Modulation of Kv4.2 Channel Expression and Gating by Dipeptidyl Peptidase 10 (DPP10) (original) (raw)
Abstract
The dipeptidyl aminopeptidase-like protein DPPX (DPP6) associates with Kv4 potassium channels, increasing surface trafficking and reconstituting native neuronal _I_SA-like properties. Dipeptidyl peptidase 10 (DPP10) shares with DPP6 a high amino acid identity, lack of enzymatic activity, and expression predominantly in the brain. We used a two-electrode voltage-clamp and oocyte expression system to determine if DPP10 also interacts with Kv4 channels and modulates their expression and function. Kv4.2 coimmunoprecipitated with HA/DPP10 from extracts of oocytes heterologously expressing both proteins. Coexpression with DPP10 and HA/DPP10 enhanced Kv4.2 current by approximately fivefold without increasing protein level. DPP10 also remodeled Kv4.2 kinetic and steady-state properties by accelerating time courses of inactivation and recovery (_τ_rec: WT = 200 ms, +DPP10 = 78 ms). Furthermore, DPP10 introduced hyperpolarizing shifts in the conductance-voltage relationship (∼19 mV) as well as steady-state inactivation (∼7 mV). The effects of DPP10 on Kv4.1 were similar to Kv4.2; however, distinct biophysical differences were observed. Additional experiments suggested that the cytoplasmic N-terminal domain of DPP10 determines the acceleration of inactivation. In summary, DPP10 is a potent modulator of Kv4 expression and biophysical properties and may be a critical component of somatodendritic _I_SA channels in the brain.
INTRODUCTION
Voltage-gated K channels belonging to the mammalian Kv4 subfamily all rapidly activate and inactivate in response to subthreshold membrane depolarization, producing transient outward K currents. Uniquely characterized by fast recovery from inactivation, Kv4 channels evidently underlie the majority of the somatodendritic A-type K currents (_I_SA) in neurons (Sheng et al., 1992; Serodio et al., 1994; Johns et al., 1997; Shibata et al., 2000; Malin and Nerbonne, 2000) and the transient outward K current in cardiomyocytes (_I_TO) (Dixon et al., 1996; Barry et al., 2002; Xu et al., 1999). By underlying _I_SA, Kv4 channels may contribute to delayed excitation, spike repolarization, and frequency of slow repetitive firing in neuronal cells (Connor and Stevens, 1971; Baxter and Byrne, 1991; Hille, 2001; Malin and Nerbonne, 2000). Furthermore, their subcellular distribution underscores their important roles in dendritic signal processing, including regulation of back-propagating action potentials, integration of synaptic inputs, and induction of long-term potentiation (Hoffman et al., 1997; Johnston et al., 2000; Watanabe et al., 2002; Christie and Westbrook, 2003; Lilliehook et al., 2003). In cardiac cells, Kv4 currents determine the initial phase of action potential repolarization (Xu et al., 1999; Guo et al., 2000).
Differences between the biophysical behaviors of the _I_SA channels in neurons and Kv4 channels in heterologous systems have long suggested that the native channel may be a multisubunit macromolecular complex comprised of Kv4 pore-forming subunits and modulatory proteins (Rudy et al., 1988; Chabala et al., 1993; Serodio et al., 1994, 1996). Indeed, recent reports confirmed that two novel proteins with previously unknown functions, Kv channel-interacting protein (KChIP) and dipeptidyl aminopeptidase-related protein (DPPX or DPP6), are the main critical components of neuronal _I_SA channels that regulate Kv4 trafficking and gating (An et al., 2000; Nadal et al., 2003). Isolated initially through molecular techniques (yeast two-hybrid screening), KChIPs form a growing subfamily of cytosolic Ca-binding proteins that specifically bind Kv4 channels and modulate their currents. The binding of calcium is achieved through EF-hand motifs located in the conserved C-terminal region, consistent with KChIPs' membership in the recoverin/neuronal calcium sensor (NCS) family. Association of KChIPs (with the exception of KChIP4a) with the cytoplasmic N-terminus of Kv4 channels typically results in increased surface expression, slower inactivation, and accelerated recovery from inactivation (An et al., 2000; Bähring et al., 2001b; Holmqvist et al., 2002).
Identified as a Kv4 accessory subunit by coimmunoprecipitation, DPP6 is an integral membrane glycoprotein found predominantly in the brain and may be involved in neuronal plasticity (Nadal et al., 2003; Wada et al., 1992; de Lecea et al., 1994). Functionally, coexpression studies in Xenopus oocytes show that DPP6 dramatically increases surface expression of Kv4 channels and reconstitutes fast gating kinetics (i.e., inactivation and recovery) found in native _I_SA (Nadal et al., 2003). The topology of DPP6 consists of a short intracellular N-terminal domain and a disproportionately long extracellular C-terminal domain (Kin et al., 2001). The two DPP6 isoforms, DPP6-S (short) and DPP6-L (long), differ solely in the length of the cytoplasmic N-terminal domain (Wada et al., 1992). Closely related to dipeptidyl peptidase-4/CD26, DPP6 belongs to the family of nonclassical serine protease that cleaves N-terminal dipeptides from oligo- and polypeptides with a penultimate prolyl residue (McDonald and Schwabe, 1977; Wada et al., 1992). Curiously, DPP6 apparently lacks enzymatic activity in part due to a serine-to-aspartate substitution in the catalytic triad located in the C-terminal aminopeptidase-like domain, although dipeptidyl peptidase activity cannot be regained by restoration of the catalytic serine (Kin et al., 2001).
Among the DPP4/CD26 gene subfamily, DPP6 is not the only member with marked expression in the brain and without enzymatic activity. Dipeptidyl peptidase 10 (DPP10), originally identified as an expressed sequence tag (EST) clone and designated as dipeptidyl peptidase-related protein 3 (DPRP3), has a similar predicted molecular mass (∼91 kDa) and amino acid composition (48–51% identity) with DPP6 (Qi et al., 2003). DPP10 contains a serine-to-glycine substitution within the catalytic triad and, like DPP6, is not associated with any DPP enzymatic activity. Northern blot analysis shows DPP10 gene expression is highest in the brain, pancreas, spinal cord, and adrenal glands, with lower levels in placenta, liver, and trachea (Qi et al., 2003; Allen et al., 2003). Based on the similarities between DPP6 and DPP10, we performed experiments to examine whether DPP10 interacts with Kv4.2 channels and functions like DPP6 in altering their expression and gating properties. Here, we first report that DPP10 associates with Kv4.2 channels in immunoprecipitation experiments. Using two-electrode voltage-clamp, we show that DPP10 produces qualitatively many of the same biophysical effects as DPP6 proteins when coexpressed with Kv4.2 in Xenopus oocytes. Still, quantitative differences exist between the effects of DPP10 and DPP6 on channel gating, especially the time course of inactivation and recovery from inactivation. Western blot analysis of N-terminal HA-tagged DPP10 reveals that in oocytes DPP10 has a lower level of N-glycosylation when compared to DPP6-S. Furthermore, electrophysiological recordings of coexpression with HA-tagged DPP10 and HA-tagged DPP6-S, as well as chimera between DPP10 and DPP6-S, indicate that the cytoplasmic N-terminus of DPPs plays an important role in influencing the time course of Kv4.2 fast inactivation. This study shows that, similar to DPP6, DPP10 may play a critical part in the assembly of the Kv4 macromolecular complex in the brain and modulate its function. Some of these results have appeared in abstract form (Jerng and Pfaffinger, 2004).
MATERIALS AND METHODS
Molecular biology
A blast-verified plasmid construct containing human dipeptidyl peptidase 10 (DPP10) was purchased from American Type Culture Collection (ATCC, Manassas, VA) with the IMAGE clone identification #4811771, which corresponds to GenBank #BC030832 for human dipeptidyl aminopeptidase-related protein 3 (DPRP3). The complete insert sequence (including 5′ untranslated region (UTR), open reading frame, 3′ UTR, and poly-A tail) was determined by automated DNA sequencing (DNA sequencing core facility at Baylor College of Medicine, Houston, TX) and perfectly matched the DNA sequence published by Qi et al. (2003). Sequence analysis using Vector NTI (InforMax, Frederick, MD) detected five potential ATG start codons within the 5′ UTR and indicated an absence of Kozak consensus sequence (shown underlined) for ribosome binding (5′-GCCACCATGG-3′) (Kozak, 1989). To prevent potential aberrant initiation of translation and encourage correct translation, a new DPP10 construct without 5′ UTR and with most of Kozak canonical sequence was generated. PCR primers (forward primer = 5′-TCC, CCG, CGG, GCC, ACC, ATG, AAC, CAA, ACT, GCC, AGC, GTG, TCC-3′, reverse primer = 5′-GAT, ACA, ACG, CTC, CAG, TAA, ACC, GAG-3′) were used to amplify a DNA fragment (bps 1–232) with nested 5′ _Sac_II site. Afterwards, the PCR product was redigested (New England Biolabs, Beverley, MA) and inserted into the _Sac_II and _Nco_I sites of DPP10 plasmid, thus removing the 5′ UTR. To create N-terminal HA-tagged DPP10 for biochemical studies, a PCR product was generated with a forward primer containing the HA sequence in-frame between the Kozak sequence and starting ATG (HA-forward = 5′- TCC, CCG, CGG, GCC, ACC, ATG, TAC, CCA, TAC, GAC, GTC, CCA, GAC, TAC, GCT, ATG, AAC, CAA, ACT, GCC, AGC, GTG, TCC-3′) and the previous reverse primer. Again, subcloning of the PCR fragment was conducted after _Sac_II and _Nco_I digestion. The sequences of HA-tagged and untagged clones were confirmed by DNA sequencing of the subcloned segment.
Human DPP6-L was obtained from ATCC as 5′ (GenBank #R87715, IMAGE #180815) and 3′ (GenBank #W96197, IMAGE #361688) partial ORF clones from brain and retina, respectively. The 5′ fragment of IMAGE #180815 was digested with _Eco_RI, blunt-ended with Klenow, and excised with _Sac_I. The fragment was then ligated into blunt-ended Not I and sticky-ended _Sac_I sites of IMAGE #180815. The final complete DPP6-L clone was then sequenced by automated sequencing and confirmed to be the complete DPP6-L. An alignment between our and published DPP6-L sequence (NM 130797) reveals 7-nt differences, of which only one produced an amino acid change (P854L). Single nucleotide polymorphism analysis at the National Center for Biotechnology Information (NCBI) showed that a leucine (L) present in our clone is the more common residue at position 854; genomic sequence results validated by multiple independent submissions to the refSNP cluster, by frequency or genotype data, and observation of alleles in at least two chromosome apiece indicated that the average allele frequency of T (coding for L854) is 0.736, and that of C (coding for P854) is 0.264 (population sample size, in number of chromosomes = 1498, NCBI). Furthermore, an expressed sequence tag (EST) clone search yielded L854 in four out of seven clones identified. Therefore, our clone appears to reflect the more common allele of the population in general.
Wada et al. had previously reported that DPP6-S differs from DPP6-L only by the cytoplasmic N-terminus (Wada et al., 1992). Thus, human DPP6-S was constructed by amplifying the sequence that corresponds to the cytoplasmic N-terminus and transmembrane domain of DPP6-S from a 5′ partial DPP6-S clone from ATCC isolated from germ cells (GenBank #AI651135, IMAGE #2304141). This fragment, containing a 5′ nested _Sap_I site, Kozak consensus sequence, and 3′ internal _Mfe_I site, was subcloned into the DPP6-L construct by replacement. An HA-tag version was also generated by the same approach. The clone was verified by sequencing the region amplified by PCR and compared against published sequence (NM 001936). As expected, our sequence varied from published sequence, with leucine 792 (L792) instead of proline (P).
