Linkage of Inflammatory Bowel Disease to Human Chromosome 6p (original) (raw)
Summary
Inflammatory bowel disease (IBD) is characterized by a chronic relapsing intestinal inflammation. IBD is subdivided into Crohn disease and ulcerative colitis phenotypes. Given the immunologic dysregulation in IBD, the human-leukocyte-antigen region on chromosome 6p is of significant interest. Previous association and linkage analysis has provided conflicting evidence as to the existence of an IBD-susceptibility locus in this region. Here we report on a two-stage linkage and association analysis of both a basic population of 353 affected sibling pairs (ASPs) and an extension of this population to 428 white ASPs of northern European extraction. Twenty-eight microsatellite markers on chromosome 6 were genotyped. A peak multipoint LOD score of 4.2 was observed, at D6S461, for the IBD phenotype. A transmission/disequilibrium test (TDT) result of_P_=.006 was detected for D6S426 in the basic population and was confirmed in the extended cohort (_P_=.004; 97 vs. 56 transmissions). The subphenotypes of Crohn disease, ulcerative colitis, and mixed IBD contributed equally to this linkage, suggesting a general role for the chromosome 6 locus in IBD. Analysis of five single-nucleotide polymorphisms in the TNFA and LTA genes did not reveal evidence for association of these important candidate genes with IBD. In summary, we provide firm linkage evidence for an IBD-susceptibility locus on chromosome 6p and demonstrate that TNFA and LTA are unlikely to be susceptibility loci for IBD.
Introduction
Inflammatory bowel disease (IBD [MIM601458, MIM266600, and MIM191390]) is clinically characterized by abdominal pain, chronic diarrhea, rectal bleeding, weight loss, intestinal stenoses, fistulas, toxic megacolon, and such associated extraintestinal manifestations as arthritis and uveitis (Podolsky 1991). It affects ∼1/1,000 individuals in Western countries, with the median age at onset being during early adulthood (Probert et al.1996; Shivananda et al.1996). The etiology of this disorder is unknown. The pathophysiology is characterized by a chronic relapsing inflammation of the gastrointestinal tract. On the basis of clinical and histopathologic features, IBD is categorized into two main subtypes: Crohn disease (CD [MIM266600]) and ulcerative colitis (UC [MIM191390]) (Hamilton 1987; Podolsky1991).
Chromosome 6 has been implicated in the etiology of IBD, by association and genetic-linkage studies dating as far back as 1972. Previously reported associations include class I (Gleeson et al.1972; van den Berg-Loonen et al.1977; Purrmann et al.1985; Biemond et al.1986), class II (Fujita et al.1984; Toyoda et al.1993; Nakajima et al.1995), and tumor necrosis factor–α (TNF-α) alleles (Bouma et al.1996; Plevy et al.1996). However, the association data for human leukocyte antigen (HLA) and TNF-α are not conclusive (Russell et al. 1975; Delpre et al. 1980; Smolen et al.1982; Satsangi et al.1996b). Recent association studies have suggested a role for HLA in susceptibility to UC (Roussomoustakaki et al. 1997; Perri et al. 1998; Uyar et al.1998; Bouma et al.1999) and CD (Forcione et al.1996). Neither the particular HLA antigen that might confer susceptibility to IBD nor the particular subphenotype that might be affected is clear.
TNF-α is a key inflammatory mediator in the pathophysiology of IBD. The ex vivo capacity of lamina propria mononuclear cells to produce TNF-α has been used to predict relapse in CD (Schreiber et al. 1999). Furthermore, therapeutic interventions targeting TNF-α with a monoclonal antibody have proved to be effective in clinical trials (Targan et al.1997). Given these data, the question of whether TNF-α gene variations are an etiologic factor in IBD is intriguing. Association of an inferred haplotype (TNFa2b1c2d4e1) of microsatellites flanking the TNF locus with CD has been described (Plevy et al. 1996) in 148 patients with IBD and in 60 control subjects, yielding P values of .01 (odds ratio [OR] 4.4) and .001 (OR 7.4) in comparison with those for control subjects and patients with UC, respectively. In a direct investigation of polymorphisms of the TNFA-gene promoter and two RFLPs of the lymphotoxin-α (LTA) gene in 154 patients with IBD and in 54 healthy control subjects, the uncommon allele 2 of the −308 TNFA promoter polymorphism was found to be decreased in UC (_P_=.044) (Bouma et al.1996). Further investigation involving the use of both the microsatellites and the known functional single-nucleotide polymorphisms of this region is therefore warranted.
Despite the several positive association findings in this genomic region and despite the strong functional evidence supporting TNF-α, questions remain regarding (1) the existence and relevance of an IBD risk factor on chromosome 6 and (2) the putative genetic role of TNF in the etiology of IBD. This uncertainty stems, in part, from the lack of linkage evidence fulfilling the significance criterion of a LOD score >3.6, as defined by Lander and Krugylak (1995). Several groups have reported suggestive evidence for linkage of IBD to chromosome 6p. In our previous genomewide linkage analysis, a LOD score of 2.1 was observed (Hampe et al.1999). Yang et al. (1999) have also reported evidence supporting a role for the chromosome 6p region, with the use of multiple regression analysis in 49 families with CD. Other previous genomewide linkage analyses, by Hugot et al. (1994) and by Satsangi et al. (1996a), did not indicate the actual significance levels attained for chromosome 6 markers, since no significant LOD score >3.6 was observed. In the recent genomewide analysis by Cho et al. (1998), a multipoint LOD score of 0.7 was observed for chromosome 6p. Satsangi et al. (1996b) have also reported a_P_ value of .017 for linkage of UC to the DRB1 locus.
