A Dual Role of Cyclin E in Cell Proliferation and Apotosis May Provide a Target for Cancer Therapy (original) (raw)
. Author manuscript; available in PMC: 2005 Dec 6.
Published in final edited form as: Curr Cancer Drug Targets. 2004 Feb;4(1):65–75. doi: 10.2174/1568009043481669
Abstract
Cyclin E is essential for progression through the G1-phase of the cell cycle and initiation of DNA replication by interacting with and activating its catalytic partner, the cyclin dependent kinase 2 (Cdk2). Rb, as well as Cdc6, NPAT, and nucleophosmin, critical components of cell proliferation and DNA replication, respectively, are targets of Cyclin E/Cdk2 phosphorylation. There are a number of putative binding sites for E2F in the cyclin E promoter region, suggesting an E2F-dependent regulation. Skp2 and Fbw7 are novel proteins, responsible for ubiquitin-dependent proteolysis of Cyclin E. The tight regulation of cyclin E expression, both at the transcriptional level and by ubiquitin-mediated proteolysis, indicates that it has a major role in the control of the G1- and S-phase transitions. Cyclin E is also transcriptionally regulated during radiation-induced apoptosis of hematopoietic cells. In addition to its biological roles, deregulated cyclin E expression has an established role in tumorigenesis. Cell cycle regulatory molecules, such as cyclin E, are frequently deregulated in different types of cancers, where overexpressed native or low molecular weight forms of Cyclin E have a significant role in oncogenesis. During apoptosis of hematopoietic cells, caspase-dependent proteolysis of Cyclin E generates a p18-Cyclin E variant. Understanding the role of Cyclin E in apoptosis may provide a novel target, which may be effective in cancer therapy. This review summarizes what is known about the biological role of cyclin E, its deregulation in cancer, and the opportunities it may provide as a target in clinical therapy.

ABBREVIATIONS: CDK = Cyclin-dependent kinases, CKI = CDK inhibitors, IR = Ionizing radiation, LMW = Low molecular weight, T380 = Threonine 380
INTRODUCTION
Cyclins bind to their catalytic partners, the cyclin-dependent kinases (CDKs), to control cell cycle progression by regulating the transitions between cell cycle phases [1–3]. The Cyclin/Cdk interaction is achieved through the cyclin box domain, which contains a conserved amino acid sequence [4, 5]. The activity of these CDKs is regulated positively by cyclins and negatively by binding of CDK inhibitors (CKIs) [6, 7]. The activation of Cyclin/Cdk complexes results in a cascade of phosphorylation of proteins that induces cell cycle progression. The human G1 cyclins, the D- and E-type cyclins, were identified functionally by screening of human cDNA libraries for sequences that could complement G1 cyclin mutations in Saccharomyces cerevisiae [8, 9]. The human Cyclin E protein consists of 395 amino acids and has an approximate molecular weight of 50 kDa [8, 9]. The cyclin E mRNA levels show a periodic pattern of expression, being synthesized during the G1-phase of the cell cycle, with levels increasing sharply in late G1, followed by accumulation of Cyclin E protein and then down regulation in S-phase, emphasizing cyclin E’s key role in the maintenance of the restriction point [9–11]; (reviewed in [12, 13]).
Recently, a novel cyclin was discovered by three independent research groups [14–16]. The encoded protein was shown to have the characteristic cyclin box motif and displayed homology to Cyclin E, and thus was named Cyclin E2. Cyclin E2 was discovered through a search of rat and murine expressed sequence tag (EST) databases using a cyclin fold profile [14], and through yeast two-hybrid assays using Cdk2 [16] and p27/Kip1 [15] proteins as bait. Since Cyclin E2 is a relatively new cyclin, little is known about its function. The full length cyclin E2 cDNA sequence encodes a 404 amino acid protein with a predicted molecular weight of 47 kDa. The protein shares 75% homology to the cyclin box of Cyclin E, with an overall homology of 47% (Fig. 1). It forms a catalytically active kinase complex with Cdk2 and has been shown to phosphorylate both histone H1 and pRb. The Cyclin E2 associated kinase levels rise as cells approach S-phase. This is due to the increase in Cdk2 kinase activity resulting from phosphorylation and activation by the Cdk activating kinase and the decline of CKI levels. The levels then fall gradually as cells pass through S-phase [14–17].
Fig. (1). Cyclin E1, E2, and their functional domains.