The putative transmembrane (TM) segments of DPP10 and DPP6-S have high amino acid identity (IAIALLVILV_CSLI__SVILL, where the gaps represent nonidentical residues). A unique _Mfe_I site exists in the nucleotide sequence of the TM domain of DPP6-S but not DPP10 due to a single base difference; thus, site-directed mutagenesis (QuikChange Mutagenesis System, Stratagene, La Jolla, CA) was used to introduce the _Mfe_I site without altering the amino acid (the underlined alanine: WKGIAIALLV). The _Mfe_I sites in DPP10 and DPP6-S were then utilized to genetically switch the cytoplasmic N-terminal domains and generate DPP10/DPP6-S and DPP6-S/DPP10 clones.
Kv4.2 and Kv4.1 genes in pBluescript were generous gifts of Drs. Lily Jan and Manuel Covarrubias, respectively. For expression and fluorescent imaging in COS-7 cells, Kv4.2 was subcloned into pEGFP-N1 vector (BD Biosciences, Palo Alto, CA) to produce C-terminal fusion with the green fluorescent protein. KChIP3 (GenBank #AI796484, ATCC), DPP10, DPP6-S, and DPP6-L were subcloned also into a pCMV-based vector for mammalian cell expression using appropriate RE digests followed by ligation.
RNA transcript synthesis and oocyte injection
For expression in oocytes, runoff cRNA transcripts were synthesized by linearizing the respective plasmids with appropriate restriction enzymes and then utilizing the mMESSAGE mMACHINE high-yield capped RNA transcription kit with the T7 RNA polymerase (Ambion, Austin, TX). Defolliculated stage V and VI Xenopus laevis oocytes were harvested and each injected with 2–4 ng of cRNA using a Nanoinjector (Drummond, Broomall, PA) and incubated at 18°C for 1–3 days in standard ND96 solution (in mM: 96 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2, and 5 HEPES, pH 7.4 adjusted with NaOH) supplemented with 5 mM Na-pyruvate and 5 _μ_g/ml gentamycin to permit channel expression.
Western blotting
After the completion of electrophysiological recordings, 10 oocytes were added to 200 _μ_l of homogenization solution (100 mM NaCl, 1% Triton-X-100, 1X protease inhibitor; Roche Diagnostics, Indianapolis, IN, 5 mM EDTA, 5 mM EGTA, and 20 mM Tris-HCl at pH 7.6) in 1.5 ml microcentrifuge tubes on ice. Oocyte lysis was accomplished by repeated suction and expulsion through 26.5-gauge needles attached to 1-ml syringes. The homogenates were centrifuged at 16,000 × g for 10 min at 4°C and separated out into three layers: light lipid (top), clear aqueous (middle), and heavy particular (bottom) layers. After centrifugation, the clear aqueous layer was quickly removed with a thin gel-loading tip attached to micropipettes, and the proteins were separated on SDS-polyacrylamide gel electrophoresis according to Sambrook et al. (1989). Homogenates for molecular weight assays using Westerns were boiled before loading, and boiling apparently led to the aggregation of Kv4.2 proteins, which ran as a high-molecular weight band with smear.
The separated proteins were transferred to Immobilon membranes (Millipore, Bedford, MA) using manufacturer's protocols. The following primary antibodies were used for Western blotting: anti-Kv4.2 antibodies (0.3 _μ_g/ml; Chemicon International, Temecula, CA) and anti-HA-antibodies (0.1 mg/ml; Roche Diagnostics). The immunoblots were probed with the primary antibodies and appropriate secondary (HRP-conjugated) antibody for 1 h each at room temperature, and the bound antibodies were detected using chemiluminescence with an ECL detection kit (Pierce, Rockford, IL).
Coimmunoprecipitation
For immunoprecipitation, oocytes expressing Kv4.2 alone or with HA/DPP10 were added to modified homogenization buffer (100 mM NaCl, 5 mM EDTA, 1% Triton X-100, protease inhibitor, and 10 mM Tris-Cl at pH 7.6) at 60 oocytes/400 _μ_l. Oocytes were disrupted by repeated suctioning through 26.5-gauge needles, and the membrane fraction was separated out from debris by centrifugation at 16,000 × g (13,000 rpm with Eppendorf tabletop centrifuge) for 15 min at 4°C. The supernatants (100 _μ_l aliquots) were incubated with goat IgG (ICN Biomedical, Irvine, CA), or goat anti-Kv4.2 antibody (C20, Santa Cruz Biotechnology, Santa Cruz, CA) or goat anti-HA antibody (Y-11-G, Santa Cruz Biotechnology) at a 20 _μ_g/ml final concentration overnight at 4°C. The mixtures were then incubated with UltraLink immobilized A/G beads (Pierce) for 2 h to bring down the antibody-antigen complexes. The beads were washed three times with the lysis buffer containing proteinase inhibitors by centrifugation (2500 × g)/resuspension. The beads were then transferred to a fresh tube, and the bound immunocomplexes were eluted using 40 _μ_l of 10% SDS with 100 mM DTT. The eluted material were mixed with 20 _μ_l of sample buffer (50 mM Tris (pH 6.8), 2.5% SDS, 15% glycerol, and 100 mM DTT), ready for separation on SDS-PAGE gels.
Fluorescent imaging of transfected COS-7 cells
Transfection and fluorescent imaging of COS-7 cells were conducted as previously described (Zhou et al., 2004). Briefly, COS-7 cells maintained in DMEM (Invitrogen, Carlsbad, CA) supplemented with fetal bovine serum (Hyclone Laboratories, Logan, UT) and 50 U/ml penicillin plus 50 mg/ml streptomycin (Invitrogen) were plated on eight-well Lab-Tek chamber slides (Nalge Nunc International, Rochester, NY) and transfected on the following day using the FuGENE 6 Transfection Reagent (Roche Diagnostics). At 40–44 h post-infection, the cells were thoroughly washed with phosphate-buffered saline (PBS) and fixed with paraformaldehyde in PBS. After several PBS washes, the slides were overlaid with coverslips using VectaShield (Vector Laboratories, Burlingame, CA) and imaged using Olympus Flowview confocal microscope (Olympus BX51 with Flowview 300; Olympus America, Melville, NY).
Electrophysiology
Whole-oocyte currents were measured at room temperature (∼22–23°C) using two-electrode voltage-clamp (Oocyte Clamp, Warner Instruments, Hamden, CT) while the oocytes were bathed in ND96 solution. Microelectrodes were pulled from borosilicate glass capillaries (TW100F-4, World Precision Instruments, Sarasota, FL) using flaming/brown micropipette puller (Model P-17, Sutter Instrument, Novato, CA) to feature a tip resistance of <1 MΩ and filled with 3 M KCl. Voltage-clamp data were digitized and lowpass-filtered (Frequency Devices, Haverhill, MA) at various frequencies. For example, for 1000-ms depolarizations, current traces were digitized at 500 _μ_s/point (2 kHz) and filtered at 1 kHz, whereas data for 200-ms depolarizations were digitized at 100 _μ_s/point (10 kHz) and filtered at 5 kHz. Data acquisition was done on a desktop PC using pClamp6 (Axon Instruments, Foster City, CA). The average offset was <2 mV after recordings, and the average leak during recordings was <0.2 _μ_A and subtracted offline by assuming Ohmic leak. The capacitative transients were subtracted either on-line using P/4 protocol (200 ms pulses) or by scaling up transients at voltages without ionic currents (at −90 mV) and subtracting them from total outward K currents.
Data analysis
Data analysis of two-electrode voltage-clamp records was primarily performed using Clampfit (Axon Instruments) with additional analysis using Origin (OriginLab, Northampton, MA). Current decays were described with the sum of two or three exponential terms using Clampfit. The left cursor was placed at the time point with the fastest change in the declining phase of the current, as determined by differentiating the current trace. The right cursor was placed at the end of the traces. Peak conductance (_G_p) was calculated as 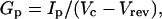 where _I_p is the peak current, _V_c is the command voltage, and _V_rev is the reversal potential (−90 to −95 mV in ND96). Peak conductance-voltage (G_p–_V) curves were fitted for comparison using the first-order Boltzmann function
where _I_p is the peak current, _V_c is the command voltage, and _V_rev is the reversal potential (−90 to −95 mV in ND96). Peak conductance-voltage (G_p–_V) curves were fitted for comparison using the first-order Boltzmann function 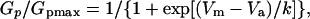 where _G_p/_G_pmax is the fraction of maximal conductance, _V_m is the given membrane potential, _V_a is the potential for half-maximal activation, and k is the slope factor. Steady-state inactivation curves were described by a simple first-order Boltzmann function as well. Statistical results were obtained using Origin and expressed as mean ± standard deviation.
where _G_p/_G_pmax is the fraction of maximal conductance, _V_m is the given membrane potential, _V_a is the potential for half-maximal activation, and k is the slope factor. Steady-state inactivation curves were described by a simple first-order Boltzmann function as well. Statistical results were obtained using Origin and expressed as mean ± standard deviation.
RESULTS
Sequence alignment of DPP10 and DPP6
Amino acid sequence comparison indicated that DPP10 is a member of the S9B subfamily of serine proteases and closely related to DPP6 and DPP4/CD26 (Qi et al., 2003). Our sequence alignment of DPP10 (GenBank Accession #BC030832), DPP6-S (GenBank Accession #M96859), and DPP6-L (GenBank Accession #M96860) generated using the AlignX function of VectorNTI program confirms that a high degree of identity exists between DPP10 and DPP6 (Fig. 1). The sequence identity of DPP10 to DPP6-S (55%) and DPP6-S (48%) as determined by AlignX closely matches those reported by Qi and co-workers. Positions of DPP6 identity are not isolated to particular regions but are instead distributed throughout the length of the DPP10 protein. The most nonconserved segment is the cytoplasmic N-terminal domain. As a S9B dipeptidyl peptidase, DPP10 possesses the consensus sequence DW(V/L)YEEE glutamate residues at positions 228–234, where the same sequence is found in DPP6-S and DPP6-L at aa 228–234 and 290–296, respectively (Fig. 1). And, consistent with it being a type II transmembrane protein, DPP10 shares with DPP6 amino acid residues within the putative transmembrane segment. Thus, the transmembrane topology of both DPP10 and DPP6 consists of an intracellular N-terminal domain, a transmembrane segment, and an extracellular C-terminal domain with numerous putative N-glycosylation sites (there are seven sites for DPP6 and nine sites for DPP10).
FIGURE 1.

Sequence comparison between DPP10 and DPP6 in human. Based on the structure of DPP4/CD26, DPP6 and DPP10 proteins consist of a cytoplasmic N-terminal domain, a transmembrane domain (TM, with underline), and a long C-terminal domain. The C-terminal domain in DPP4/CD26 has been partitioned into a glycosylation-rich, a cysteine-rich, and a catalytic region (Mentlein, 1999). Putative potential N-glycosylation sites in DPP6 and DPP10 are indicated in bold and underline, and the conserved catalytic residues are indicated by asterisks. Note that the DPP4 catalytic serine (GKSYGG) in DPP10 and DPP10 is respectively replaced by aspartate (D) and glycine (G). Residues identical between DPP6 and DPP10 are highlighted by shading. The S9B dipeptidyl peptidase consensus sequence DW(V/L)YEEE is indicated with underline and S9B. Transcripts of DPP10 and the two isoforms of DPP6, DPP6-S (short) and DPP6-L (long), are present predominantly in the brain, as detected by Northern hybridization (Qi et al., 2003; Allen et al., 2003).
Curiously, DPP10 and DPP6 share additional unique functional and sequence features. Previously studies have shown that both DPP10 and DPP6 are members of serine dipeptidyl peptidases incapable of peptidase activity, signified by substitution of the catalytic serine (in the GXSYGG sequence) by glycine in DPP10 and aspartate in DPP6 (Kin et al., 2001; Qi et al., 2003). Furthermore, both DPP10 and DPP6 possess a long stretch of acidic, negatively charged residues at the C-terminal tail (5 aa in DPP10, 6 aa in DPP6) which is not found in DPP4/CD26. The degree of sequence identity and common features suggest that DPP10 may perform functions similar to DPP6, including physical association and modulation of Kv4 channel expression and gating.