Given our prior suggestive linkage results for the HLA region (Hampe et al. 1999), which were obtained by genomewide analysis, we hypothesized that this region may harbor a gene predisposing to IBD. To test this hypothesis, we increased the marker density in this region by adding an additional 11 microsatellite markers and by expanding the study population to a total of 428 ASPs. In view of the previous functional and genetic evidence implicating TNF-α, we tested five known functional single-nucleotide polymorphisms in the TNFA and LTA genes in the expanded population.
Families and Methods
Family Ascertainment and Phenotypes
A family cohort recruited by an international group of IBD investigators at the Charite University Hospital, Berlin; the I Department of Medicine at the Christian-Albrechts-University Kiel, Kiel, Germany; King's College School of Medicine, Guy's Hospital, and St. Mark's Hospital, London; Academic Medical Center, Amsterdam; and other central European centers was investigated in this study. Forty-six percent of the families were of German origin, 6% were from the Netherlands, and 48% were recruited in the United Kingdom. Informed, written consent was obtained from all study participants. Recruitment protocols were approved by institutional review committees at each participating center. Diagnosis of IBD and classification as CD or UC were determined by the use of standard diagnostic criteria, as described elsewhere (Truelove and Pena1976; Lennard-Jones1989). The basic cohort has been used in previous studies within the collaborative group (Curran et al.1998; Hampe et al.1998, 1999; Olavesen et al., in press). Ascertainment criteria were determined prior to the initiation of patient collection. It was required that clinical, radiological, and endoscopic (type and distribution of lesions) examinations unequivocally confirm the diagnosis of either UC or CD. Histology findings had to either confirm or be compatible with this diagnosis. In instances of uncertainty, the diagnosis of indeterminate colitis was assigned, and the patient was excluded from the study. For families originating in the United Kingdom and the Netherlands, medical records for all patients were reviewed by one or more of the principal investigators. For families of German origin, patients were directly examined by one or more of the principal investigators, when possible. Alternatively, two written records containing a detailed disease history and the results of all diagnostic procedures were obtained for each patient and were reviewed by the principal investigators. A venous blood sample was obtained from the affected siblings and their parents, if possible. An overview of the family cohort is given intable 1.
Table 1.
Number of Sibships and ASPs in Both the Family Cohort Used in the Basic Study and the Extended Cohort [Note]
| No. of Sibships/(ASPsa) in Each Cohort Studied, According to Disease Type | ||||||||
|---|---|---|---|---|---|---|---|---|
| Basic Cohort | Extended Cohort | |||||||
| Sibship Size | CD | UC | Mixedb | Total | CD | UC | Mixedb | Total |
| 2 | 114 (114) | 78 (78) | 38 (38) | 230 (230) | 150 (150) | 102 (102) | 47 (47) | 299 (299) |
| 3 | 14 (42) | 12 (36) | 9 (27) | 35 (105) | 14 (42) | 13 (39) | 10 (30) | 37 (111) |
| 4 | 1 (6) | 2 (12) | 3 (18) | 1 (6) | 2 (12) | 3 (18) | ||
| Total | 129 (162) | 90 (114) | 49 (77) | 268 (353) | 165 (198) | 115 (141) | 59 (89) | 339 (428) |
Genotyping
Genomic DNA was prepared from whole blood by use of the Puregene system (Gentra Systems). Twenty-eight highly polymorphic microsatellite markers on chromosome 6 were genotyped by fluorescent methods, as described by Hall and Nanthakumar (1997). Primer sequences were derived from The Genome Database or from the literature (Partanen and Koskimes1988). In brief, individual DNA samples were arrayed in 96-well microtiter plates and were amplified by PCR with the appropriate primers. Product length of the microsatellite markers was determined, on denaturing polyacrylamide gels, by electrophoresis done with the use of ABI 377 automated DNA sequencers. Allele analyses and individual allele calling were performed as described elsewhere (Hall and Nanthakumar1997; Idury and Cardon1997).
Single-nucleotide polymorphisms in the TNF genes were typed with the use of TAQman technology from PE Biosystems. This method uses the 5′-exonuclease activity of the Taq polymerase in a combination of PCR and competitive hybridization (Holland et al.1991; Livak et al.1995). Probes and primers were designed with the use of Primer Express software (PE Biosystems). Oligonucleotides and reaction conditions are given intable 2. A map indicating marker positions in the TNF gene region is given infigure 1. PCR reactions were performed in an ABI 9700, and fluorescence results were determined with the use of an ABI 7700 sequence-detector single-point measurement. Genotype errors resulting from non-Mendelian segregation in pedigrees were detected and corrected as described by Hall and Nanthakumar (1997).
Table 2.