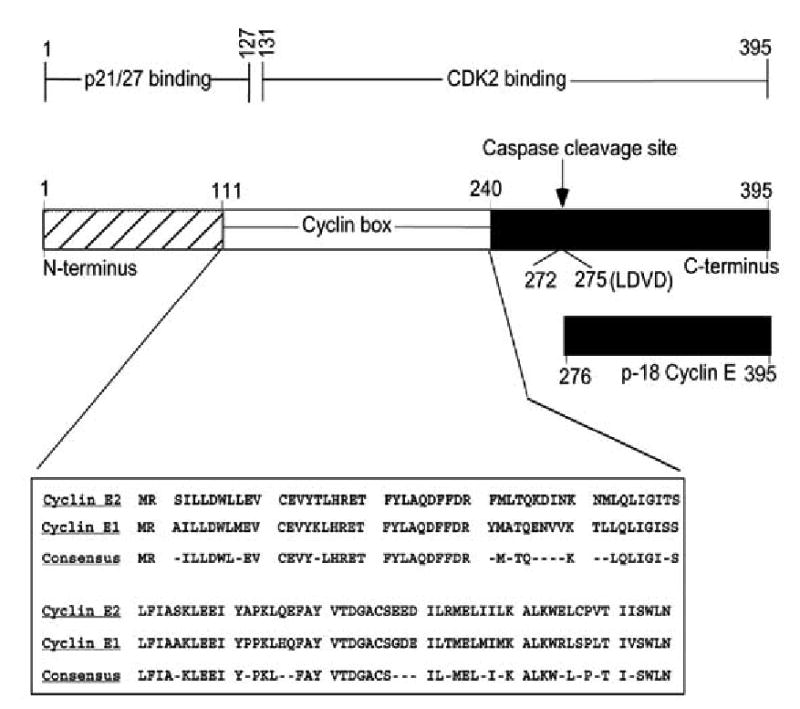
The functional domains through which Cyclin E1 interacts with p21, p27, and Cdk2 are shown as suggested [116]. Cdk2 interacts with most of the cyclin box (amino acid residues 111 to 240) and the C-terminus of cyclin E (amino acid residues 131 to 395) The entire N-terminus and partially the cyclin box of cyclin E (amino acid residues 1 to 127) are responsible for p21 and p27 binding The position of the caspases cleavage site (LDVD) and the resulting p-18 cyclin E fragment are indicated. The amino acid homology between cyclin E1 and cyclin E2, in their cyclin box, is shown below.
Cyclin E2 has the same ability as Cyclin E to rescue a lethal S. cerevisiae triple CLN mutant, demonstrating its ability to bind and activate Cdc28 in yeast [14]. Ectopic expression of Cyclin E2 increases the percentage of cells in S-phase, as does Cyclin E, due to premature phosphorylation events [14, 17]. Since Cyclin E2 shares so much similarity with Cyclin E, in terms of homology and Cdk2-associated kinase activity, it has been suggested that their functions are also similar.
Ectopic and constitutive overexpression of Cyclin E, but not of Cyclin D1 or A, can lead to accelerated G1 progression [18–20] and chromosome instability in certain types of cells [21]. Moreover, overexpression and Cyclin E activation result in redundancy of functional Cyclin E/Cdk2 in breast tumor cells. Constitutively expressed Cyclin E may be the major cyclin which complexes with p107 and E2F throughout the cell cycle of cancer cells [22]. All phenotypic observations of Cyclin D1 deficiency can also be rescued, and normal development in Cyclin D1-dependent tissues are restored in mice. This demonstrates that Cyclin E can functionally replace Cyclin D1, and it suggests that Cyclin E is the major downstream target of Cyclin D1 [23]. While Cyclin E complexes phosphorylate the protein product of the retinoblastoma tumor suppressor gene, pRb, which is also phosphorylated by the D-type Cyclin/Cdk4/6 complexes [24], it is likely that other substrates exist during late G1 (Table 1). Moreover, this Cyclin E/Cdk2 catalytic complex is capable of phosphorylating pRb when Cyclin/D/Cdk4/Cdk6 complexes become inactivated by p16 overexpression [22]. Unlike cyclin D, cyclin E remains essential in the absence of pRb, since: (i) its inducible expression in fibroblasts accelerates G1/S progression without affecting the kinetics of pRb phosphorylation [25]; (ii) unlike the D-type cyclins, cyclin E is essential for cell-cycle progression in pRb-deficient cells [19]; (iii) ectopic expression of cyclin E bypasses pRb-mediated cell cycle arrest [26, 27]; and (iv) cyclin E is required for S-phase entry in Drosophila [28]. These findings highlight a fundamental difference between the Cyclin D and Cyclin E complexes, strongly suggesting that other key rate-limiting substrates exist for Cyclin E/Cdk2 [26, 27].