DPP10 in Xenopus oocytes: expression, glycosylation, and coimmunoprecipitation with Kv4.2
To determine if DPP10 physically associates with Kv4.2 channels, DPP10 was genetically fused at the N-terminus with the HA-tag, a nine-amino acid peptide sequence (YPYDVPDYA) present in the human influenza virus hemaglutinin protein. The resulting protein, HA/DPP10, was heterologously expressed in Xenopus oocytes with and without Kv4.2, and the membrane preparations from these oocytes were used in immunoprecipitation experiments to examine their possible interaction. Fig. 2 A shows that anti-HA antibody works effectively on HA/DPP10 oocyte membrane preps and immunoprecipitated HA/DPP10 proteins, identifying an ∼97 kDa band on Western blots (upper center panel), but does not pick up any background signal in oocytes expressing only Kv4.2 (upper left panel). Inversely, anti-Kv4.2 antibody specifically detects Kv4.2 in extracts containing Kv4.2 but not HA/DPP10 (lower left and center panels). As the figure indicates, the expressed Kv4.2 migrated on SDS gels as three protein populations with differing apparent masses, despite having been denatured by 7.5% SDS and reduced by 100 mM DTT. The population with the lowest molecular mass (∼65 kDa) likely corresponds to the Kv4.2 monomeric protein, which has an expected mass of 70 kDa. The other, higher-molecular mass populations appear to represent protein aggregates, and boiling before sample loading can result in the appearance of only these forms (Fig. 2 B, upper panels). When HA/DPP10 and Kv4.2 are coexpressed, immunoprecipitation using anti-HA antibody brings down both HA/DPP10 and Kv4.2. Likewise, anti-Kv4.2 antibody effectively coimmunoprecipitates both Kv4.2 and HA/DPP10 when expressed together. The coimmunoprecipitation of Kv4.2 and DPP10 demonstrates that they associate with each other.
FIGURE 2.

Physical association between DPP10 and Kv4.2 and determination of glycosylation level of DPP10. (A) Coimmunoprecipitation of DPP10 with Kv4.2 in Xenopus oocytes. Membrane preps of oocytes expressing Kv4.2 alone (left blots, upper and lower), HA-tagged DPP10 (HA/DPP10) alone (middle blots, upper and lower), and both (right blots, upper and lower) were immunoprecipitated using goat IgG, goat monoclonal anti-Kv4.2 antibody, or goat monoclonal anti-HA antibody (as indicated). The immunoprecipitated proteins were separated out on 6% SDS-PAGE gels and transferred onto Immobilon membranes. The immunoblots were probed using rabbit anti-Kv4.2 antibody (lower blots) or rat anti-HA antibody (upper blots), followed by secondary anti-rabbit and anti-rat antibodies conjugated to horseradish peroxidase (HRP). The Kv4.2 and anti-HA antibodies were highly specific in our assays, as seen in upper left and lower middle blots. HA/DPP10 ran at ∼97 kDa. Kv4.2 bands represents monomeric and aggregate forms. (B) DPP10 is less glycosylated when compared to DPP6-S (lower blots). A subset of oocytes injected with Kv4.2, Kv4.2 + HA/DPP6-S, and Kv4.2 + HA/DPP10 cRNAs for expression were treated with tunicamycin (tu: 2.5 mM) by injection and incubation in tunicamycin-containing ND96. Whole-oocyte homogenates were separated on SDS-PAGE, transferred onto Immobilon, and probed with anti-Kv4.2 and anti-HA antibodies. Coexpression with HA/DPP10 or HA/DPP6-S does not significantly affect Kv4.2 protein levels, but tunicamycin treatment produces marked reduction (upper blots). Xo = uninjected oocyte.
As shown in Fig. 2 A, HA/DPP10 exhibits a molecular mass larger than expected. In fact, an ∼8 kDa difference exists between the observed 97 kDa size versus the 89 kDa protein predicted from amino acid sequence. This difference is likely due to N-glycosylation, which has been demonstrated for DPP6 (Kin et al., 2001). N-linked glycosylation, for most plasma membrane proteins, occurs cotranslationally in the endoplasmic reticulum (ER) to maximize access to the target asparagine (N) residue by the catalytic glycosyl transferase enzyme (Alberts et al., 1994). The transferase searches for sequences N-X-S or N-X-T (where X is any amino acid except proline) and attaches a single species of precursor oligosaccharide that subsequently undergoes trimming in the ER and extensive modification in the Golgi apparatus. To demonstrate that indeed the shift in molecular mass is due to N-linked glycosylation, oocytes injected with Kv4.2 and HA/DPP10 were injected and incubated with 2.5 _μ_g/ml tunicamycin to prevent glycosylation. Injected oocytes were allowed to incubate for 3 days before their collection and processing for Western blot analysis. Indeed, tunicamycin treatment decreased the size of HA/DPP10 to approximately the predicted size of 89 kDa (Fig. 2 B, lower right panel). In addition, tunicamycin treatment also resulted in a marked decrease in the levels of both Kv4.2 and HA/DPP10 (Fig. 2 B, upper left and lower right panels).
The same treatment was also applied to HA/DPP6-S to allow comparison of the contribution of glycosylation to the total protein mass of DPP10 and DPP6-S. For HA/DPP6-S, its length of 813 aa predicts a protein with a size of ∼90 kDa, but experimental data reproducibly showed that HA/DPP6 exhibits a 25 kDa shift in molecular mass to ∼115 kDa, identical to that observed by Nadal and colleagues for the native DPP6 protein (Fig. 2 B, lower left panel). As with HA/DPP10, the presence of tunicamycin shifted the protein back to its expected size on SDS-PAGE. In addition to reducing steady-state levels of HA/DPP6-S, HA/DPP10, and Kv4.2 proteins, tunicamycin also greatly decreased the current expression by Kv4.2 channels complexed with HA/DPP10 (by ∼73%) or HA/DPP6-S (by ∼83%) (data not shown). These results indicate that post-translational modification resulting from N-glycosylation accounts for ∼8 kDa (8.2%) and ∼25 kDa (21.7%) of protein mass in DPP10 and DPP6-S, respectively, and suggest that DPP10, despite having more putative extracellular N-linked glycosylation sites than DPP6, appears to be less glycosylated due possibly to either fewer attached oligosaccharide or differing modification in the Golgi apparatus.
DPP10 increases surface expression and dramatically alters Kv4.2 channel biophysical properties
We also examined the potential effects of DPP10 on Kv4.2 expression and function in Xenopus oocytes. The results of the quantitative and statistical analyses are reported in Table 1 for experiments related to Kv4.2. Oocytes injected with both Kv4.2 and DPP10 cRNAs (1:1 molar ratio) expressed ∼4–6 times more outward currents than with Kv4.2 cRNA alone, as determined by comparisons of currents at +50 mV (Fig. 3 B). The mechanism by which DPP10 and DPP6 increase Kv4.2 current does not appear to be due to an increase in steady-state level of total Kv4.2 proteins, since our Western results did not detect a dramatic increase in the amount of Kv4.2 proteins (Fig. 2 B). It has previously been shown that DPP6-S promotes the trafficking of Kv4.2 to the cell surface in CHO cells (Nadal et al., 2003); therefore, the subcellular localization of Kv4.2 proteins was studied in COS-7 cells transfected with cDNAs encoding C-terminal EGFP-tagged Kv4.2 with and without DPP10 cDNA. For comparison, Kv4.2 was also transiently coexpressed in COS-7 cells with DPP6-S, DPP6-L, and KChIP3. Kv4.2 proteins expressed alone are mostly localized to the perinuclear ER/Golgi compartments, consistent with published reports (Fig. 4 A; An et al., 2000). When coexpressed with KChIP3 (Fig. 4 D), DPP6-S (Fig. 4 B), or DPP6-L (Fig. 4 C), as expected Kv4.2 proteins redistributed away from the ER/Golgi and onto the cell surface, prominently defining the cell margins. Coexpression of Kv4.2 with DPP10 mediated effects similar to those of DPP6 and KChIP, although to a lesser degree (Fig. 4 E). In agreement with the surface trafficking data, DPP6-S increased Kv4.2 currents magnitude ∼13–25 times, significantly more than that observed with DPP10 (Table 1; Nadal et al., 2003). These results clearly demonstrate that elevated Kv4.2 functional expression in the presence of DPP10 reflects enhanced surface trafficking of Kv4.2 proteins.
TABLE 1.
Biophysical properties of Kv4.2 channels coexpressed with dipeptidyl peptidase 10 and 6 (DPP10, DPP6-S) in Xenopus oocytes
| Property | Kv4.2 | Kv4.2 + DPP10 | Kv4.2 + HA/DPP10 | Kv4.2 + DPP6-S | Kv4.2 + HA/DPP6-S |
|---|---|---|---|---|---|
| Expression (fold increase, mean ± SE) | — | 5.9 ± 1.5* (n = 4) | 4.9 ± 1.9† (n = 3) | 13.4 ± 1.1* (n = 5) | 48.2 (n = 1) |
| Activation | |||||
| Time-to-peak (At +50 mV) (ms) | 8.0 ± 0.0 | 3.1 ± 0.6* | 4.9 ± 1.0*‡ | 3.4 ± 0.5* | 3.2 ± 0.3* |
| Peak G_–_V relation | |||||
| _V_0.5 (mV) | −0.1 ± 3 | −18.9 ± 4* | −28.6 ± 4*‡ | −22.1 ± 3* | −25.8 ± 3* |
| Slope (mV/_e_-fold) | 20.6 ± 2.5 (n = 5) | 17.5 ± 2.3 (n = 4) | 18.2 ± 2.2 (n = 7) | 16.2 ± 1.2* (n = 4) | 17.0 ± 1.1 (n = 3) |
| Inactivation | |||||
| Time course (At +50 mV) | |||||
| _τ_-fast (ms) | 19 ± 1 | 6 ± 3* | 28 ± 2* | 14 ± 2 | 26 ± 3 |
| _τ_-intermediate (ms) | 60 ± 3 | 23 ± 10* | 55 ± 15 | ||
| _τ_-slow (ms) | 382 ± 25 | 280 ± 71* | 164 ± 20* | 891 ± 169* | 494 ± 149 |
| _W_-fast | 59 ± 2 | 69 ± 7 | 78 ± 2* | 80 ± 1* | 86 ± 1* |
| _W_-intermediate | 24 ± 2 | 19 ± 5 | 8 ± 2* | ||
| _W_-slow | 9 ± 1 | 4 ± 1* | 12 ± 1* | 6 ± 1 | 6 ± 1 |
| _W_-non-inactivating | 8 ± 0 (n = 3) | 8 ± 1 (n = 9) | 10 ± 1 (n = 4) | 6 ± 0* (n = 4) | 8 ± 0 (n = 3) |
| Steady-state inactivation | |||||
| _V_0.5 (mV) | −63.9 ± 2 | −70.2 ± 2* | −69.6 ± 1* | −71.2 ± 2* | −65.6 ± 1‡ |
| Slope (mV/_e_-fold) | 5.1 ± 0.3 (n = 6) | 4.8 ± 0.2 (n = 5) | 3.7 ± 0.1*‡ (n = 6) | 4.1 ± 0.3 (n = 8) | 3.5 ± 0.2*‡ (n = 3) |
| Low-voltage inactivation τ (ms) | |||||
| At _V_0.5 −5 mV | 1275 ± 276 | 191 ± 40* | ND | 258 ± 27* | ND |
| At _V_0.5 | 1356 ± 170 | 211 ± 11* | ND | 344 ± 67* | ND |
| At _V_0.5 +5 mV | 1266 ± 85 (n = 3–4) | 168 ± 25* (n = 3) | ND | 378 ± 63* (n = 3) | ND |
| Recovery (at −100 mV) | |||||
| _τ_-rec (ms) | 201 ± 21 (n = 4) | 79 ± 13* (n = 4) | 88 ± 3* (n = 3) | 56 ± 13* (n = 6) | 34 ± 6* (n = 3) |
FIGURE 3.