Overview of Oligonucleotides Used for TAQman Assays Detecting Polymorphisms in TNF Genes[Note]
| Polymorphismand Type ofProbe or Primer | Oligonucleotidesa | Concentration(nM) | PCRConditions |
|---|---|---|---|
| TNF-β: _Nco_I: Forward Reverse Probe 1 Probe 2 | GGGCCTTGGTGGGTTTGGTTAAGGGGACAAGATGCAGTCAGAGAAFam-TCTGTTTCTGCCATGATTCCTCTCTGTTCC-TamraTet-CTGTTTCTGCCATGGTTCCTCTCTGTTCC-Tamra | 900900200200 | 50 cycles at 65°C annealing and 1-min extension |
| _Asn_26: Forward Reverse Probe 1 Probe 2 | CCCGTCAGCACCCCAAGATGTGGGAGGTCAGGTGGATGTTTACC Fam-TTGCCCACAGCACCCTCAAACCTG-TamraTet-TGCCCACAGCAACCTCAAACCTGC-Tamra | 900900100100 | 40 cycles at 65°C annealing and 1-min extension |
| TNF-α: −308: Forward Reverse Probe 1 Probe 2 | CCTGCATCCTGTCTGGAAGTTAGAAGTGGGCCACTGACTGATTTGTGTGTFam-AACCCCGTCCCCATGCCCCTC-TamraTet-AACCCCGTCCTCATGCCCCTCAA-Tamra | 1,0001,000100100 | 45 cycles at 70°C annealing and 1-min extension |
| −244: Forward Reverse Probe 1 Probe 2 | CCTCCAGGGTCCTACACACAACAAGCATCAAGGATACCCCTCFam-CCCAGAAGACCCCCCTCAGAATC-TamraTet-CCAGAAGACCCCCCTCGGAATC-Tamra | 900900100100 | 40 cycles at 60°C annealing and 30-s extension |
| −238: Forward Reverse Probe 1 Probe 2 | CAGTGGCCCAGAAGACCCAGCATCAAGGATACCCCTCACFam-AATCGGAGCAGGGAGGATGGG-TamraTet-AATCAGAGCAGGGAGGATGGGGA-Tamra | 900300100100 | 40 cycles at 60°C annealing and 30-s extension |
Figure 1.
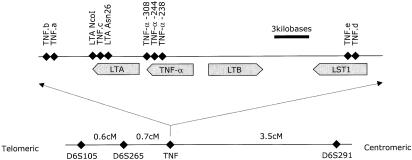
Overview of TNF region: relative positions of the TNF microsatellites a–e and of the single-nucleotide polymorphisms of the TNFA and LTA genes and their promoters (top). The physical distances are approximate. Distances in the top panel are derived fromGenBank record Y14768. The linkage map of the wider area (bottom) shows genetic map distances, as derived from the analyzed data set.
To facilitate inclusion of markers from different genetic maps (e.g.,Centre d'Etude du Polymorphisme Humain andCooperative Human Linkage Center), marker order and distances were calculated with the use of genotype data from the 339 pedigrees. The genetic map was constructed by use of the automated mapping program MultiMap, version 2.0 (Matise et al. 1994).
Statistical Analysis
Genetic analyses were conducted with the use of the two aforementioned standard diagnostic categories: CD and UC. A third category, ALL, contains CD/CD, UC/UC, and CD/UC (mixed) ASPs. The ALL category therefore represents IBD as a single phenotype for analysis. Allele frequencies for each marker were calculated from the cohort genotype data for all individuals. Each marker was analyzed in ASPs, by means of both two-point and multipoint nonparametric allele-sharing tests, with the use ofMAPMAKER/SIBS with the “weighted pairs” option (Kruglyak and Lander1995). For multipoint analysis, LOD scores were computed at 1-cM intervals along the chromosome. The mean information content across chromosome 6p was 85%.
Association statistics were calculated by the TDTLIKE program from the ANALYZE software package (Terwilliger 1995). Tested alleles are restricted to ⩾10 observed transmissions. The program algorithm provides P values corrected for the testing of multiple alleles.
Results
Previous analyses involving the use of 17 microsatellite markers in the basic cohort of 353 ASPs revealed suggestive evidence of linkage in the HLA region on chromosome 6p. A peak LOD score of 2.1 was observed close to marker D6S276 (Hampe et al. 1999). To further investigate this interval, 11 additional microsatellites in the peak region between D6S309 and D6S257 were analyzed. The multipoint maximum-likelihood-score (MLS) curves for UC, CD, and the combined, ALL phenotype are shown in figure 2. For the IBD phenotype, the maximum MLS of 2.9 is located <1 cM from D6S461. The complete two-point results for all analyzed markers are shown in table 3. In the total IBD population, a maximum two-point LOD score of 2.12 was observed for the TNFa microsatellite marker, and, in the UC subpopulation, a LOD score of 2.27 was seen at D6S461. The subphenotypes of CD and UC and the mixed populations (which show UC and CD in the same family) contribute approximately equally to the linkage results.
Figure 2.
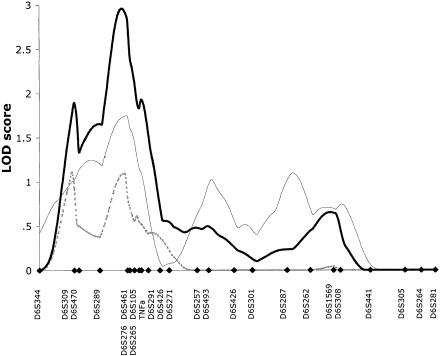
Multipoint MLS curves for chromosome 6 from the basic family data set. Multipoint analyses were performed with use of theMAPMAKER/SIBS program. Results for CD (dashed line), UC (thin unbroken line), and ALL (thick unbroken line) are shown. Genetic distances between markers were estimated from the data set. Marker positions are denoted by blackened diamonds.