Table 1.
Interacting Partners of Cyclin E and their Biological Role
| Interacting Proteins | Biological Function | Ref. |
|---|---|---|
| Id2, Id3 | Modulate transcription factors | [52][51] |
| Cdc25A | Activates Cyclin E/Cdk2 | [63] |
| NPAT | Synthesis of histones | [36] |
| Nucleophosmin, B23 | Centrosome duplication | [37][38] |
| PCNA | Modulates DNA replication | [114] |
| P21, p27 | Inhibitors of Cyc E/Cdk2 activity | [6, 7] |
| CDC6 | Initiation of DNA replication | [33] |
| CDK2 | Catalytic partner of Cyclin E1/E2 | [115] |
| SAP155/SF3B1 | Processing of pre-mRNA splicing | [54][53] |
| BAF155, SW1/SNF | Remodeling of the chromatin complex (transcriptional regulation) | [43] |
Role of Cyclin E in DNA Replication
A number of biological functions have been attributed to Cyclin E, based mainly on its interaction with and phosphorylation of these substrates (Table 1). Thus, Cyclin E/Cdk2 complexes have been shown to play a key and essential role in the initiation of DNA replication [29], in addition to the cell cycle transition [19, 20]. The proteins that regulate the replication machinery, such as the origin recognition complex (ORC), are highly conserved from yeast to humans and are necessary to recruit a prereplication protein, Cdc6, and the minichromosome maintenance complex (MCM) to chromatin [30]. After the recruitment of the MCM complex, Cdc6 is removed from replication origins [31, 32]. In Xenopus, Cyclin E/Cdk2 binds chromatin by its direct association with the amino-terminus of Cdc6. This interaction is required for the initiation of DNA replication [33]. As replication proceeds, Cyclin E persists on chromatin, potentially explaining its ability to block re-replication. In mitosis, the Cyclin E-chromatin association is abrogated by Cyclin E phosphorylation and then re-established upon exit from mitosis by dephosphorylation, allowing initiation of a new round of DNA replication [33]. In the Xenopus system, Cyclin E/Cdk2 binds chromatin as a ternary complex with the Cdk2 inhibitor p27/Xic1, a relative family member of the human CDK inhibitors p27/Kip1 and p21/Waf1 [34]. NPAT is also the substrate of Cyclin E/Cdk2 both in vitro and in vivo. Its levels peak at the G1/S boundary and, when overexpressed, it accelerates S-phase entry [35]. The control of NPAT by Cyclin E/Cdk2 allows the induction of histone gene transcription at the beginning of S-phase [35, 36]. The cell cycle regulated phosphorylation of p220/NPAT by Cyclin E/Cdk2 provides strong evidence for the direct role of cyclin E in DNA replication. Centrosome duplication in Xenopus egg extracts was shown to also require the activity of Cyclin E/Cdk2. Cyclin E/Cdk2-specific phosphorylation of nucleophosmin causes it to dissociate from the centrosome, thereby allowing centriole disorientation and splitting to occur [37]. The MMps1p-like kinase is another substrate of Cyclin E/Cdk2, required to regulate centrosome duplication. This kinase is unstable if Cdk2 is inhibited [38].
Constitutive activation of Cyclin E/Cdk2 results in uncoupling of the initiation of centrosome and DNA duplication leading to unscheduled initiation of centrosome duplication prior to S-phase entry [39]. Cyclin E overexpression, together with loss of p53 gene, increases the frequency of centrosome hyperamplification synergistically in tumors of p53 null-mice, as well as in tissue culture cells. In a structural analysis of some human breast tumors, the centrosomes of most tumors were found to have significant changes compared to that of normal breast tissue [40]. These studies indicate that deregulation of Cyclin E could lead to genomic instability in human cancers, at least, due to its key regulatory role in correct and timely centrosome duplication.