Effects of DPP10 on Kv4.2 current in Xenopus oocytes. (A) Whole-oocyte currents of Kv4.2 and Kv4.2 coexpressed with DPP10. Families of current traces were elicited by depolarizing steps from −100 mV to +70 mV at 10 mV steps for 1 s, but only the initial 250 ms is shown. Inset shows the current waveform at +50 mV during the initial 25 ms. (B) Comparison of Kv4.2 current expression with and without DPP10 coexpression. Each data point set (connected by line) represents results from single-day sessions (n = 5). As indicated in Table 1, DPP10 increased Kv4.2 current expression on average by ∼5.9-fold. (C) Voltage-dependence of time-to-peak. Measurements taken from traces from respective voltages and plotted against the membrane potential. (D) Voltage-dependence of peak conductance and steady-state inactivation. The peak conductance-voltage relationship was calculated from current-voltage relationship according to the method described in Materials and Methods. The steady-state inactivation protocol consists of a 10-s prepulse that varied between −100 and −30 mV in 10-mV increments, followed by a test pulse at +50 mV. The time between episode starts was 30 s. (E) The time course of recovery from inactivation at −100 mV, measured using a double-pulse protocol. From a holding potential of −100 mV, a +50 mV depolarization of 1 s (prepulse) activated and inactivated the channels. A second pulse to +50 mV (test pulse) was delivered after a variable interpulse interval (shown on panel) at −100 mV. Non-inactivating current at the end of the prepulse was subtracted from peak values of prepulse and test pulse, and subsequently the adjusted test pulse value was divided by that of the prepulse to produce the fractional recovery. The inter-episode interval for the recovery protocol was 15 s. •, Kv4.2; ○, Kv4.2 + DPP10. Data points shown as mean ± standard deviation.
FIGURE 4.

Coexpression of DPP10 changes the subcellular localization of Kv4.2 expressed in COS-7 cells by promoting surface trafficking. COS-7 cells were transiently transfected with EGFP-Kv4.2 cDNA alone (A) or along with DPP6-S (B), DPP6-L (C), KChIP3 (D), or DPP10 (E) cDNA. The EGFP signals were imaged by confocal microscopy, showing the distribution of total Kv4.2 proteins. When expressed alone, Kv4.2 proteins accumulate in the perinuclear ER/Golgi compartments (A). Coexpression of DPP10 (E) redistributes Kv4.2 proteins, decreasing ER/Golgi accumulation and increasing cell surface expression. Similar Kv4.2 redistribution is observed with coexpression of DPP6-S (B), DPP6-L (C), and KChIP3 (D). Scale bars, 20 _μ_m (as indicated).
Aside from increasing current expression of Kv4.2 channels, DPP10 dramatically changed the waveform of Kv4.2 currents. Families of whole-oocyte currents were elicited by depolarizing steps from −100 to +70 mV with 10-mV increments for 1 s, but only the initial 250-ms portion is shown in Fig. 3 A. When compared to Kv4.2 expressed alone, Kv4.2 current in the presence of DPP10 rises and decays very rapidly, resulting in marked decrease in time required to reach maximal current amplitude (time-to-peak) (Fig. 3 A). The decreased time-to-peak displays very little voltage-dependence in the entire voltage range tested (from −30 mV to +70 mV) (Fig. 3 C). The potassium conductance at peak current was calculated for each voltage by using the modified form of Ohm's law (as described in Materials and Methods) and plotted as a function of voltage. The peak conductance-voltage relationship (peak G_–_V) was described with first-order Boltzmann functions, and the results showed that the midpoint voltage to maximal conductance (_V_0.5) for DPP10 coexpression exhibited a ∼19 mV hyperpolarizing shift when compared to Kv4.2 control (Kv4.2: −0.1 ± 3.0 mV, n = 5; Kv4.2 + DPP10: −18.9 ± 4.2 mV, n = 4) (Fig. 3 D). The leftward shift along the voltage axis occurred with minor increase in the steepness of the slope, although the finding was not statistically significant. We also studied the effects of DPP10 on the steady-state inactivation of Kv4.2 channels using a two-step protocol with a conditioning pulse of 10-s duration, which was immediately followed by a 250-ms test pulse at +50 mV. The results showed that DPP10 produced an ∼6–7 mV hyperpolarizing shift (Kv4.2: −63.9 ± 1.8 mV, n = 6; Kv4.2 + DPP10: −70.2 ± 1.5, n = 5) with a small but significant decrease in slope. A leftward shift in G_–_V larger than that of steady-state inactivation indicates that DPP10 increases the fraction of channels available for activation at threshold or subthreshold potentials.
Two-pulse protocols were used to examine the recovery from inactivation of channels coexpressed with DPP10. Inactivation was induced by the initial depolarization at +50 mV, and a second depolarization to +50 mV was used to determine the fraction of channel that recovered after a variable interval of rest at −100 mV. At −100 mV, Kv4.2 channels in oocytes typically recover monoexponentially with a time constant of ∼200 ms, and coexpression with DPP10 significantly accelerated the rate of recovery at −100 mV at ∼2.5–3-fold (_τ_-rec = 78.8 ± 13 ms, n = 4) (Fig. 3 E).
DPP10 modifies Kv4.1 biophysical properties differently from Kv4.2
DPP10 also modulated the biophysical properties of Kv4.1 channels, as reported in Table 2. Similar to Kv4.2, coexpression of DPP10 with Kv4.1 channels decreased the time-to-peak current and sharply accelerated current decay (Fig. 5 A). The dramatic speeding up of inactivation resulting from association with DPP10 can be clearly seen from normalized traces at +50 mV (Fig. 5 B). As with Kv4.2, DPP10 markedly decreased the values and voltage-dependence of time-to-peak (Fig. 5 C). Furthermore, coexpression of DPP10 with Kv4.1 channels also accelerated recovery from inactivation at −100 mV approximately threefold (_τ_-rec: Kv4.1 = 275 ± 40 ms, n = 4; Kv4.1 ± DPP10 = 108 ± 4.8 ms, n = 3) (Fig. 5 E).
TABLE 2.
Biophysical properties of Kv4.1 channels coexpressed with dipeptidyl peptidase 10 and 6 (DPP10, DPP6-S) in Xenopus oocytes
| Property | Kv4.1 | Kv4.1 + DPP10 | Kv4.1 + DPP6-S |
|---|---|---|---|
| ACTIVATION | |||
| Time-to-peak (@ +50 mV) (ms) | 9.1 ± 2.2 | 3.5 ± 0.7* | 6.2 ± 1.8 |
| Peak G_–_V relation | |||
| _V_0.5 (mV) | −16.0 ± 1.2 | −1.3 ± 1.4* | −31.9 ± 2.6* |
| Slope (mV/_e_-fold) | 14.0 ± 0.5 (n = 4) | 19.4 ± 0.3* (n = 4) | 16.1 ± 2.3 (n = 3) |
| INACTIVATION | |||
| Time course (@ +50 mV) | |||
| _τ_-fast (ms) | 16 ± 3 | 11 ± 1* | 17 ± 7 |
| _τ_-intermediate (ms) | 99 ± 15 | 57 ± 10* | 108 ± 24 |
| _τ_-slow (ms) | 370 ± 46 | 394 ± 21 | 342 ± 63 |
| _W_-fast | 12 ± 3 | 49 ± 9* | 8 ± 2 |
| _W_-intermediate | 24 ± 7 | 20 ± 3 | 22 ± 5 |
| _W_-slow | 56 ± 9 | 25 ± 9* | 58 ± 5 |
| _W_-non-inactivating | 9 ± 3 (n = 6) | 9 ± 0 (n = 5) | 11 ± 1 (n = 4) |
| Steady-state inactivation | |||
| _V_0.5 (mV) | −66.9 ± 1.8 | −70.7 ± 2.7* | −83.6 ± 1.9* |
| Slope (mV/_e_-fold) | 4.8 ± 0.4 (n = 5) | 7.1 ± 0.8* (n = 5) | 6.8 ± 0.4* (n = 8) |
| RECOVERY (@ −100 mV) | |||
| _τ_-rec (ms) | 275 ± 40 (n = 4) | 108 ± 5* (n = 3) | 137, 127 (n = 2) |
FIGURE 5.

DPP10 affects Kv4.1 kinetics and steady-state properties. (A) TEVC recordings of currents from oocytes expressing Kv4.1 and Kv4.1 + DPP10. Current traces were elicited by depolarizing steps from −100 to +60 mV at 10 mV steps for 1-s duration, with the first 500 ms shown here. (Inset) Current waveforms at +50 mV during the initial 25 ms. (B) Normalized and superimposed traces of Kv4.1 (thick) and Kv4.1 + DPP10 (thin) currents at +50 mV, showing the dramatic acceleration in the time course of inactivation in the presence of DPP10 (C) Time-to-peak measurements for voltages ranging from −40 mV to +80 mV. (D) Voltage-dependence of peak conductance and steady-state inactivation. (E) Recovery from inactivation at −100 mV as determined by double-pulse protocol. The experiments were conducted and analyzed as described in Fig. 3. • = Kv4.1, ○ = Kv4.1 + DPP10. Data points shown as mean ± standard deviation.
However, not all Kv4.1 biophysical properties were affected in the same way as Kv4.2. DPP10 did not produce a significant hyperpolarizing shift in the steady-state inactivation of Kv4.1 channels (_V_0.5: Kv4.1 = −66.9 ± 1.8 mV, n = 5; Kv4.1 ± DPP10 = −70.7 + 2.7 mV, n = 5) (Fig. 5 D). In a phenomenon unseen with DPP10 coexpressed with Kv4.2 channels, DPP10 significantly decreased the voltage-sensitivity of inactivation of Kv4.1 as indicated by the decrease in the curve's slope (slope: Kv4.1 = 4.84 ± 0.4 mV/_e_-fold, n = 5; Kv4.1 + DPP10 = 7.08 ± 0.8 mV/_e_-fold, n = 5). Furthermore, as an additional surprise, the conductance-voltage (G_–_V) relationship exhibited a significant rightward shift along the voltage axis (_V_0.5: Kv4.1 = −16.0 ± 1.2 mV, n = 4; Kv4.1 + DPP10 = −1.3 ± 1.4 mV, n = 4) (Fig. 5 D). Like steady-state inactivation, the voltage-sensitivity of G_–_V relationship was also decreased by DPP10 (slope: Kv4.1 = 14.0 ± 0.5 mV/_e_-fold, n = 4; Kv4.1 + DPP10 = 19.4 ± 0.3 mV/_e_-fold, n = 4). These data together suggest that the association of DPP10 with Kv4.1 affects steady-state properties differently from those of Kv4.2; in particular, DPP10 significantly decreases the general voltage-sensitivity of Kv4.1 channel gating.