Table 3.
Two-Point LOD Scores and TDT Results for Loci in Chromosome 6 Linkage Region [Note]
| Results for Loci in the Chromosome 6 Regionof Two Study Populations | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Basic Population | Extended Population | ||||||||
| Two-PointLOD Scorea | Two-PointLOD Scorea | ||||||||
| Locus | Theta | CD | UC | ALL | TDTb (P Value) | CD | UC | ALL | TDTb (P Value) |
| D6S309 | .014 | .49 | .28 | .72 | >.05 | ||||
| D6S470 | .101 | .18 | .38 | .56 | >.05 | .14 | .09 | .36 | >.05 |
| D6S289 | .114 | .13 | .46 | .78 | >.05 | .10 | .17 | .60 | >.05 |
| D6S461 | .005 | .01 | 2.27 | 1.91 | .018 (ALL) | .20 | .88 | 2.37 | >.05 |
| D6S276 | .030 | .22 | 1.29 | 1.68 | >.05 | ||||
| D6S105 | .006 | .76 | 1.75 | 1.84 | >.05 | 1.52 | 1.11 | 2.80 | >.05 |
| D6S265 | .007 | .33 | .16 | .50 | >.05 | 1.34 | .00 | 1.17 | >.05 |
| TNFb | <.001 | >.05 | |||||||
| TNFa | <.001 | .29 | 1.70 | 2.12 | >.05 | 1.19 | 1.22 | 3.36 | >.05 |
| LTA_Nco_I | <.001 | .00 | .00 | .65 | >.05 | .00 | .00 | .94 | >.05 |
| TNFc | <.001 | >.05 | >.05 | ||||||
| LTA_Asn_26 | <.001 | .00 | .03 | .70 | >.05 | .00 | .01 | .96 | >.05 |
| TNFA–308 | <.001 | .00 | .00 | .50 | >.05 | .36 | .19 | .75 | >.05 |
| TNFA–238 | <.001 | .20 | .01 | .48 | >.05 | .01 | .02 | .59 | >.05 |
| TNFd | <.001 | >.05 | >.05 | ||||||
| TNFe | .035 | >.05 | >.05 | ||||||
| D6S291 | .054 | .08 | .47 | .62 | >.05 | .36 | .48 | 1.07 | >.05 |
| D6S426 | .046 | .20 | .00 | .10 | .006 (ALL) | .40 | .02 | .46 | .004 (ALL) |
| D6S271 | .119 | .07 | .11 | .58 | >.05 | .19 | .35 | 1.46 | >.05 |
| D6S257 | .046 | .00 | .56 | .03 | >.05 | .00 | .72 | .21 | >.05 |
When 11 markers were added in this linkage region, the maximum information content increased from 80% to 95%. Genotyping of further markers was therefore not expected to improve the linkage information or the magnitude of linkage results. To increase power to detect linkage, the study cohort was expanded to 428 ASPs. Five microsatellite markers covering 10 cM of the peak linkage, as well as five flanking markers, were genotyped in the additional families. Nonparametric linkage analysis revealed strengthened linkage evidence and a peak multipoint LOD score of 4.2 at marker D6S461 in the total IBD cohort. The MLS curves for CD, UC, and the ALL cohorts are presented in figure 3. The maximum two-point LOD score of 3.36 was observed for the microsatellite TNFa in the ALL cohort.
Figure 3.
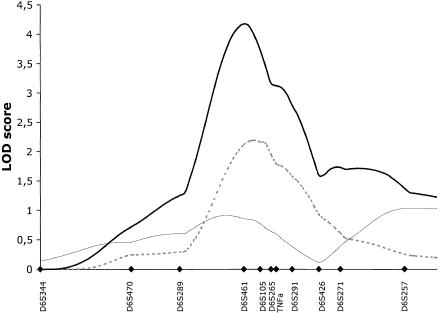
Multipoint MLS curves for markers typed in the peak region of chromosome 6p. Markers were typed in the extended family set of 428 ASPs. Multipoint analyses were performed with use of theMAPMAKER/SIBS program. Results for CD (dashed line), UC (thin unbroken line), and ALL (thick unbroken line) are shown. Genetic distances between markers were estimated from the data set. Marker positions are denoted by blackened diamonds.
On the basis of the strong functional evidence linking the TNFA and LTA molecules to IBD pathogenesis and on the basis of their location near the linkage peak, these genes were chosen for further investigation. Five known single-nucleotide polymorphisms in the TNFA and LTA genes (table 2) and the five TNF microsatellites were therefore genotyped in the expanded population of 428 ASPs. The combination of the microsatellite alleles TNFa2b1c2d4e1 had been observed at an increased frequency in CD, in a previous case-control study (Plevy et al.1996). In our extended cohort, this allele combination was present in 8.1% of patients with CD, in 10.1% of patients with UC, and in 12.3% of unaffected parents (_P_>.10). Allele 2 of the “−244” TNFA promoter polymorphism (Weissensteiner and Lanchbury 1997) was observed in only three individuals. This finding suggests that this polymorphism is not critically involved in the etiology of IBD, and, therefore, it was excluded from the linkage and association analysis. The association analysis of the single-nucleotide polymorphisms revealed no significant findings. All P values were >.25. The microsatellite markers were also analyzed, by use of the TDT test, for evidence of association. A single suggestive TDT result of _P_=.006 was observed for marker D6S426 (χ2=10.3; 81 vs. 45 transmissions) at the proximal flank of the linkage curve in the basic cohort. This result was strengthened by the addition of families in the extended population, yielding a P value of .004 (χ2=11.0; 97 vs. 56 transmissions). The complete TDT results for all markers are given intable 3.