Transcriptional Regulation of Cyclin E
The cyclin E promoter contains several putative binding sites for the E2F transcription factor. A variant E2F binding site is a cyclin E repressor module responsible for the periodic down-regulation of the cyclin E promoter until the growing cells have reached the late G1- phase [41]. This site induces transcriptional repression by binding to a large complex, containing E2F4, DP1, and a pocket protein, which functions to delay the expression of cyclin E until late G1 [42]. pRb forms a repressor complex with a histone deacetylase (HDAC) and the SNF2-like (BRG1 and hbrm) component of the mammalian hSW1/SNF nucleosome remodeling complex, starting from the end of S-phase until late G1 [43]. The components of the mammalian hSW1-SNF complex (BAF155 and BRG1) interact with Cyclin E and modulate the ability of BRG1 to induce growth arrest [44]. The SW1-SNF complexes have an important role in transcriptional regulation, altering the chromatin structure by relieving the transcription from the nucleosome-mediated repression, thereby, opening access to the activators of transcription. The phosphorylation of pRb by Cyclin D/Cdk4 abrogates its interaction with HDAC and transactivates cyclin E and, thereby, overcomes the G1 arrest [43]. Transcription of cyclin A and cdk1 is repressed by the complex containing pRb and hSW1/SNF. Cyclin E/Cdk2 can phosphorylate pRB or the hSW1/SNF component after Cyclin E reaches a certain level, when the interaction of pRB-hSW1/SNF is disrupted. Deregulation of any of the components of this transcriptional complex could lead to the unscheduled expression of cyclin E, which is very common in cancer cells. We have previously shown that constitutive levels of several E2F1 target genes are increased in Rb-deficient fibroblasts [45]. Similarly, Cyclin E levels are increased constitutively when Rb has been inactivated by the HPV16-E7 oncogene expression in human foreskin fibroblasts [46].
Also, Cyclin E/Cdk2 kinase levels are induced following genotoxic stress in human colon cancer cells by: (i) fluoropyramidines in HT29 but not SW620 cells [47]; or (ii) low concentrations of actinomycin D in HPV16-E7 expressing RKO cells [48]. Upon initial characterizations of Cyclin E2, it has been shown that there may be a strong correlation between the functional inactivation of pRb and elevated levels of Cyclin E2 in tumor-derived cell lines and primary MEFs lacking pRb [14, 49, 50]. The Id family members and SAP155 represent additional substrates of Cyclin E/Cdk2 (Table 1). Ids negatively regulate the basic-helix-loop-helix (bHLH) transcription factors through the formation of heterodimers that cannot bind DNA. The phosphorylation of Ids at serine residue by Cyclin E/Cdk2 nullifies the negative regulation by Ids [51, 52]. Moreover, Cyclin E precipitates U2snRNA and the spliceosome through the specific interaction of SAP155. Possibly, cyclin E regulates the pre-mRNA processing, mediating the interaction with SAP155 [53, 54].
The above reports suggest that the major transcription factor responsible for cyclin E induction is E2F. However, E2F1 has recently been reported to be upregulated in response to DNA damage in a manner analogous to p53, with its levels being unchanged following DNA damage of epithelial cells with mutated p53 [55]. In contrast, we found that cyclin E was upregulated in cells with mutated p53 expressed either endogenously (RPMI-8226 myeloma cells) or following its stable transfection [56]. Northern and run-on analyses have indicated that the cyclin E levels induced by radiation result mostly from transcriptional regulation. Cyclin E induction is not restricted to radiation and the hematopoietic cell lines we have investigated. Cyclin E levels were also increased following treatment with chemotherapeutic agents, such as the topoisomerase I inhibitor VP16 [56]. In addition, we recently found that cyclin E is also upregulated in other cell types, such as those of prostate and epithelial cell types. Interestingly, we have seen that, in prostate cancer cells, the regulation of Cyclin E2 expression following radiation differs from that of Cyclin E (DuPree, E., Mazumder, S., Hissong, J, and Almasan A., unpublished). Although further work is required to establish whether E2F plays a regulatory role in radiation-induced cyclin E expression, it is quite possible that cyclin E is regulated differently by mitogens and radiation.
