend back to a common relatively ancient founder. Several distinct haplotypes carrying this mutation have been observed. However, at the intragenic level, the single nucleotide–polymorphism motifs converged. Those investigators concluded that an ancestor common to all the E148Q carriers may have lived some 1,500–2,000 years ago, a time span not unrealistic for this part of the world. For these reasons, we believe that our failure to identify a homozygosity region does not detract from the significance of our findings. Another possible explanation for our findings is related to the frequency of the mutation. A commonly occurring mutation could very well enter through different pathways even in a sibship, especially through affected parents (Veske et al. 1996). There are several examples of such sibships in our study.
To test whether this new locus is of any relevance to the general population, we performed a second, independent study in MZ and DZ twins. We verified earlier findings that LDL, HDL, total cholesterol, triglycerides, and BMI are all strongly influenced by genetic variance (Knoblauch et al. 1997). An IBD linkage analysis in DZ twins yielded strong evidence in support of the 13q locus as being a QTL for lipid concentrations, as well as for BMI. Moreover, the effect of this locus on LDL cholesterol was estimated to be ∼26%. We believe that these studies are unique, since we not only mapped a heretofore unknown gene but also proved, in principle, its effects and relevance in a completely unrelated, normal population.
Our results extend earlier studies indicating the existence of a cholesterol-lowering gene. Hobbs et al. (1989) described a similar unusual kindred with FH, in which one-third of affected relatives had normal LDL levels. Those investigators concluded that the low-cholesterol phenotype was most consistent with a single dominant gene suppressing the hypercholesterolemic effect of the LDL-receptor mutation in the family that they studied. They also excluded the LDL-receptor gene and the apo B-100 and apo E genes, by linkage analysis. Nora et al. (1985) also described a family with FH with normal cholesterol values in obligate-heterozygous patients with FH. Conceivably, by expansion of these pedigrees, a linkage analysis to verify and extend our results could be performed.
To test for linkage, we first performed an affecteds-only analysis on the core pedigree, which yielded a promising LOD score of 2.31 at D13S170. We next performed a parametric two-point linkage analysis in the core pedigree, assuming complete penetrance and including unaffected individuals according to our phenotypic definitions, which yielded two closely neighboring markers with LOD scores >3 at markers D13S254 and D13S129. We then examined the extended pedigree according to a conventional, parametric, two-point-linkage technique, which yielded a LOD score of 5.22 at marker D13S129. Three-point analysis using FASTLINK gave a LOD score of 5.67 at a location close to D13S129. A GENEHUNTER analysis gave a LOD score of 4.50 in the area of this same marker. An affected-sib-pair approach using the MLB statistic provided a LOD score of 4.82, corresponding to a nominal P value of 1.25×10-6. We believe that our approach, using several methods, provides robust evidence that the cholesterol-lowering gene resides on chromosome 13q.
Since LDL values are a continuous trait, our definition of being affected or unaffected by the cholesterol-lowering gene is potentially arbitrary until we identify the responsible gene mutation. We took care to consider the effects of age, gender, and BMI on LDL values. Our affected cutoff point is in accord with the accepted definition of FH among close relatives, for persons age >18 years, which is 165 mg/dl (Williams et al. 1996). Additional support in favor of our phenotypic definition comes from the analysis of corrected LDL concentrations of Druze and Christian-Lebanese FH-heterozygous individuals, who consume a diet very similar to that of the Moslem-Arab individuals whom we studied. The FH-heterozygous persons defined as affected for the cholesterol-lowering gene in our study had corrected LDL concentrations within the range seen in persons without FH who were from the Druze and Christian-Lebanese families with FH that have been described elsewhere (Leitersdorf et al. 1991). In contrast to the situation for individuals with FH in China (Sun et al. 1994) and Tunisia (Slimane et al. 1993), who, because of the ingestion of a low-fat diet, can have similarly low LDL values, we can rule out any dietary effect. The diet of the family with FH examined by us does not differ from the usually high-fat diet ingested by other people from this area. Their diet is no different from that ingested by the Christian-Arab population residing in Lebanon and described initially by Khachadurian (1964). Interestingly, Khachadurian observed that, with the exception of four FH-homozygous patients from a single family, 50 other FH-homozygous patients all had cholesterol values >600 mg/dl. The families that he studied had a null mutation similar to the mutation in the family that we studied, which results in an almost completely nonfunctioning LDL receptor. Finally, in the family that we studied, lipid values from persons not affected by the putative cholesterol-lowering gene are in the expected range, as in other families with FH in Israel.
The potential arbitrariness of our phenotype—and the clue that the allele in question might be common—led us first to demonstrate that the 13q locus is indeed a QTL in the pedigree in question and then to prove that the 13q locus has relevance to lipid concentrations in the general population. Relevance for the general population can usually not be shown for monogenic diseases. FH, for example, is caused by literally hundreds of separate mutations in the LDL-receptor gene (Hobbs et al. 1992). However, linkage of the LDL receptor–gene locus to LDL serum concentrations in normal subjects or in patients without FH and with coronary disease has not been convincingly shown (Knoblauch et al. 1997).