DPP10 accelerates Kv4.2 inactivation at high and low voltages
Acceleration of inactivation is a prominent and unmistakable feature of DPP10 coexpression with Kv4.2 and Kv4.1 channels. As shown previously with Kv4.2 channels by Nadal et al. in 2003, acceleration of inactivation time course is also a distinct effect of modulation by DPP6-S (also known as DPPX-S). To compare the relative degree of the acceleration that accompanies coexpression of DPP10 and DPP6-S, we normalized traces under strong depolarization (+50 mV) for Kv4.2 control, Kv4.2 + DPP10, and Kv4.2 + DPP6-S (Fig. 6 A). As seen with the traces, the current trace for Kv4.2 + DPP10 reaches peak amplitude at the same time as that of Kv4.2 + DPP6-S, but the DPP10 trace decays significantly faster. To describe the changes in the kinetics of high-voltage inactivation that underlie the observed accelerated inactivation, we used the sums of exponential fits to quantify the time courses of inactivation over a period of 1 s seen with both DPP10 and DPP6-S. The development of macroscopic inactivation in Kv4 channels is complex and often described by using the sum of three exponential terms (Pak et al., 1991; Baldwin et al., 1991; Jerng and Covarrubias, 1997; Jerng et al., 1999; Bähring et al., 2001a; Beck et al., 2002). Simulations of Kv4.1 and Kv4.2 experimental inactivation data using kinetic modeling have provided important insights into the potential functional significance of these different exponential processes (see Discussion). We have also observed that the time course of inactivation of Kv4.2 without and with DPPs necessitated the use of three exponential terms. At +50 mV, inactivation of Kv4.2 alone is well fit with time constants of 18.7 ± 1.2 ms (fast phase: weight = 58.9 ± 2.4), 60.0 ± 2.8 ms (intermediate phase: weight = 24.3 ± 1.9), and 382 ± 25 ms (slow phase: weight = 8.7 ± 0.7) (n = 3) (Fig. 6 B; Table 1). Coexpression with DPP10 dramatically accelerated fast and intermediate phases of inactivation, decreasing their time constants by nearly threefold to 6.3 ± 2.8 ms and twofold to 23.4 ± 10 ms, respectively (n = 9). The time constant of the slow phase, as well as the proportions of the three phases and steady-state current, showed little or no significant changes. Thus, it appears that the decreased time-to-peak associated with DPP10 results from accelerated inactivation rates as well as activation.
FIGURE 6.

Quantitative analysis of Kv4.2 inactivation at high and low voltages. (A) Normalized and superimposed traces of Kv4.2, Kv4.2 + DPP10, and Kv4.2 + DPP6-S currents at high voltage (+50 mV). (B) The effects of DPP10 and DPP6-S on Kv4.2 inactivation at +50 mV. Sum of three exponential terms was necessary to properly describe the current decays over 1 s, generating time constants and fractional amplitudes for fast (fast), intermediate (inter), and slow (slow) components. The fractional amplitude of the non-inactivating, steady-state (S-S) current is also shown. • = Kv4.2, ○ = Kv4.2 + DPP10, ▵ = Kv4.2 + DPP6-S. (C) The time course of pre-open, low-voltage inactivation of Kv4.2 and Kv4.2 + DPP10 measured at _V_0.5 and _V_0.5 ± 5 mV of steady-state inactivation using the protocol described in the inset. In brief, from the holding potential of −100 mV, a 100-ms pulse to +50 mV was delivered to obtain the control current. This pulse was followed by a return to −100 mV for 1 s to allow full channel recovery. The membrane was then stepped to a prepulse potential (around _V_0.5 of steady-state inactivation) for a variable duration before depolarizing again to +50 mV for 500 ms to test amount of available, activatable channels. Single exponential function was used to describe the data. (D) The voltage-dependence of pre-open inactivation at ∼_V_0.5.
A different phenomenon took place with the time course of inactivation in the presence of DPP6-S (Fig. 6 B; Table 1). DPP6-S accelerated the fast phase of inactivation to a lesser degree when compared to DPP10 (DPP6-S: 13.8 ± 1.8 ms, n = 4). With DPP6-S, the intermediate phase of inactivation showed little-to-no change in its kinetics, whereas the slow phase interestingly displayed slower inactivation (DPP6-S: 891 ± 169 ms; n = 4). Unlike DPP10 coexpression, DPP6-S coexpression with Kv4.2 significantly increased the weight of the fast phase (DPP6-S: 80.0 ± 1.2, n = 4) with compensatory decreases mostly restricted to the intermediate phase of inactivation. Therefore, whereas acceleration in the time course of inactivation results from DPP6 coexpressions, DPP6's effect primarily derives from increasing the weight of the fast component by ∼33% and not by accelerating inactivation rates. Thus, these results suggest that at high voltage DPP10 and DPP6-S modify Kv4.2 differently to produce the observed acceleration of inactivation.
Kv4 channels underlie the subthreshold somatodendritic currents in neurons, and their subthreshold inactivation may be attributed to channel transitions into pre-open, closed-inactivated states at low voltages (Jerng et al., 1999; Beck and Covarrubias, 2001; Bähring et al., 2001a). As potentially critical components of _I_SA channels that accelerate high-voltage inactivation, DPP10 may also speed up low-voltage inactivation. Using a pulse protocol described in the inset of Fig. 6 C, pre-open inactivation was measured for both Kv4.2 alone and Kv4.2 coexpressed with DPP10. Measurements were taken at the voltage where half of the channels are inactivated under steady-state condition, as well as 5 mV above and 5 mV below. For control Kv4.2, the voltages were −68 mV, −63 mV, and −58 mV, and for Kv4.2 + DPP10, the voltages were −75 mV, −70 mV, and −65 mV. As Fig. 6 C shows, DPP10 dramatically accelerated the low-voltage, pre-open inactivation. As measured by single exponential fits, the time constants for Kv4.2 and Kv4.2 + DPP10 inactivation at _V_0.5 were 1356 ± 170 ms (n = 4) and 211 ± 11 ms (n = 3), respectively. Similar sixfold increases in rates were obtained for 5 mV above and below the _V_0.5 voltage. Measurement of pre-open inactivation in the presence of DPP6-S also showed a marked decrease in the time constant (Table 1), resulting in an ∼4–5-fold change. When compared with the acceleration of recovery from inactivation (DPP10: ∼2.5-fold; DPP6-S: ∼3.5-fold), the effect of this dramatic acceleration of closed-state inactivation dominates, and consequently the voltage-dependence of steady-state inactivation exhibits the ∼7 mV hyperpolarizing shift.
Role of cytoplasmic N-terminus: HA-tagging slows time course of inactivation
An important clue as to how DPP10 accelerates Kv4.2 inactivation at high voltage derived from functional consequences of HA-tagging at the DPP10 cytoplasmic N-terminus. N-terminal HA-tagging of DPP10 and DPP6-S allowed us to determine the molecular mass and relative level of protein expressed, but it also permitted the investigation of the structure-function relationship of the distinct N-terminal portions of DPP10 and DPP6 in channel gating. Coexpression of HA/DPP10 with Kv4.2 led to insignificant change in current expression as compared to coexpressions with untagged DPP10 (Table 1). HA/DPP10 increased expression by ∼4.9-fold, where DPP10 increased expression by 4–6-fold. However, preliminary data showed that HA-tagging of DPP6-S resulted in a further fivefold increase in current expression, to 55-fold over Kv4.2 alone (Table 1). Tagging of DPP6-S suggests that the cytoplasmic N-terminus may be involved in surface expression of DPPs, as suggested by reports indicating that this domain is important in targeting yeast and rat DPPs to the surface (Roberts et al., 1989; Hong and Doyle, 1990).
The time course of inactivation is significantly affected by the N-terminal HA-tagging of both DPP10 and DPP6 (Fig. 7, A and B; Table 1). For DPP6-S, HA-tagging does not change the time-to-peak values established by the Kv4.2-DPP6 association. However, with DPP10, the time-to-peak value slightly increased from 3.1 ± 0.6 (untagged, n = 4) to 4.9 ± 1.0 (HA-tagged, n = 7). HA-tagging of both DPP10 and DPP6 significantly slowed the kinetics of inactivation, which is most dramatic with DPP10 (Fig. 7, A and B, left panels). Curve fitting using exponential functions showed that coexpression of HA-tagged DPPs modified Kv4.2 in a way that two-exponential terms were sufficient to describe the current decay. Although it is not possible to precisely correlate exponential components derived from three- and two-exponential fits and compare their rates and factional amplitudes, we can provide a rough estimate of the degree of slowing by comparing only the dominant fast inactivating component. The time constant of the fast component for DPP6-S and DPP10 respectively increased ∼1.8 and ∼3.5-fold by HA-tagging. The slowing of the fast phase of inactivation is accompanied by an increase of its contribution from ∼80 to 86% in DPP6-S, or from ∼70 to 80% in DPP10.
FIGURE 7.

Effects of HA-tagging DPP10 and DPP6-S at the N-terminus. (A, left panel) Kv4.2, Kv4.2 + DPP10, and Kv4.2 + HA/DPP10 currents at +50 mV, after normalization. (B, left panel) Kv4.2, Kv4.2 + DPP6-S, and Kv4.2 + HA/DPP6-S current traces at +50 mV after normalization. (Middle panels) Comparison of peak conductance-voltage relationships. (Right panels) Comparison of recovery from inactivation at −100 mV.
The attachment of an HA-tag to the cytoplasmic N-terminus of DPP10 and DPP6-S produced differing effects on their ability to modify channel steady-state properties. HA/DPP10 had a small but significant effect on the conductance-voltage relationship compared to that of untagged DPP10, where no significant change was seen with HA/DPP6-S (Fig. 7, A and B, middle panels). As shown in Table 1, DPP10 and DPP6-S brought about similar amounts of hyperpolarizing shifts in the G_–_V relationship: the membrane voltages (_V_0.5) for half-maximal conductance were −22.1 ± 2.9 mV for DPP6-S and −18.9 ± 4.2 mV for DPP10. HA-tagging of DPP6-S did not significantly affect the _V_0.5 value (−25.8 ± 2.5 mV); however, HA-tagging of DPP10 introduce further hyperpolarizing shift to −28.6 ± 3.9 mV. The slopes of the G_–_V curves did not change as a result of HA-tagging of the DPP10 or DPP6-S N-termini, but interestingly HA/DPP10 and HA/DPP6-S shifts G_–_V relationships to a similar degree. Further illustrating differing effects of HA-tagging, HA/DPP10 produced no additional effects on the extent of hyperpolarizing shift in steady-state inactivation over that of DPP10, but placing an HA-tag on DPP6-S significantly lessened the degree of the shift (Table 1). Finally, recovery from inactivation was not affected by HA-tagging of DPP10 or DPP6-S (Fig. 7, A and B, right panels). In summary, the attachment of an HA-tag on the cytoplasmic N-terminal domain of either DPP10 or DPP6-S markedly slowed the kinetics of inactivation.
DPP10 and DPP6-S chimeras show that the cytoplasmic N-terminal domains determine the level of inactivation acceleration
To investigate further the role of the cytoplasmic N-terminus in inactivation modulation, we created chimeric constructs between DPP10 and DPP6-S, interchanging their cytoplasmic N-terminal domains. As Fig. 8 A clearly shows, the DPP10/DPP6 chimera containing the DPP10 N-terminal domain conferred DPP10-like accelerated inactivation to Kv4.2 channels. Conversely, the DPP6/DPP10 chimera with the DPP6 N-terminal domain gives inactivation acceleration more like that of DPP6-S (Fig. 8 B). Other than time course of inactivation, expression and functional effects of DPP10 and DPP6-S are nearly indistinguishable and were found in the chimeric mutants (data not shown). In combination with the HA/DPP10 and HA/DPP6 results, it would appear that the N-terminal cytoplasmic domain of DPP10 plays a role in providing acceleration to Kv4.2 inactivation.
FIGURE 8.

Probing the role of the DPP10 cytoplasmic N-terminus. DPP10/6 chimera consists of DPP10 cytoplasmic N-terminal domain fused to the transmembrane and extracellular C-terminal domain of DPP6-S. In DPP6/10, the N-terminal domain of DPP10 is replaced by that of DPP6. (A and B, left and middle panels) Families of current traces from oocytes expressing Kv4.2 + DPP10, Kv4.2 + DPP6-S, Kv4.2 + DPP10/6, and Kv4.2 + DPP6/10 provoked by voltage steps from −100 mV to +80 mV at 10 mV steps over 100 ms. The holding potential was −100 mV. Capacitance and leak were subtracted online by using P/4 protocol. (Right panels) Normalized traces at +50 mV superimposed for comparison.