Post-Transcriptional Regulation of Cyclin E
Cyclin E periodicity is not only determined at the transcriptional level, but it is also maintained by post-translational regulation through ubiquitin-dependent proteolysis [57, 58]. In normal proliferating cells, Cyclin E has a short half-life of less than 30 min, but this can be extended to more than 2 hrs by the addition of pharmacologic inhibitors of the proteasome. Turnover of Cyclin E by this proteasome pathway is regulated and modulated by the binding of Cyclin E to Cdk2 and also by site-specific auto-phosphorylation of Cyclin E. The unbound Cyclin E is degraded by the proteasome, and binding to Cdk2 protects it from degradation. Cyclin E/Cdk2 activity reverses the stabilizing effect of complex assembly. By site-directed mutagenesis, it has been shown that the threonine (T380A) mutation of Cyclin E prevents it from ubiquitin-mediated proteolytic degradation. Skp2 was thought to be the F-box protein responsible for Cyclin E degradation since skp2 gene inactivation by homologous recombination results in p27 and Cyclin E accumulation [59, 60]. Since p27 is the predominant substrate of Skp2, its accumulation leads indirectly to the inhibition of Cyclin E/Cdk2 kinase activity and autophosphorylation of Cyclin E on T380, preventing its recognition by the Cyclin E-specific F-box protein. Another protein, Fbw7, has been discovered recently and shown to specifically target ubiquitin-mediated proteolysis of Cyclin E [61, 62]. The interaction of Cyclin E with this F-box protein depends on Cyclin E phosphorylation at T380 and T62. Interestingly, some tumor cell lines that have high levels of cyclin E also have mutations in the Fbw7 gene or express low levels of its mRNA, suggesting that Fbw7 may function as a tumor suppressor. Cross-talk between the regulation of cyclin E transcription and that of Cyclin E protein stability results in fine tuning of Cyclin E levels, emphasizing its crucial role in cell cycle regulation and predicting deleterious effects of a constitutively high level of expression of cyclin E as seen in many cancer cells. In contrast, CDC25A, an upstream regulator of Cyclin E, is a phosphatase that removes the inhibitory phosphate group from Cdk2 during the G1- to S-phase transition [63], and its expression is regulated by E2F and other transcription factors. In normal cells, CDC25A is negatively regulated by radiation through checkpoint-mediated ubiquitination. In checkpoint deficient cells, CDC25A is overexpressed, leading to radioresistant DNA synthesis [64]. Cyclin E2, like Cyclin E, is highly unstable and subject to proteolysis. A proteasome inhibitor, LLnL, has been shown to increase the levels of Cyclin E2 [15–17]. Also, Cyclin E2 complex formation stabilizes the protein, increasing its expression level. This can be observed through co-transfection with either Cdk2 or p27/Kip1 [14, 15, 17]. A several-fold increase in Cyclin E2 levels can be caused by mutating its Cdk2 phosphorylation site [16, 17].
Cell Cycle Regulators in Apoptosis
Apoptosis, a universal genetic program of cell death in higher eukaryotes, is a basic process involved in cellular development and differentiation [65]. Apoptosis may be essential for the prevention of tumor formation, and its deregulation is widely believed to be involved in pathogenesis of many human diseases, including cancer (reviewed in [66]). In almost all instances, deregulated cell proliferation and suppressed cell death together provide the underlying platform for neoplastic progression [67]. In cancer, called a disease of deregulated cell proliferation [68], one principal target is the late G1 cell cycle regulated by pRb [69]. Defects in this pathway, which may be universal in human cancer, include deletions of the Rb gene itself and deregulation of the CDKs that phosphorylate and functionally inactivate pRb, either through direct overactivation of CDKs or through genetic loss of their inhibitors [68]. We have shown that absence of pRb is capable of activating an apoptotic response associated with p53 stabilization and that increased expression of its target genes, such as cyclin E, is dependent on E2F transcription. Moreover, we have shown that Cyclin E levels are also regulated by genotoxic stress and that Cyclin E activation plays a functional role in apoptosis of hematopoietic cells [56].
The activation of caspases is a common and critical regulator of the execution phase of apoptosis, triggered by many factors, including genotoxic agents, such as γ-irradiation or treatment with anti-cancer agents [70–72]. Once cells are committed to cell death, apoptogenic factors, the best known of which is cytochrome c, are released from mitochondria to initiate the caspase cascade [71, 73]. Cytochrome c acts as a cofactor to stimulate the complexing of Apaf-1 with Caspase-9 [74], which then initiates activation of the caspases cascade. Caspases are synthesized as inactive precursors, which are activated by proteolytic cleavage to generate active enzymes. They further proteolytically cleave proteins crucial for the maintenance of the cellular cytoskeleton, DNA repair, signal transduction, and cell cycle control. There are many in vivo caspase substrates, including transcription factors, kinases, enzymes involved in DNA repair, and cytoskeletal proteins [reviewed in [75]]. In addition, several proteins essential for cell cycle regulation, such as pRb [76, 77], MDM2 [78], PITSLRE [79], p21/Waf1/Cip1, and p27/Kip1 [80], are also caspase targets.