Discussion
In this study, we have provided linkage evidence for the existence of an IBD-susceptibility locus on chromosome 6p. The peak multipoint LOD score of 4.2 surpasses the LOD-score criterion of 3.6 suggested for establishment of linkage in a complex genetic disorder (Lander and Kruglyak 1995). The suggestive linkage findings of other investigators provide further support for this finding. From these data, we conclude that there is a major disease-risk determinant for IBD in the region surrounding the HLA complex on chromosome 6p. The equal contribution of the subphenotypes of families with CD, UC, and mixed CD/UC to the linkage result in the ALL category (figs.2 and3) suggests that the underlying gene may be important for both CD and UC. The overlap with the linkages for insulin-dependent diabetes mellitus, psoriasis, and other autoimmune disorders in the HLA region may point to common factors that may play a general role in the predisposition to dysregulated immune responses (Nair et al. 1997; Becker et al.1998; Concannon et al.1998). Alternatively, an IBD-specific gene may eventually be identified in this region.
Substantial functional evidence and successful clinical trials have implicated TNF-α in the pathogenesis of IBD (Targan et al.1997; Schreiber et al.1999). In experimental systems, variations in the TNFA promoter have been associated with a differential capacity to produce TNF-α in response to inflammatory stimuli (Kroeger et al. 1997; Wilson et al.1997). The −308 TNF-α promoter polymorphism has been clearly linked to susceptibility to malaria and meningococcal infection (McGuire et al.1994; Nadel et al.1996). The capacity to produce TNF-α and IL-1β in lamina propria mononuclear cells has been used to predict relapse in CD (Schreiber et al.1999). On the basis of both the significant functional evidence implicating TNF-α in the pathogenesis of IBD and the location at the center of our linkage peak, this gene was selected for further investigation. Previous studies have defined common variants in the TNF genes. We investigated five known TNFA and LTA polymorphisms that might have a functional relevance for association to IBD. No association of the TNF single-nucleotide polymorphisms or the microsatellites was detected in the expanded cohort of 428 ASPs. We conclude from these data that variations in TNF-α do not, therefore, predispose to IBD but, rather, likely act as an important secondary mediator in the inflammation process. The use of TNF polymorphisms in the stratification or reclassification of IBD does not, therefore, seem to be warranted
There is a very high density of immunoregulatory genes in the region implicated by the linkage. These genes—in addition to TNF-α—include, among others, the HLA antigens, members of the NFκB inflammatory signal-transduction pathway, heat-shock proteins, antigen-processing members of the ubiquitin complex, and the complement family. Each of the members of these gene families will have to be examined for genetic evidence implicating it in the disease process. It is anticipated that this process will be extremely difficult because of the exceptionally high density of immunoregulatory genes in this genomic area.
Interestingly, a positive TDT association of_P_=.004 is observed for the anonymous microsatellite marker D6S426, which is located at the proximal boundary of the linkage region. The corresponding two-point LOD score at this location is .46 in the total population with IBD. Given the multiple association tests performed in this study (table 3), this result may represent a false-positive finding and, thus, should not be overinterpreted. Alternatively, this result may point to a disease-gene location on the proximal flank of the linkage peak. This would correspond to a more proximal linkage-peak location, which has been observed in other studies (Silverberg et al.1999). A difference in location between the linkage peak and the actual gene location would not be unexpected in a complex disorder (Kruglyak and Lander1996).
In summary, we have presented firm linkage evidence for the existence of an IBD-susceptibility gene in the HLA region. TNF-α, although clearly an important inflammatory mediator in IBD, does not appear to play an important role in the genetic predisposition to the disease. The single point association at D6S426 warrants further investigation.
Acknowledgments
The authors thank the physicians, the patients with IBD, and their families for participating in this study. The cooperation of the German Crohn's and Colitis Association (DCCV e. V.) and, from Germany, Profs. Raedler (Hamburg) and Kruis (Köln), Drs. Theuer (Heilbronn) and Meckler (Gedern), Prof. Lochs, Dr. Wedel, and T. Herrmann (Berlin), Dr. Herchenbach (Recklinghausen), Prof. Scheurlen (Würzburg), Dr. Demharter (Augsburg), Dr. Simon (Munich), Dr. Purrmann (Moers), Dr. Jessen (Kiel), Dr. Zehnter (Dortmund), Drs. Lübke and Weismüller (Koblenz), Dr. Eiche (Denkendorf), Dr. Schönfelder (Aachen), and Prof. Fleig (Halle); from Austria, Drs. Wewalka (Linz) and Knofloch (Wels); and from the United Kingdom, Drs. Hodgson, Sanderson, Pounder, Forbes, and Forgacs (London), Dr. Bird (Maidstone) Dr. Hines (Haywards-Heath), Drs. Cairns and Ireland (Brighton), Dr. Barrison (St. Albans), and Dr. Smith-Lang (Sidcup), is gratefully acknowledged. The authors appreciate the many stimulating discussions and gracious support offered by Drs. Emtage and Daniels, and they further acknowledge the expert technical efforts of Jonalyn Matusalem, Larenia Pedriguez, and Hye Jin Yang and the expert Macintosh support provided by Carl Manaster. The authors wish to express their appreciation for the dedicated help of Dan Prestridge, who will always stay in our memories. This work was supported by Axys Pharmaceuticals Inc., the National Association for Colitis and Crohn's Disease (United Kingdom), The Crohn's in Childhood Research Association (United Kingdom), the Sir Halley Stewart Trust, Deutsche Forschungsgemeinschaft grant Schr 512/5-1, Training and Mobility of Research Network grant ERB-4061-PL-97-0389 from the European Union, by Mucosaforschungsgesellschaft, a MedNet “Chronisch-entzündliche Darmerkrankungen” from the German Federal Department for Research and Education (BmBF), and an educational grant from Arzneimittelwerk Dresden (to J.H.).