DZ twins are a particularly powerful sib-pair model, because of identical ages and a shared environment, at least during childhood. Sample size can be sharply reduced, without a loss of power, when DZ-twin siblings are examined. The utility of DZ twins in the quantitative sib-pair linkage–analysis approach to genes relevant to cardiovascular disease was recently demonstrated by Austin et al. (1998), who found linkage between the microsomal triglyceride-transfer–protein gene locus and plasma triglyceride concentrations. In addition, our earlier twin study (Knoblauch et al. 1997) showed evidence for linkage between the macrophage-scavenger–receptor gene locus and HDL concentrations. Our linkage results are almost two orders of magnitude below the criteria suggested by Lander and Kruglyak (1995), who, for a successful replication of an earlier result, called for a P value of ⩽.01.
We have yet to identify potentially interesting candidate genes in this chromosomal area. As part of an effort to dissect the genetic factors involved in cholesterol homeostasis, a genomewide analysis with a mouse model revealed several new candidate genes (Welch et al. 1996). However, none of these loci are syntenic to the homologous human chromosome (13q). We believe that low cholesterol levels in the affected individuals in this pedigree may be caused by a single gene. Although this hypothesis is speculative, we suggest that this gene may act on lipoprotein assembly in the liver. Functional studies will be required in order to test this issue. Elucidation of this gene may give new insight into mechanisms of lipoprotein regulation and protection from atherosclerosis and could lead to a new class of cholesterol-lowering agents.
Acknowledgments
Support was provided by a grant-in-aid from the Sarah and Moshe Mayer Foundation for Research (to E.L.) and support from PE Biosystems and LION Bioscience AG (to H.S.).
References
- Abel L, Müller-Myhsok B (1998) Robustness and power of the maximum likelihood binomial (MLB) and the maximum likelihood score (MLS) methods in multipoint linkage analysis of affected sibship data. Am J Hum Genet 63:638–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aksentijevich I, Torosyan Y, Samuels J, Centola M, Pras E, Chae JJ, Oddoux C, et al (1999) Mutation and haplotype studies of familial Mediterranean fever reveal new ancestral relationships and evidence for a high carrier frequency with reduced penetrance in the Ashkenazi Jewish population. Am J Hum Genet 64:949–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcais A, Abel L (1999) Maximum-Likelihood-Binomial method for genetic model-free linkage analysis of quantitative traits in sibships. Genet Epidemiol 17:102–117 [DOI] [PubMed]
- Austin MA, Talmud PJ, Luong L-A, Haddad L, Day INM, Newman B, Edwards LK, et al (1998) Candidate-gene studies of the atherogenic lipoprotein phenotype: a sib-pair linkage analysis of DZ women twins. Am J Hum Genet 62:406–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL (1986) A receptor-mediated pathway for cholesterol homoestasis. Science 232:34–47 [DOI] [PubMed]
- Busjahn A, Knoblauch H, Faulhaber H-D, Uhlmann R, Hoehe M, Schuster H, Luft FC (1999) The QT interval is linked to two long-QT syndrome loci in normal subjects. Circulation 99:3161–3164 [DOI] [PubMed]
- Collins DR, Knott TJ, Pease RJ, Powell LM, Wallis SC, Robertson S, Pullinger CR, et al (1988) Truncated variants of apolipoprotein B cause hypobetalipoproteinemia. Nucleic Acids Res 16:8361–8375 [DOI] [PMC free article] [PubMed]
- Cottingham RW Jr, Idury RM, Schäffer AA (1993) Faster sequential genetic linkage computations. Am J Hum Genet 53:252–263 [PMC free article] [PubMed]
- Davis CG, Lehrman MA, Russell DW, Anderson RG, Brown MS, Goldstein JL (1986) The J.D. mutation in familial hypercholesterolemia: amino acid substitution in cytoplasmic domain impedes internalization of LDL receptors. Cell 45:15–24 [DOI] [PubMed]
- Eaves LJ, Neale MC, Maes H (1996) Multivariate multipoint linkage analysis of quantitative trait loci. Behav Genet 26:519–525 [DOI] [PubMed]
- Friedewald WT, Levy RI, Fredrickson DS (1972) Friedewald formula: estimation of the concentration of low density lipoprotein cholesterol in plasma without the use of preparative ultracentrifugation. Clin Chem 18:499–502 [PubMed]