DISCUSSION
We have demonstrated that DPP10 associates with Kv4 channels and consequently increases Kv4 current and modifies functional properties in a heterologous expression system. The manners and degrees by which DPP10 alters the kinetics and voltage-dependence of Kv4 gating depend on the isoform of Kv4 pore-forming subunit present. This study shows that, in addition to DPP6, DPP10 reconstitutes native neuronal _I_SA properties to heterologously expressed Kv4 currents and suggests that DPP10 may be a principal constituent of the neuronal _I_SA macromolecular complex that regulates firing frequency and dendritic signal integration. Our investigation also suggests that the cytoplasmic N-terminal domain of DPP10 determines the acceleration of Kv4 inactivation.
Coexpression of Kv4 and DPP10 reconstitute _I_SA functional properties
Does DPP10 indeed colocalize and interact with Kv4 channels in vivo and thereby influence _I_SA expression and function in the brain? Northern hybridization, RT-PCR, and in situ hybridization indicate that Kv4 (Kv4.1, Kv4.2, and Kv4.3) mRNA and protein exhibit pronounced distribution throughout the brain, but especially in the cerebellum, thalamus, cerebral cortex, hippocampus, substantia nigra, and olfactory bulbs (Isbrandt et al., 2000; Serodio and Rudy, 1998). DPP10 transcripts are strongly expressed in the brain as well, concentrated in the cerebral cortex, medulla, occipital lobe, frontal lobe, and temporal lobe (Qi et al., 2003; Allen et al., 2003). Furthermore, DPP10 EST clones have been derived from hypothalamus, hippocampus, and whole brain (Qi et al., 2003). The overlapping expression patterns of Kv4 and DPP10 in the cerebral cortex and hippocampus suggests that significant Kv4-DPP10 interactions may occur in these important regions. Interestingly, DPP10 shows high expression in the cerebral cortex but very little expression in the cerebellum, whereas DPP6 shows converse expression patterns, with more expression in the cerebellum and less expression in the cerebral cortex (Allen et al., 2003; de Lecea et al., 1994). This may perhaps explain why Nadal and colleagues, in their coimmunoprecipitation from rat cerebellum, did not identify DPP10 as an auxiliary modulator of Kv4 channels. An alternative explanation may be that DPP10 associates with Kv4 channels with a lower affinity and thus were unrecovered during the immunoprecipitation process, despite attempts to preserve their interactions by chemical crosslinking (Nadal et al., 2003). However, that appears unlikely, considering the strength of signals detected in our coimmunoprecipitation studies conducted without crosslinking (Fig. 2 A).
The biophysical effects of DPP10, including accelerated time courses of inactivation and recovery from inactivation and hyperpolarizing shifts in the voltage-dependence of steady-state activation and inactivation, results in Kv4 channels properties very similar to that of most neuronal _I_SA. This suggests that DPP10, along with DPP6, is likely a necessary component of the _I_SA channel needed for proper function. The speeding-up of the kinetic parameters would permit channel gating to respond quickly to changing membrane potentials, and most significantly, the differential shifts of channel activation and inactivation along the voltage axis translate into a larger “window current,” or percentage of active _I_SA at subthreshold voltages. Increasing the availability of _I_SA would have a marked impact on the ability of _I_SA to impede back-propagating action potentials and reduce basal dendritic excitability (Hoffman et al., 1997).
DPP10 increases surface expression of Kv4.2 channels
The amount of Kv4.2 current generated at a given potential is proportional to the number of available channels at the cell surface, the probability of channel opening, and the intrinsic single-channel conductance. In turn, the availability of functional surface channels depends on the production of pore-forming _α_-subunits and the proper assembly of channels, followed by their trafficking to the plasma membrane and subsequent degradation or recycling. Our immunocytochemical, electrophysiological, and biochemical data show that DPP10 mediates the redistribution of Kv4.2 proteins to the cell surface in mammalian cells and increases the expression of Kv4.2 current in oocyte without apparently increasing the total amount of Kv4.2 protein. Similar findings were observed with the related DPP6-S, consistent with an earlier report showing that DPP6-S facilitates Kv4.2 trafficking and targeting (Nadal et al., 2003). Although we have not precisely quantitated the contribution of enhanced trafficking to the increases in Kv4.2 current, it is clear that biophysical changes also play a role, albeit likely a minor one. DPP10 lowers the voltage for activation ∼10 mV without slope change, leading to fractional increases in current upon depolarization. Furthermore, it has recently been shown that DPP6-S increases the single-channel conductance of Kv4.3 channel from 4.0 pS to 6.8 pS, an increase of ∼70% (Rocha et al., 2004). However, it remains to be seen if coassembly of Kv4 channels with DPP10 affects single-channel conductance.
Glycosylation may affect a protein's proper folding and stability in intracellular organelles, routing to the cell surface and turnover, and/or their function (Lennarz, 1983). For example, in rat DPP4/CD26, mutation of a critical asparagine involved in N-glycosylation (N319Q) abolishes enzymatic activity, cell-surface expression, and dimerization (Fan et al., 1997). Although the effects we have observed with tunicamycin are consistent with such a role for DPP10 and DPP6-S glycosylation in the facilitation of Kv4 surface expression, further studies are needed to rule out potential direct negative effects of tunicamycin on DPP and Kv4.2 protein levels. Our results do clearly show that N-glycosylation of DPP10 increases the molecular mass ∼12 kDa less than DPP6-S (comparing ∼8 kDa to ∼20 kDa, respectively) in the oocyte expression system, consistent with published works showing no or minimal glycosylation of DPP10 heterologously expressed in PEAK cells or isolated from human brain (Qi et al., 2003; Allen et al., 2003). Since the more glycosylated DPP6-S on average increases expression slightly more than DPP10 (4–6-fold vs. ∼13-fold, respectively) and removal of glycosylation reduced current more in DPP6-S than DPP10, glycosylation may be a factor in the ability of DPP10 to enhance Kv4 surface expression.
Molecular basis of Kv4-DPP10 association and subsequent gating modulation by DPP10
The demonstration that both DPP10 and DPP6 interact with Kv4 channels suggests that the regions of substantial sequence identity (TM and extracellular domain: see Fig. 1) feature structural elements sufficiently similar to permit physical interaction and gating modulation. Although the atomic structures of DPP10 and DPP6 remains unsolved, three-dimensional structures of other members of the prolyl oligopeptidase (POP) gene family have been determined, including cytosolic POP from pig and extracellular portions of DPP4/CD26 from human and rat (Fulop et al., 1998; Rasmussen et al., 2002; Oefner et al., 2003; Ludwig et al., 2003; Hiramatsu et al., 2003). The soluble POP and DPP4/CD26 monomers both form barrel-shaped structures comprised of a _β_-propeller domain and an α/_β_-hydrolase domain, despite sharing only 12% amino acid identity and 23% conservation. An unexpected feature of the basic structural organization is that opposing (N- and C-terminal) ends of the POP and DPP4/CD26 (extracellular part) polypeptides contain elements that contribute to the α/_β_-hydrolase domain, as the C-terminal end loops back and assembles with an N-terminal portion (Fulop et al., 1998; Rasmussen et al., 2002; Oefner et al., 2003). Interestingly, this spatial arrangement positions the enzymatic domain juxtaposed with the plasma membrane and suggests a potential region of α_–_β subunit interaction for future studies.
What are the molecular bases for the modulation of Kv4 channel properties by DPP10 and DPP6? The carbohydrate trees attached to DPP6 heterologously expressed in COS-1 cells and DPP4/CD26 from seminal plasma contain negatively charged N-acetylneuraminic (sialic) acid residues (Kin et al., 2001; Lambeir et al., 1997), and the voltage-dependent parameters of Kv4 gating may conceivably be modified by alterations of the effective surface charge caused by these external negative charges near the cell surface. Indeed, a published report shows that N-glycosylation of Kv1.1 channels results in faster activation kinetics, negatively shifted _V_0.5 of activation, and steeper slope of G_–_V relationship (Watanabe et al., 2003). Furthermore, glycosylation of skeletal muscle sodium channel (SkM1, Nav1.4) influences channel gating as reduction of sialylation or deletion of likely glycosylation sites leads to ∼11 mV depolarizing shifts in G_–_V, steady-state inactivation, and voltage-dependence of time constants of activation and inactivation (Bennett et al., 1997; Bennett, 2002). However, our data is inconsistent with glycosylation of DPPs mediating the observed gating changes because DPP10 affects gating with significantly lower level of glycosylation than DPP6-S, the amounts and/or directions of voltage shift in G_–_V and steady-state inactivation are not identical, and prevention of N-glycosylation by tunicamycin does not eliminate channel modulation (data not shown).
Our electrophysiological data indicate that association with DPPs perturbs the physical changes underlying Kv4 inactivation. Previous studies have shown that Kv4 inactivation involves multiple processes (Jerng and Covarrubias, 1997; Jerng et al., 1999; Bähring et al., 2001a). The fastest inactivation process involves occlusion of the inner pore by the autoinhibitory N-terminal domain (Gebauer et al., 2004), whereas a slower inactivation process occurs with closed channels, or channels flickering closed at depolarized potentials, and appears to be a result of uncoupling of voltage-sensor movement and channel opening (Jerng et al., 1999; Bähring et al., 2001a; Beck and Covarrubias, 2001; Beck et al., 2002). This “pre-open” inactivation is sensitive to mutations in the inner vestibular region (Jerng et al., 1999). A possible mechanism for this “unresponsiveness” is hinted at by a study of spHCN channels that suggests an uncoupling of voltage-sensor S4 from the channel activation gate occurs at the channels inner vestibule (Shin et al., 2004).
The acceleration of Kv4 inactivation by DPP10 or DPP6 appears to primarily involve modifications of the slower pre-open inactivation process rather than the faster pore-occlusion process. First, the most striking effect of DPP protein coexpression is the dramatic acceleration of low-voltage, pre-open inactivation of Kv4.2 channels. DPP10 and DPP6-S accelerate pre-open inactivation by ∼7- and ∼3–4-fold, respectively (Table 1). Deletion studies on Kv4.2 channels have shown that pre-open inactivation is not significantly affected by deletion of the N-terminus and thus does not involve a traditional pore-blocking mechanism (Bähring et al., 2001a). Second, a previous study on DPP6-S has shown dramatic acceleration of inactivation of Kv4.2 channels coexpressed with KChIP1 (Nadal et al., 2003). KChIPs bind the Kv4.2 N-terminus, sequestering this inactivation domain from performing pore-blocking functions (Zhou et al., 2004). Thus, DPPs must have mechanisms available to accelerate Kv4 channel inactivation separate from enhancing the function of the Kv4.2 inactivation peptide.
An allosteric model of Kv4 gating with significant pre-open (closed-state) inactivation can explain changes in inactivation gating induced by coexpression with DPP10 and DPP6 (Fig. 9; also see Beck and Covarrubias, 2001; Bähring et al., 2001a; Beck et al., 2002). The main features of the models are:
- The opening equilibrium (C ↔ O) is weakly voltage-dependent and reverse-biased (consistent with flickery Kv4 openings in single channel records), leading to significant contribution of pre-open closed-state inactivation (C → _I_C) at all voltages (Baldwin et al., 1991; Jerng et al., 1999; Beck and Covarrubias, 2001).
- Channels must close first before undergoing final and complete inactivation (O → C → _I_C) at the end of a long pulse, resulting in typically monoexponential recovery kinetics.
- Inactivation may occur from closed states along the activation pathway (_C_0–_C_4) to account for rapid recovery at hyperpolarized potentials (Beck and Covarrubias, 2001; Bähring et al., 2001a). A second open-inactivated state (_I_D) is included to match the number of experimentally observed inactivating components (one aggregate of closed-inactivated states and two open-inactivated states) (Beck and Covarrubias, 2001; Bähring et al., 2001a; Beck et al., 2002) (Fig. 9).
FIGURE 9.