Apoptotic targets of CDKs are predicted, given reports of an interplay between the cell cycle control processes and apoptosis [81, 82], and that the apoptosis regulatory proteins themselves can directly impinge on the cell cycle machinery [83–86]. Clearly, induction of apoptosis by various stimuli has been shown to require activation of Cdk2 [87, 88], whereas forced expression of CKIs in cultured cells [56], [88], neurons [89], or during myocyte differentiation [90] prevents apoptosis. Importantly, similar observations were made in noncycling developing thymocytes, in which Cdk2 was activated; conversely, Cdk2 inhibition abrogates apoptosis [91]. Moreover, conditional cyclin A expression was reported to be involved in apoptosis of fibroblasts [92], whereas ectopic expression of cyclin D1 in rat fibroblasts under serum starvation lead to apoptotic cell death [93].
Our studies have revealed that cyclin E plays an important role in apoptosis of hematopoietic cells, in addition to its reported key regulatory role in the control of the G1- to S-phase transition and the initiation of DNA replication. Based on our studies [56, 94], we propose a dual role for cyclin E in apoptosis of hematopoietic cells (Fig. (2)). Initially, we found a substantial induction of cyclin E mRNA, accompanied by increased production of Cyclin E protein and Cyclin E/Cdk2 kinase activity in multiple myeloma and lymphoma cells following radiation. This increase of Cyclin E levels might be implicated in the initiation phase of apoptosis. Consistent with a role of cyclin E in apoptosis, its overexpression in hematopoietic cells greatly sensitizes these cells to radiation, while its inhibition by a dominant-negative Cdk2 blocks cell death [56].
Fig. (2). Regulation and role of of cyclin E in cell cycle control.

The orderly progression of cells is controlled by sequential association of G1-cyclins (D and E) to their catalytic subunits, the cyclin-dependent kinases (Cdk4, Cdk6, and Cdk2). Following genotoxic stress, p53 is activated leading to increased levels of its transcriptional targets, such as p21 [117]. p21 then binds and inhibits the Cyclin/Cdk complexes. In cells of hematopoietic origin, and at least in some epithelial cells, Cyclin E levels are also increased by genotoxic stress agents The retinoblastoma protein (pRb), present in its unphosphorylated form, is bound to the transcription factor E2F before cells enter S-phase. pRb is phosphorylated by Cyclin E/Cdk2 and, thereby, E2F is released, leading to transactivation of genes essential for cell cycle progression, such as dihydrofolate reductase (DHFR), thymidylate synthase (TS) [118] and possibly cyclin E. Additional Cyclin E targets are shown in Table 1. The effects of Cyclin E can be prevented by expression of a dominant-negative Cdk2 [56] or pharmacologic inhibitors, such as flavopiridol.
Recently, we found that the native p50-Cyclin E is proteolytically cleaved and thus converted to a p18 C-terminal Cyclin E fragment, which becomes the most abundant form of Cyclin E during the course of apoptosis, induced by radiation and chemotheraputic agents, such as VP16. Cyclin E cleavage results in abrogation of its binding to Cdk2 and, therefore, inactivation of its associated kinase activity and cell cycle function [94]. The conversion of Cyclin E from a p50- to a p18-fragment may be a general process, as it is produced in all hematopoietic tumor cell lines we have examined and following treatment with multiple genotoxic stress agents which trigger apoptosis. The p18-Cyclin E, generated at a later stage by caspase-mediated proteolytic cleavage, might participate, directly or indirectly, in the amplification of the apoptotic response initiated by caspase activation. Overexpression of Bcl-2 not only prevents apoptosis but also completely inhibits the expression of p18-Cyclin E, indicating that p18-Cyclin E is directly associated with apoptosis. Cyclin E is inactivated by proteolytic cleavage, as the p18-Cyclin E, generated during apoptosis through a more severe truncation of Cyclin E, can no longer bind Cdk2 and thus is lacking any associated active kinase activity. Thereby, caspase-dependent proteolytic cleavage is an additional mechanism used by hematopoietic tumor cells to regulate the cellular functions of Cyclin E during apoptosis. Our recent examination of potential interacting partners of this Cyclin E fragment has revealed an unexpected connection to DNA repair. Thus we have identified binding of p18-Cyclin E to Ku70, a critical component in Non-homologous End-joining Repair (NHEJ), which represents the major DNA-damage repair pathway following genotoxic stress, such as ionizing radiation. Cyclin E interaction with Ku70 may inactivate the DNA repair machinery in cells undergoing apoptosis (Fig. (3)). Augmenting apoptotic responses and minimizing DNA repair in tumor cells, while at the same time facilitating effective DNA repair in normal cells, is the ultimate goal in clinical therapy. Cyclin E may be unique amongst most proteins in accomplishing all these tasks.