Electronic-Database Information
Accession numbers and URLs for data in this article are as follows:
- Centre d'Etude du Polymorphisme Humain,http://www.cephb.fr/
- Cooperative Human Linkage Center, The,http://lpg.nci.nih.gov/CHLC/
- GenBank,http://www.ncbi.nlm.nih.gov/Genbank/GenbankOverview.html (for genetic map distances in fig. 1)
- Genome Database, The,http://www.gdb.org/ (for primer sequences)
- MAPMAKER/SIBS site:ftp://ftp-genome.wi.mit.edu/distribution/software/sibs/
- MultiMap Home Page,http://linkage.rockefeller.edu/multimap/
- Online Mendelian Inheritance in Man (OMIM),http://www.ncbi.nlm.nih.gov/Omim/ (for IBD [MIM 601458], CD [MIM 266600], and UC [MIM 191390])
References
- Becker KG, Simon RM, Bailey-Wilson JE, Freidlin B, Biddison WE, McFarland HF, Trent JM (1998) Clustering of non-major histocompatibility complex susceptibility candidate loci in human autoimmune diseases. Proc Natl Acad Sci USA 95:9979–9984 [DOI] [PMC free article] [PubMed]
- Biemond I, Burnham WR, D'Amaro J, Langman MJ (1986) HLA-A and -B antigens in inflammatory bowel disease. Gut 27:934–941 [DOI] [PMC free article] [PubMed]
- Bouma G, Crusius JB, Garcia-Gonzalez MA, Meijer BU, Hellemans HP, Hakvoort RJ, Schreuder GM, et al (1999) Genetic markers in clinically well defined patients with ulcerative colitis (UC). Clin Exp Immunol 115:294–300 [DOI] [PMC free article] [PubMed]
- Bouma G, Xia B, Crusius JB, Bioque G, Koutroubakis I, Von Blomberg BM, Meuwissen SG, et al (1996) Distribution of four polymorphisms in the tumour necrosis factor (TNF) genes in patients with inflammatory bowel disease (IBD). Clin Exp Immunol 103:391–396 [DOI] [PMC free article] [PubMed]
- Cho JH, Nicolae DL, Gold LH, Fields CT, LaBuda MC, Rohal PM, Pickles MR, et al (1998) Identification of novel susceptibility loci for inflammatory bowel disease on chromosomes 1p, 3q, and 4q: evidence for epistasis between 1p and IBD1. Proc Natl Acad Sci USA 95:7502–7507 [DOI] [PMC free article] [PubMed]
- Concannon P, Gogolin-Ewens KJ, Hinds DA, Wapelhorst B, Morrison VA, Stirling B, Mitra M, et al (1998) A second-generation screen of the human genome for susceptibility to insulin-dependent diabetes mellitus. Nat Genet 19:292–296 [DOI] [PubMed]
- Curran ME, Lau KF, Hampe J, Schreiber S, Bridger S, Macpherson AJS, Cardon LR, et al (1998) Genetic analysis of inflammatory bowel disease in a large European cohort supports linkage to chromosomes 12 and 16. Gastroenterology 115:1066–1071 [DOI] [PubMed]
- Delpre G, Kadish U, Gazit E, Joshua H, Zamir R (1980) HLA antigens in ulcerative colitis and Crohn's disease in Israel. Gastroenterology 78:1452–1457 [PubMed]
- Ferencik S, Lindemann M, Horsthemke B, Grosse-Wilde H (1992) A new restriction fragment length polymorphism of the human TNF-B gene detected by _Asp_HI digest. Eur J Immunogenet 19:425–430 [DOI] [PubMed]
- Forcione DG, Sands B, Isselbacher KJ, Rustgi A, Podolsky DK, Pillai S (1996) An increased risk of Crohn's disease in individuals who inherit the HLA class II DRB3*0301 allele. Proc Natl Acad Sci USA 93:5094–5098 [DOI] [PMC free article] [PubMed]
- Fujita K, Naito S, Okabe N, Yao T (1984) Immunological studies in Crohn's disease. I. Association with HLA systems in the Japanese. J Clin Lab Immunol 14:99–102 [PubMed]
- Gleeson MH, Walker JS, Wentzel J, Chapman JA, Harris R (1972) Human leucocyte antigens in Crohn's disease and ulcerative colitis. Gut 13:438–440 [DOI] [PMC free article] [PubMed]
- Hall J, Nanthakumar E (1997) Automated fluorescent genotyping. In: Boyle AL (ed) Current protocols in human genetics. Vol 2. John Wiley & Sons, New York, pp 2.8.1–2.8.19 [DOI] [PubMed] [Google Scholar]
- Hamilton SR (1987) The differential diagnosis of idiopathic inflammatory disease by colorectal biopsy. Int J Colorectal Dis 2:113–117 [DOI] [PubMed]
- Hampe J, Hermann B, Bridger S, MacPherson AJ, Mathew CG, Schreiber S (1998) The interferon-gamma gene as a positional and functional candidate gene for inflammatory bowel disease. Int J Colorectal Dis 13:260–263 [DOI] [PubMed]