- Fulker DW, Cherny SS (1996) An improved multipoint sib-pair analysis of quantitative traits. Behav Genet 26:527–532 [DOI] [PubMed]
- Gould AL, Rossouw JE, Santanello NC, Heyse JF, Furberg CD (1998) Cholesterol reduction yields clinical benefit: impact of statin trials. Circulation 97:946–952 [DOI] [PubMed]
- Grundy SM (1997) Cholesterol and coronary heart disease. Arch Intern Med 157:1177–1184 [PubMed]
- Heath S (1997) Markov chain Monte Carlo segregation and linkage analysis for oligogenic models. Am J Hum Genet 61:748–760 [DOI] [PMC free article] [PubMed]
- Hixon JE, Vernier DT (1990) Apo E genotyping: restriction isotyping of human apolipoprotein E by gene amplification and cleavage with _Hha_I. J Lipid Res 31:545–548 [PubMed]
- Hobbs HH, Brown MS, Goldstein JL (1992) Molecular genetics of the LDL receptor gene in familial hypercholesterolemia. Hum Mutat 1:445–466 [DOI] [PubMed]
- Hobbs HH, Leitersdorf E, Leffert CC, Cryer DR, Brown MS, Goldstein JL (1989) Evidence for a dominant gene that suppresses hypercholesterolemia in a family with defective low density lipoprotein receptors. J Clin Invest 84:656–664 [DOI] [PMC free article] [PubMed]
- Khachadurian AK (1964) The inheritance of essential familial hypercholesterolemia. Am J Med 37:402–410 [DOI] [PubMed] [Google Scholar]
- Knoblauch H, Busjahn A, Münter S, Nagy Z, Faulhaber H-D, Schuster H, Luft FC (1997) Heritability analysis of lipids and three gene loci in twins link the macrophage scavenger receptor to high-density lipoprotein cholesterol concentrations. Arterioscler Thromb Vasc Biol 17:2054–2060 [DOI] [PubMed]
- Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES (1996) Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet 58:1347–1363 [PMC free article] [PubMed]
- Kruglyak L, Lander ES (1995) Complete multipoint sib-pair analysis of qualitative and quantitative traits. Am J Hum Genet 57:439–454 [PMC free article] [PubMed]
- Lander ES, Kruglyak L (1995) Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet 11:241–246 [DOI] [PubMed]
- Lathrop GM, Lalouel JM, Julier C, Ott J (1984) Strategies for multilocus linkage analysis in humans. Proc Natl Acad Sci USA 81:3443–3446 [DOI] [PMC free article] [PubMed]
- Leitersdorf E, Friedlander Y, Bard JM, Fruchart JC, Eisenberg S, Stein JC (1991) Diverse effects of ethnicity on plasma lipoprotein (a) levels in heterozygous patients with familial hypercholesterolemia. J Lipid Res 32:1513–1519 [PubMed]
- Neale MC (1997) Mx: statistical modeling, 4th ed. Virginia Commonwealth University, Richmond [Google Scholar]
- Neale MC, Cardon LR (1992) Methodology for genetic studies of twins and families. Kluwer Academic, Dordrecht, the Netherlands [Google Scholar]
- Nora JJ, Lortscher RM, Spangler RD, Bilheimer DW (1985) I. Familial hypercholesterolemia with "normal" cholesterol in obligate heterozygotes. Am J Med Genet 22:585–591 [DOI] [PubMed]
- Slimane MN, Pousse H, Maatoug F, Hammami M, Ben-Farhat MH (1993) Phenotypic expression of familial hypercholesterolemia in central and southern Tunisia. Atherosclerosis 104:153–158 [DOI] [PubMed]
- Sprecher DL, Schaefer EJ, Kent KM, Gregg RE, Zech LA, Hoeg MM, McManus B, et al (1984) Cardiovascular features of homozygous familial hypercholesterolemia: analysis of 16 patients. Am J Cardiol 54:20–30 [DOI] [PubMed]
- Sun XM, Patel DD, Webb JC, Knight BL, Fan LM, Cai HJ, Soutar AK (1994) Familial hypercholesterolemia in China: identification of mutations in the LDL-receptor gene that result in a receptor-negative phenotype. Arterioscler Thromb 14:85–94 [DOI] [PubMed]
- Veske A, Oehlmann R, Younus F, Mohyuddin A, Müller-Myhsok B, Mehdi SQ, Gal A (1996) Autosomal recessive non-syndromic deafness locus maps to chromosome 21q22 in a large consanguineous kindred from Pakistan. Hum Mol Genet 5:165–168 [DOI] [PubMed]
- Welch CL, Xia YR, Shechter I, Farese R, Mehrabian M, Mehdizadeh S, Warden CH, et al (1996) Genetic regulation of cholesterol homeostasis: chromosomal organization of candidate genes. J Lipid Res 37:1406–1421 [PubMed]
- Williams RR, Hamilton-Craig I, Kostner GM, Hegele RAA, Hayden MR, Pimstone SN, Faergeman O, et al (1996) MEDPED: an integrated genetic strategy for preventing early deaths: genetic approaches to noncommunicable diseases. Springer-Verlag, Berlin and Heidelberg [Google Scholar]