Kinetic state diagram describing the allosteric model for Kv4 inactivation. In this model (Beck and Covarrubias, 2001; Bähring et al., 2001a), Kv4 channels can inactivate either from the open state or preferentially from closed states along the activation pathway. The coupling between closed-state inactivation and voltage-dependent activation (transitions from _C_0 to _C_4) is defined by the allosteric factor (f), resulting in increased likelihood of inactivation occurring from later closed states. Transitions between states are represented by forward and reverse arrows.
Computer simulations suggest that the rate constant _k_IC sets the measured time constant of recovery from inactivation (Bähring et al., 2001a). The rate of low-voltage inactivation, represented by _k_CI, primarily sets the intermediate time constant of inactivation at high-voltage (Bähring et al., 2001a). Furthermore, at high-voltage, the fast decay component that involves pore block of open channels is controlled by the rate constant _k_OI. Finally, the slow component of inactivation is set by the rate constants for transitions between _I_O and _I_D (_k_ID and _k_DI) (Bähring et al., 2001a).
Accordingly, acceleration of closed-state inactivation by DPP10 and DPP6-S should be evident in accelerated inactivation of the intermediate phase (_τ_-intermediate = 60 ± 3 ms at +50 mV; Table 1) at high voltage. Reductions of the intermediate time constant at +50 mV by approximately sevenfold by DPP10 and ∼3–4-fold by DPP6-S results in time constants of ∼8.6 ms and ∼15 ms, respectively. These values are smaller than that of Kv4.2 _τ_-fast (19 ± 1 ms) and match those of _τ_-fast when Kv4.2 is coexpressed with DPP10 (6 ± 3 ms) and DPP6-S (14 ± 2 ms) (Table 1). A similar phenomenon occurs with Kv4.1 channels coexpressed with DPP10 or DPP6-S. Preliminary data shows that DPP6-S accelerated Kv4.1 closed-state inactivation (at _V_0.5 of steady-state inactivation) by approximately sevenfold (Rocha et al., 2004), while producing a similar approximately ninefold and approximately sixfold change between _τ_-intermediate of Kv4.1 and _τ_-fast of Kv4.1+DPP10 and Kv4.1+DPP6-S (Table 2). These results suggest that when Kv4 channels are coexpressed with DPP10 or DPP6-S, closed-state inactivation at high voltages (C → O → C → _I_C) may occur faster than that of open-state inactivation (C → O → _I_O) and hence determine the fast phase of inactivation. The remaining fractions of inactivation in the presence of DPPs therefore occurs with channel transitions through the open-state inactivation and closing, before ultimately residing at the closed-inactivated state at the end of the pulse (Fig. 9). While it is not experimentally possible to correlate the these remaining components with specific kinetic states, our data suggests that open-state inactivation may also be altered by interaction with DPPs.
We propose that DPP10 and DPP6-S, acting through their cytoplasmic N-terminal domains, differentially modulate inactivation primarily by affecting the intracellular structures underlying closed-state inactivation, the channel inner vestibule (S4–S5 linker, lower portions of S5–S6; Jerng et al., 1999). However, we do not rule out other scenarios, such as one where accelerated inactivation results from faster open-state inactivation, with the intracellular domain of DPP10 or DPP6 acting directly or indirectly on the inactivation particle or its putative receptor on the inner pore (Gebauer et al., 2004). In conclusion, although the ability of DPP cytoplasmic N-terminus to determine inactivation is consistent with current understanding of the structures and kinetics of Kv4 inactivation, the precise molecular basis of the interaction between Kv4 and DPPs and the different fast inactivation caused by different DPPs remains unknown and warrants further detailed studies.
Final remarks
The neuronal somatodendritic A-type K channel is an assembly of pore-forming and accessory subunits, leading to a greater supermolecular complex (Nadal et al., 2003). The machineries necessary for ion permeation and channel gating are located in the Kv4 tetramer, and the association of KChIPs or DPPs modifies Kv4 gating behavior, reconstitutes certain functional properties of _I_SA, and thus is likely critical to _I_SA physiology. This study, along with published works, provides a general impression of the relative physiological importance of DPPs and KChIPs in binary complexes with Kv4 in brain (refer to Nadal et al., 2003, An et al., 2000, and this study). Both DPPs and KChIPs facilitate current expression, leftward-shift the G_–_V relationships, and accelerate recovery from inactivation. However, DPPs dramatically increases the rate of pre-open inactivation more than KChIPs (DPPs: ∼4–7-fold; KChIP1: ∼2.5-fold) and dramatically accelerate inactivation (this study; Beck et al., 2002), with significant consequences. The dominance of pre-open inactivation over recovery in DPP coexpression produces a negative shift in steady-state inactivation, whereas, in KChIP, coexpression recovery dominates over pre-open inactivation and leads to a positive shift. Thus, DPPs play an especially critical role in establishing the overall subthreshold character and fast inactivation of the neuronal somatodendritic A-type channels. Furthermore, robust close-state inactivation in the presence of DPPs likely determines fast inactivation of _I_SA since, in the ternary complex (Kv4-KChIP-DPP), KChIP binding would effectively remove or slow the innate rapid open-state inactivation (Beck et al., 2002; Zhou et al., 2004).
Structurally, the subunit stoichiometry of DPP assembly with Kv4 channels is currently unknown, but KChIP physically caps the Kv4 N-terminus and assembles with Kv4 with a 4:4 subunit stoichiometry (Zhou et al., 2004; Kim et al., 2003). Coimmunoprecipitation from native tissue and heterologous coexpression studies have shown that Kv4 channels likely bind both KChIP and DPP6 proteins simultaneously and that the ternary complex exhibits biophysical properties distinct from those of DPP10-, DPP6-, and KChIP1-Kv4 binary complexes (Nadal et al., 2003). The fact that native _I_SA can vary substantially in their biophysical properties between different types of neurons and different regions of the brain suggests that the interplay between Kv4-specific accessory subunits such as KChIPs and DPPs likely form the critical basis of variability in native _I_SA currents. Since KChIPs has been shown to reset or “normalize” the properties of Kv4 channels (Beck et al., 2002), the ability of DPP10 and DPP6 to differentially modulate Kv4 expression and inactivation rate is likely crucial to the function of neuronal _I_SA. Although many questions regarding DPP regulation of Kv4 channel remain to be investigated, the discovery of DPP10 modulation of Kv4 channels and the studies of the functional roles of its glycosylation and N-termini have allowed us to begin to understand how these accessory subunits play a critical part in these important somatodendritic A-type K channels.
Acknowledgments
We thank Dr. Lily Jan for the Kv4.2 gene and Dr. Manuel Covarrubias for the Kv4.1 gene. H.H.J. also thanks Rachel W. Kung for her valued assistance in the preparation of this manuscript.
This work was supported by National Institutes of Health Postdoctoral Training Grant 133003317 (to H.H.J.), RO1 NS-31583, and P01NS37444.
References
- Alberts, B., D. Bray, J. Lewis, M. Raff, K. Roberts, and J. D. Watson. 1994. Molecular Biology of the Cell. Garland Publishing, New York, NY.
- Allen, M., A. Heinemann, E. Noguchi, G. Abecasis, J. Broxholme, C. P. Ponting, S. Bhattacharyya, J. Tinsley, Y. Zhang, R. Holt, E. Y. Jones, N. Lench, A. Carey, H. Jones, N. J. Dickens, C. Dimon, R. Nicholls, C. Baker, L. Xue, E. Townsend, M. Kabesch, S. K. Weiland, D. Carr, E. von Mutius, I. M. Adcock, P. J. Barnes, G. M. Lathrop, M. Edwards, M. F. Moffatt, and W. O. Cookson. 2003. Positional cloning of a novel gene influencing asthma from chromosome 2q14. Nat. Genet. 35:258–263. [DOI] [PubMed] [Google Scholar]
- An, W. F., M. R. Bowlby, M. Betty, J. Cao, H. P. Ling. G. Mendoza, J. W. Hinson, K. I. Mattson, B. W. Strassle, J. S. Trimmer, and K. J. Rhodes. 2000. Modulation of A-type potassium channels by a family of calcium sensors. Nature. 403:553–556. [DOI] [PubMed] [Google Scholar]
- Bähring, R., L. M. Boland, A. Varghese, M. Gebauer, and O. Pongs. 2001a. Kinetic analysis of open- and closed-state inactivation transitions in human Kv4.2 A-type potassium channels. J. Physiol. 535:65–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bähring, R., J. Dannenberg, H. C. Peters, T. Leicher, T., O. Pongs, and D. Isbrandt. 2001b. Conserved Kv4 N-terminal domain critical for effects of Kv channel-interacting protein 2.2 on channel expression and gating. J. Biol. Chem. 276:23888–23894. [DOI] [PubMed] [Google Scholar]
- Baldwin, T. J., M.-L. Tsaur, G. A. Lopez, Y. N. Jan, and L. Y. Jan. 1991. Characterization of a mammalian cDNA for an inactivating voltage-sensitive K+ channel. Neuron. 7:471–483. [DOI] [PubMed] [Google Scholar]
- Barry, D. M., H. Xu, R. B. Schuessler, and J. M. Nerbonne. 2002. Functional knockout of the transient outward current, long-QT syndrome, and cardiac remodeling in mice expressing a dominant-negative Kv4 α subunit. Circ. Res. 90:497–499. [DOI] [PubMed] [Google Scholar]
- Baxter, D. A., and J. H. Byrne. 1991. Ionic conductance mechanisms contributing to the electrophysiological properties of neurons. Curr. Opin. Neurobiol. 1:105–112. [DOI] [PubMed] [Google Scholar]
- Beck, E. J., and M. Covarrubias. 2001. Kv4 channels exhibit modulation of closed-state inactivation in inside-out patches. Biophys. J. 81:867–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck, E. J., M. Bowlby, W. F. An, K. J. Rhodes, and M. Covarrubias. 2002. Remodeling inactivation gating of Kv4 channels by KChIP1, a small-molecular-weight calcium-binding protein. J. Physiol. 538:691–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett, E., M. S. Urcan, S. S. Tinkle, A. G. Koszowski, and S. R. Levinson. 1997. Contribution of sialic acid to the voltage dependence of sodium channel gating. A possible electrostatic mechanism. J. Gen. Physiol. 109:327–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett, E. S. 2002. Isoform-specific effects of sialic acid on voltage-dependent Na+ channel gating: functional sialic acids are localized to the S5–S6 loop of domain I. J. Physiol. 538:675–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabala, L. D., N. Bakry, and M. Covarrubias. 1993. Low-molecular-weight poly(A)+ mRNA species encode factors that modulate gating of a non-Shaker A-type K+ channel. J. Gen. Physiol. 102:713–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie, J. M., and G. L. Westbrook. 2003. Regulation of backpropagating action potentials in mitral cell lateral dendrites by A-type potassium currents. J. Neurophysiol. 89:2466–2472. [DOI] [PubMed] [Google Scholar]
- Connor, J. A., and C. F. Stevens. 1971. Prediction of repetitive firing behaviour from voltage-clamp data on an isolated neurons soma. J. Physiol. 213:31–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lecea, L., E. Soriano, J. R. Criado, S. C. Steffensen, S. J. Henriksen, and J. G. Sutcliffe. 1994. Transcripts encoding a neural membrane CD26 peptidase-like protein are stimulated by synaptic activity. Mol. Brain Res. 25:286–296. [DOI] [PubMed] [Google Scholar]
- Dixon, J. E., W. Shi, H. S. Wang, C. McDonald, H. Yu, R. S. Wymore, I. S. Cohen, and D. McKinnon. 1996. Role of the Kv4.3 K+ channel in ventricular muscle. A molecular correlate for the transient outward current. Circ. Res. 79:659–668. [DOI] [PubMed] [Google Scholar]
- Fan, H., W. Meng, C. Kilian, S. Gram, and W. Reutter. 1997. Domain-specific N-glycosylation of the membrane glycoprotein dipeptidylpeptidase IV (CD26) influences its subcellular trafficking, biological stability, enzymatic activity, and protein folding. Eur. J. Biochem. 246:243–251. [DOI] [PubMed] [Google Scholar]
- Fulop, V., Z. Bocskel, and L. Polgar. 1998. Prolyl oligopeptidase: an unusual _β_-propeller domain regulates proteolysis. Cell. 94:161–170. [DOI] [PubMed] [Google Scholar]
- Gebauer, M., D. Isbrandt, K. Sauter, B. Callsen, A. Nolting, O. Pongs, and R. Bähring. 2004. N-type inactivation features of Kv4.2 channel gating. Biophys. J. 86:210–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, W., H. Li, B. London, and J. M. Nerbonne. 2000. Functional consequences of elimination of _I_to,f and _I_to,s: early after depolarizations, atrioventricular block, and ventricular arrhythmias in mice lacking Kv1.4 and expressing a dominant-negative Kv4 α subunit. Circ. Res. 87:73–79. [DOI] [PubMed] [Google Scholar]
- Hille, B. 2001. Ion Channels of Excitable Membranes. Sinauer Associates, Sunderland, MA.