Fig. (3). Role of cyclin E in apoptosis.
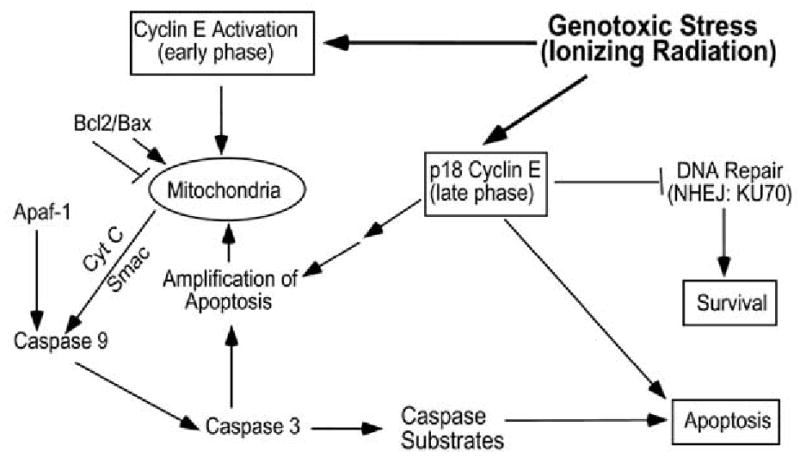
Following genotoxic stress, such as ionizing radiation, increased levels of cyclin E and the associated kinase activity are responsible for phosphorylating a critical cellular target implicated in apoptosis. Such a target may be associated with mitochondria, the critical cellular site for activation of apoptosis. Mitochondria releases apoptogenic factors, such as cytochrome C and Smac. Cytochrome C, in the presence of ATP and the adaptor molecule Apaf-1, recruits and activates caspase 9 within the apoptosome [119], leading to further activation of the effector caspases, such as caspase 3. Activated caspase 3 can function to activate a feed-back loop, leading to further cytochrome c release and amplification of the apoptosis process [71]. During the late stages of apoptosis, a truncated Cyclin E is generated through its caspase-mediated proteolytic cleavage. The resulting p18-Cyclin E may trigger apoptosis by amplifying the mitochondrial death process. It may also block cell survival by preventing the DNA repair process, since p18-Cyclin E can bind to Ku70, a critical component in non-homologous end-joining (NHEJ), the major pathway for DNA repair following genotoxic stress.
Deregulated Cyclin E Expression in Cancer: a Target for Therapy?
Normal cell proliferation is under strict regulation, governed by checkpoints located at distinct positions in the cell cycle. The deregulation of these checkpoint events and the molecules associated with them may transform a normal cell into a cancer cell. One of these checkpoints, whose deregulation results in transformation, occurs at the G1/S boundary. The G1/S transition is regulated by the Restriction point (R point), which is a point of no return, such that, if the cell passes that point, it no longer needs mitogens and is committed to completion of DNA replication and the cell cycle [95]. Any type of deregulation of the G1/S transition, along with the disappearance of the R-point, is a hallmark of cancer, leading to uncontrolled cell proliferation. The periodic appearance of cyclin E coincides precisely with the timing of the R point. In contrast to normal cells, mitotic cyclins appear prior to G1 cyclins in tumor cells [96]. Another report, however, suggests instead the converse, that passage through R point is a prerequisite for cyclin E accumulation [5]. This study found that the postmitotic G(1) cells that had not yet reached R were negative for cyclin E accumulation, while cells that had passed R accumulated cyclin E at variable times (1 to 8 h) after passage through R and 2 to 5 h before entry into S.
As these cyclins and CDKs are important components of cell cycle control and cell proliferation, alterations or mutational changes in the expression of the corresponding genes leads to oncogenesis [97], a defective or abnormal regulation of the cell cycle that is a main cause of human cancer [68, 95]. Recent reports support a significant relationship between dysregulation of cyclin E and progression of cancer. Cyclin E has been shown to be deregulated and overexpressed in several solid tumors, including breast, colon, and prostate carcinomas [68, 98–100] (reviewed in [13]). Overexpression of Cyclin E results in accelerated G1 progression and chromosome instability [18–20].