- Hampe J, Schreiber S, Shaw SH, Lau KF, Bridger S, Macpherson AJS, Cardon LR, et al (1999) A genomewide analysis provides evidence for novel linkages in inflammatory bowel disease in a large European cohort. Am J Hum Genet 64:808–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland PM, Abramson RD, Watson R, Gelfand DH (1991) Detection of specific polymerase chain reaction product by utilizing the 5′----3′ exonuclease activity of_Thermus aquaticus_ DNA polymerase. Proc Natl Acad Sci USA 88:7276–7280 [DOI] [PMC free article] [PubMed]
- Hugot JP, Laurent-Puig P, Gower-Rousseau C, Caillat-Zucman S, Beaugerie L, Dupas JL, Van Gossum A, et al (1994) Linkage analyses of chromosome 6 loci, including HLA, in familial aggregations of Crohn disease. Am J Med Genet 52:207–213 [DOI] [PubMed]
- Idury RM, Cardon LR (1997) A simple method for automated allele binning in microsatellite markers. Genome Res 7:1104–1109 [DOI] [PMC free article] [PubMed]
- Kroeger KM, Carville KS, Abraham LJ (1997) The −308 tumor necrosis factor-alpha promoter polymorphism effects transcription. Mol Immunol 34:391–399 [DOI] [PubMed]
- Kruglyak L, Lander ES (1995) Complete multipoint sib-pair analysis of qualitative and quantitative traits. Am J Hum Genet 57:439–454 [PMC free article] [PubMed]
- ——— (1996) Limits on fine mapping of complex traits. Am J Hum Genet 58:1092–1093 [PMC free article] [PubMed]
- Lander E, Kruglyak L (1995) Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet 11:241–247 [DOI] [PubMed]
- Lennard-Jones JE (1989) Classification of inflammatory bowel disease. Scand J Gastroenterol Suppl 170:2–6 [DOI] [PubMed]
- Livak KJ, Flood SJ, Marmaro J, Giusti W, Deetz K (1995) Oligonucleotides with fluorescent dyes at oposite ends provide a quenched probe system useful for detecting PCR product and nucleic acid hybridization. PCR Methods Appl 4:357–362 [DOI] [PubMed]
- Matise TC, Perlin M, Chakravarti A (1994) Automated construction of genetic linkage maps using an expert system (MultiMap): a human genome linkage map. Nat Genet 6:384–390 [DOI] [PubMed]
- McGuire W, Hill AVS, Allsopp CEM, Greenwood BM, Kwiatkowski D (1994) Variation in the TNF-alpha promoter region associated with susceptibility to cerebral malaria. Nature 371:508–510 [DOI] [PubMed]
- Messer G, Spengler U, Jung MC, Honold G, Blomer K, Pape GR, Riethmuller G, et al (1991) Polymorphic structure of the tumor necrosis factor (TNF) locus: an_Nco_I polymorphism in the first intron of the human TNF-beta gene correlates with a variant amino acid in position 26 and a reduced level of TNF- beta production. J Exp Med 173:209–219 [DOI] [PMC free article] [PubMed]
- Nadel S, Newport MJ, Booy R, Levin M (1996) Variation in the tumor necrosis factor-alpha gene promoter region may be associated with death from meningococcal disease. J Infect Dis 174:878–880 [DOI] [PubMed]
- Nair RP, Henseler T, Jenisch S, Stuart P, Bichakjian CK, Lenk W, Westphal E, et al (1997) Evidence for two psoriasis susceptibility loci (HLA and 17q) and two novel candidate regions (16q and 20p) by genome-wide scan. Hum Mol Genet 6:1349–1356 [DOI] [PubMed]
- Nakajima A, Matsuhashi N, Kodama T, Yazaki Y, Takazoe M, Kimura A (1995) HLA-linked susceptibility and resistance genes in Crohn's disease. Gastroenterology 109:1462–1467 [DOI] [PubMed]
- Olavesen MG, Hampe J, Mirza MM, Saiz R, Lewis CM, Bridger S, Teare D, et al. Analysis of single nucleotide polymorphisms in the interleukin 4 receptor gene for association with inflammatory bowel disease. Immunogenetics (in press) [DOI] [PubMed] [Google Scholar]
- Partanen J, Koskimies S (1988) Low degree of DNA polymorphism in in the HLA-linked lymphotoxin (tumor necrosis factor beta) gene. Scand J Immunol 28:313–316 [DOI] [PubMed]
- Perri F, Annese V, Piepoli A, Napolitano G, Lombardi G, Ciavarella G, Di Giorgio G, et al (1998) HLA antigens and pANCA define ulcerative colitis as a genetically heterogeneous disorder. Ital J Gastroenterol Hepatol 30:56–61 [PubMed]
- Plevy SE, Targan SR, Yang H, Fernandez D, Rotter JI, Toyoda H (1996) Tumor necrosis factor microsatellites define a Crohn's disease-associated haplotype on chromosome 6. Gastroenterology 110:1053–1060 [DOI] [PubMed]
- Podolsky DK (1991) Inflammatory bowel disease (1). N Engl J Med 325:928–937 [DOI] [PubMed]