- Hiramatsu, H., K. Kyono, Y. Higashiyama, C. Fukushima, H. Shima, S. Sugiyama, K. Inaka, A. Yamamoto, and R. Shimizu. 2003. The structure and function of human dipeptidyl peptidase IV, possessing a unique eight-bladed _β_-propeller fold. Biochem. Biophys. Res. Comm. 302:849–854. [DOI] [PubMed] [Google Scholar]
- Hoffman, D. A., J. C. Magee, C. M. Colbert, and D. Johnston. 1997. K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature. 387:869–875. [DOI] [PubMed] [Google Scholar]
- Holmqvist, M. H., J. Cao, R. Hernandez-Pineda, M. D. Jacobson, K. I. Carroll, M. A. Sung, M. Betty, P. Ge, K. J. Gilbride, M. E. Brown, M. E. Jurman, D. Lawson, P. S. Distefano, and W. F. An. 2002. Elimination of fast inactivation in Kv4 A-type potassium channels by an auxiliary subunit domain. Proc. Natl. Acad. Sci. USA. 99:1035–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong, W., and D. Doyle. 1990. Molecular dissection of the NH2-terminal signal/anchor sequence of rat dipeptidyl peptidase IV. J. Cell. Biol. 111:323–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isbrandt, D., T. Leicher, R. Waldschutz, X. Zhu, U. Luhmann, U. Michel, K. Sauter, and O. Pongs. 2000. Gene structures and expression profiles of three human KCND (Kv4) potassium channels mediating A-type currents _I_TO and _I_SA. Genomics. 64:144–154. [DOI] [PubMed] [Google Scholar]
- Jerng, H. H., and M. Covarrubias. 1997. K+ channel inactivation mediated by the concerted action of the cytoplasmic N- and C-terminal domains. Biophys. J. 72:163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerng, H. H., M. Shahidullah, and M. Covarrubias. 1999. Inactivation gating of Kv4 potassium channels: molecular interactions involving the inner vestibule of the pore. J. Gen. Physiol. 113:641–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerng, H. H., and P. Pfaffinger. 2004. Dipeptidyl peptidase 10 upregulates surface expression and modulates biophysical properties of Kv4.2 channels. Biophys. J. 86:129a (Abstr.). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johns, D. C., H. B. Nuss, and E. Marban. 1997. Suppression of neuronal and cardiac transient outward currents by viral gene transfer of dominant-negative Kv4.2 constructs. J. Biol. Chem. 272:31598–31603. [DOI] [PubMed] [Google Scholar]
- Johnston, D., D. A. Hoffman, J. C. Magee, N. P. Poolos, S. Watanabe, C. M. Colbert, and M. Migliore. 2000. Dendritic potassium channels in hippocampal pyramidal neurons. J. Physiol. 525:75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, L. A., J. Furst, M. H. Butler, S. Xu, N. Grigorieff, and S. A. Goldstein. 2003. _I_to channels are octomeric complexes with four subunits of each Kv4.2 and K+ channel-interacting protein 2. J. Biol. Chem. 279:5549–5554. [DOI] [PubMed] [Google Scholar]
- Kin, Y., Y. Misumi, and Y. Ikehara. 2001. Biosynthesis and characterization of the brain-specific membrane protein DPPX, a dipeptidyl peptidase IV-related protein. J. Biochem. 129:289–295. [DOI] [PubMed] [Google Scholar]
- Kozak, M. 1989. The scanning model for translation: an update. J. Cell Biol. 108:229–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambeir, A., J. F. D. Pereira, P. Chacon, G. Vermeulen, K. Heremans, B. Devreese, J. Van Beeumen, I. De Meester, and S. Scharpe. 1997. A prediction of DPP IV/CD26 domain structure from a physio-chemical investigation of dipeptidyl peptidase (CD26) from human seminal plasma. Biochim. Biophys. Acta. 1340:215–226. [DOI] [PubMed] [Google Scholar]
- Lennarz, W. 1983. Overview: role of intracellular membrane systems in glycosylation of proteins. Methods Enzymol. 98:91–97. [DOI] [PubMed] [Google Scholar]
- Lilliehook, C., O. Bozdagi, J. Yao, M. Gomez-Ramirez, N. F. Zaidi, W. Wasco, S. Gandy, A. C. Santucci, V. Haroutunian, G. W. Huntley, and J. D. Buxbaum. 2003. Altered A_β_ formation and long-term potentiation in a calsenilin knock-out. J. Neurosci. 23:9097–9106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig, K., S. Yan, H. Fan, W. Reutter, and C. Bottcher. 2003. The 3D structure of rat DPPIV/CD26 as obtained by cryo-TEM and single particle analysis. Biochem. Biophys. Res. Commun. 304:73–77. [DOI] [PubMed] [Google Scholar]
- Malin, S. A., and J. M. Nerbonne. 2000. Elimination of the fast transient in superior cervical ganglion neurons with expression of Kv4.2W362F: molecular dissection of _I_A. J. Neurosci. 20:5191–5199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald, J. K., and C. Schwabe. 1977. Proteinases in Mammalian Cells and Tissues. North-Holland Publishing, Amsterdam, The Netherlands. 371–376.
- Mentlein, R. 1999. Dipeptidyl-peptidase IV (CD26)-role in the inactivation of regulatory peptides. Regul. Pept. 85:9–24. [DOI] [PubMed] [Google Scholar]
- Nadal, M. S., A. Ozaita, Y. Amarillo, E. Vega-Saenz de Miera, Y.E. Vega-Saenz de Miera, Y. Ma., W. Mo, E. M. Goldberg, Y. Misumi, Y. Ikehara, T. A. Neubert, and B. Rudy. 2003. The CD26-related dipeptidyl aminopeptidase-like protein DPPX is a critical component of neuronal A-type K+ channel. Neuron. 37:449–461. [DOI] [PubMed] [Google Scholar]
- Oefner, C., A. D'Arcy, A. Mac Sweeney, S. Pierau, R. Gardiner, and G. E. Dale. 2003. High-resolution structure of human apo dipeptidyl peptidase IV/CD26 and its complex with 1-[({2-[5-iodopyridin-2-yl)amino]-ethyl}amino)-acetyl]-2-cyano-(S)-pyrrolidine. Acta Crystallogr. 59:1206–1212. [DOI] [PubMed] [Google Scholar]
- Pak, M. D., K. Baker, M. Covarrubias, A. Butler, A. Ratcliffe, and L. Salkoff. 1991. mShal, a subfamily of A-type K+ channel cloned from mammalian brain. Proc. Natl. Acad. Sci. USA. 88:4386–4390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi, S. Y., P. J. Riviere, J. Trojnar, J.-L. Junien, and K. O. Akinsanya. 2003. Cloning and characterization of dipeptidyl peptidase 10, a new member of an emerging subgroup of serine proteases. Biochem. J. 373:179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen, H. B., S. Branner, F. C. Wiberg, and N. Wagtmann. 2002. Crystal structure of human dipeptidyl peptidase IV/CD26 in complex with a substrate analog. Nature Struct. Biol. 10:19–25. [DOI] [PubMed] [Google Scholar]
- Roberts, C. J., G. Pohlig, J. H. Rothman, and T. H. Stevens. 1989. Structure, biosynthesis, and localization of dipeptidyl aminopeptidase B, an integral membrane glycoprotein of the yeast vacuole. J. Cell. Biol. 108:1363–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha, C. A., M. Nadal, B. Rudy, and M. Covarrubias. 2004. Inactivation gating of Kv4 K+ channels interacting with the dipeptidyl-aminopeptidase-like protein (DPPX). Biophys. J. 86:536a (Abstr.). [Google Scholar]
- Rudy, B., J. H. Hoger, H. A. Lester, and N. Davidson. 1988. At least two mRNA species contribute to the properties of rat brain A-type potassium channels expressed in Xenopus oocytes. Neuron. 1:649–658. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular Cloning. A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Serodio, P., C. Kentros, and B. Rudy. 1994. Identification of molecular components of A-type channels activating at subthreshold potentials. J. Neurophysiol. 79:1081–1091. [DOI] [PubMed] [Google Scholar]
- Serodio, P., E. Vega-Saenz de Miera, and B. Rudy. 1996. Cloning of a novel component of A-type K+ channels operating at subthreshold potentials with unique expression in heart and brain. J. Neurophysiol. 75:2174–2179. [DOI] [PubMed] [Google Scholar]
- Serodio, P., and B. Rudy. 1998. Differential expression of Kv4 K+ channel subunits mediating subthreshold transient K+ (A-type) current in rat brain. J. Neurophysiol. 79:1081–1091. [DOI] [PubMed] [Google Scholar]
- Sheng, M., M. L. Tsaur, Y. N. Jan, and L. Y. Jan. 1992. Subcellular segregation of two A-type K+ channel proteins in rat central neurons. Neuron. 9:271–284. [DOI] [PubMed] [Google Scholar]
- Shibata, R., K. Nakahira, K. Shabasaki, Y. Wakazono, K. Imoto, and K. Ikenaka. 2000. A-type K+ current mediated by the Kv4 channel regulates the generation of action potential in developing cerebellar granule cells. J. Neurosci. 20:4145–4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin, K. S., C. Maertens, C. Proenza, B. S. Rothberg, and G. Yellen. 2004. Inactivation in HCN channels results from reclosure of the activation gate; desensitization to voltage. Neuron. 41:737–744. [DOI] [PubMed] [Google Scholar]
- Wada, K., N. Yokotani, C. Hunter, K. Doi, R. J. Wenthold, and S. Shimasaki. 1992. Differential expression of two distinct forms of mRNA encoding members of a dipeptidyl aminopeptidase family. Proc. Natl. Acad. Sci. USA. 89:197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe, I., H.-G. Wang, J. Sutachan, J. Zhu, E. Recio-Pinto, and W. B. Thornhill. 2003. Glycosylation affects rat Kv1.1 potassium channel gating by a combined surface potential and cooperative subunit interaction mechanism. J. Physiol. 550:51–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe, S., D. A. Hoffman, M. Migliore, and D. Johnston. 2002. Dendritic K+ channels contribute to spike-timing dependent long-term potentiation in hippocampal pyramidal neurons. Proc. Natl. Acad. Sci. USA. 99:8366–8371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, H., H. Li, and J. M. Nerbonne. 1999. Elimination of the transient outward current and action potential prolongation in mouse atrial myocytes expressing a dominant negative Kv4 α subunit. J. Physiol. 519:11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, W., Y. Qian, K. Kunjilwar, P. J. Pfaffinger, and S. Choe. 2004. Structural insights into the functional interaction of KChIP1 with Shal-Type K+ channels. Neuron. 41:573–586. [DOI] [PubMed] [Google Scholar]