- Probert CS, Jayanthi V, Rampton DS, Mayberry JF (1996) Epidemiology of inflammatory bowel disease in different ethnic and religious groups: limitations and aetiological clues. Int J Colorectal Dis 11:25–28 [DOI] [PubMed]
- Purrmann J, Korsten S, Bertrams J, Miller B, Lapsien B, Munch H, Reis HE, et al (1985) HLA haplotype study in familial Crohn disease. Z Gastroenterol 23:432–437 [PubMed]
- Roussomoustakaki M, Satsangi J, Welsh K, Louis E, Fanning G, Targan S, Landers C, et al (1997) Genetic markers may predict disease behavior in patients with ulcerative colitis. Gastroenterology 112:1845–1853 [DOI] [PubMed]
- Russell AS, Percy JS, Schlaut J, Sartor VE, Goodhart JM, Sherbaniuk RW, Kidd EG (1975) Transplantation antigens in Crohn's disease: linkage of associated ankylosing spondylitis with HL-Aw27. Am J Dig Dis 20:359–361 [DOI] [PubMed]
- Satsangi J, Parkes M, Louis E, Hashimoto L, Kato N, Welsh K, Terwilliger JD, et al (1996_a_) Two stage genome-wide search in inflammatory bowel disease provides evidence for susceptibility loci on chromosomes 3, 7 and 12. Nat Genet 14:199–202 [DOI] [PubMed]
- Satsangi J, Welsh KI, Bunce M, Julier C, Farrant JM, Bell JI, Jewell DP (1996_b_) Contribution of genes of the major histocompatibility complex to susceptibility and disease phenotype in inflammatory bowel disease. Lancet 347:1212–1217 [DOI] [PubMed]
- Schreiber S, Nikolaus S, Hampe J, Hämling J, Koop I, Lochs H, Raedler A (1999) Tumor necrosis factor alpha and interleukin 1-beta in relapse of Crohn's disease. Lancet 353:459–461 [DOI] [PubMed]
- Shivananda S, Lennard-Jones J, Logan R, Fear N, Price A, Carpenter L, van Blankenstein M (1996) Incidence of inflammatory bowel disease across Europe: is there a difference between north and south? results of the European Collaborative Study on Inflammatory Bowel Disease (EC-IBD). Gut 39:690–697 [DOI] [PMC free article] [PubMed]
- Silverberg MS, Steinhart AH, McLeod RS, Griffitths A, Mirea L, Bull SB, Greenberg GR, et al (1999) Evidence for linkage between Crohn's disease (CD) and a locus near the major histocompatibility complex (MHC) on chromosome 6 in a Canadian inflammatory bowel disease (IBD) population. Gastroenterology 116:A820 [Google Scholar]
- Smolen JS, Gangl A, Polterauer P, Menzel EJ, Mayr WR (1982) HLA antigens in inflammatory bowel disease. Gastroenterology 82:34–38 [PubMed]
- Targan SR, Hanauer SB, van Deventer SJ, Mayer L, Present DH, Braakman T, DeWoody KL, et al (1997) A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn's disease: Crohn's Disease cA2 Study Group. N Engl J Med 337:1029–1035 [DOI] [PubMed]
- Terwilliger JD (1995) A powerful likelihood method for the analysis of linkage disequilibrium between trait loci and one or more polymorphic marker loci. Am J Hum Genet 56:777–787 [PMC free article] [PubMed]
- Toyoda H, Wang SJ, Yang HY, Redford A, Magalong D, Tyan D, McElree CK, et al (1993) Distinct associations of HLA class II genes with inflammatory bowel disease. Gastroenterology 104:741–748 [DOI] [PubMed]
- Truelove SC, Pena AS (1976) Course and prognosis of Crohn's disease. Gut 17:192–201 [DOI] [PMC free article] [PubMed]
- Uyar FA, Imeryuz N, Saruhan-Direskeneli G, Ceken H, Ozdogan O, Sahin S, Tozun N (1998) The distribution of HLA-DRB alleles in ulcerative colitis patients in Turkey. Eur J Immunogenet 25:293–296 [DOI] [PubMed]
- van den Berg-Loonen EM, Dekker-Saeys BJ, Meuwissen SG, Nijenhuis LE, Engelfriet CP (1977) Histocompatibility antigens and other genetic markers in ankylosing spondylitis and inflammatory bowel diseases. J Immunogenet 4:167–175 [DOI] [PubMed]
- Weissensteiner T, Lanchbury JS (1997) TNFB polymorphisms characterize three lineages of TNF region microsatellite haplotypes. Immunogenetics 47:6–16 [DOI] [PubMed]
- Wilson AG, Symons JA, McDowell TL, McDevitt HO, Duff GW (1997) Effects of a polymorphism in the human tumor necrosis factor alpha promoter on transcriptional activation. Proc Natl Acad Sci USA 94:3195–3199 [DOI] [PMC free article] [PubMed]
- Yang H, Plevy SE, Taylor K, Tyan D, Fischel-Ghodsian N, McElree C, Targan SR, et al (1999) Linkage of Crohn's disease to the major histocompatibility complex region is detected by multiple non-parametric analyses. Gut 44:519–526 [DOI] [PMC free article] [PubMed]