The most common chemotherapeutic agents used to prevent the growth of cancer cells are also toxic to normal cells. In order to identify a novel cell cycle target that would selectively target the cancer cells, the challenging strategies could be the determination of the expression pattern as well as the mechanism of the target in normal versus tumor cells, and thereby, therapeutic application of that target could be achieved. Pharmacologic inhibitors of the CDKs, such as flavopiridol and UCN-01, are currently in clinical trials. While demonstrating clinical activity, neither acts specifically only against Cdk2. Other more specific Cdk2 inhibitors are currently in preclinical development [101–104]. In some tumors, cyclin E gene amplification and protein accumulation are late events, whereas in other neoplasms, the increase of cyclin E is observed during the early stages of malignancy. Even though some mutations have been detected, accumulation of Cyclin E protein mostly reflects amplification of the gene. In addition, cyclin E is sometimes modified by posttranscriptional mechanisms so that increased protein expression does not reflect the mutations in the cyclin E gene. The question of whether cyclin E is merely a link in the chain of events that leads to cell proliferation or it is the driving force for cell replication is difficult to ascertain, as it may be tumor dependent.
Immunolabeling has localized Cyclin E to the nucleus in the majority of the neoplasms studied. Although the protein is both synthesized and degraded in the cytoplasm, it is translocated to the nucleus, where it exerts its functions. In the nucleus, the nascent Cyclin E associates with its Cdk2 partner, activating its serine-threonine kinase activity shortly before entry into S-phase [11, 105]. Accumulation of Cyclin E in the cytoplasm either reflects increased synthesis, decreased degradation, or failure of transport to the nucleus. Loss of p27 expression and overexpression of Cyclin E or Cdk2 are significantly associated with malignancy in ovarian cancers. Patients with high cyclin E overexpression have a lower survival rate, with those having the p27(−)/cyclin E(++)/cdk2(++) phenotype being significantly associated with a poor overall survival. This study concluded that low p27 and high Cyclin E expression may be a valuable marker for patients with ovarian tumor. Immunohistochemical studies on 105 newly diagnosed lymphomas showed a correlation between p27 and Cyclin E expression. Low p27 and high Cyclin E expression were significantly associated with a poor prognosis [106]. One of the malignant changes associated with increasing levels of cyclin E is the morphological change from normal breast tissue, through ductal carcinoma in situ (DCIS), to invasive ductal carcinoma. There are many more cyclin E-positive cells in high grade DCIS than in those of low grade. In invasive breast cancer, cyclin E was found to be disproportionately expressed compared to other cell proliferation markers, indicating that cyclin E deregulation is the contributor to, rather than a consequence of, increased cell division [98, 107]. Moreover, poor prognosis has been associated with cyclin E expression in node-negative patients, although a relation with negative estrogen receptors may be a confounding variable in these reports [108].
Splice variants that have structural or functional abnormalities in the cyclin box have also been identified. There is considerable evidence supporting a link between cyclin E dysregulation and tumorigenesis. Particularly, in nearly all breast cancer cell lines and patient tumors, the biochemically hyperactive low molecular weight (LMW) forms of Cyclin E are very common. These LMW proteins can efficiently bind to Cdk2, with the bound Cdk2 complexes having active kinase activity [98, 109, 110]. In fact, there is an increased level of kinase activity in those cells, which leads to faster cell cycle progression and proliferation [111, 112]. These quantitative and qualitative alterations in cyclin E expression become more prominent with advanced stage breast cancer [98]. A recent large-scale study has found that a high Cyclin E total level or high levels of the LMW forms of Cyclin E were significantly correlated with poor outcome [100]. The hazard ratio for death from breast cancer for patients with high Cyclin E levels, as compared to those with low total Cyclin E levels, was about 8-fold higher than the hazard ratios associated with other independent clinical and pathological risk factors. Levels of total Cyclin E and LMW Cyclin E in these tumor tissues, correlated strongly with survival in these patients [100].
There can be significant changes of Cyclin E or Cdk2 levels during carcinogenesis. During progression from the primary to the lymph node-metastatic foci, the levels of Cyclin E protein remain the same, while Cdk2 levels increase significantly [113]. However, during a similar transition from the primary to the liver-metastatic foci, Cyclin E levels are apparently reduced, and those of Cdk2 diminish almost completely. Additionally, the decrease of Cyclin E is significantly associated with large tumor size and lymph nodal metastasis in primary carcinomas, with large tumor size and hepatic metastasis being strongly related to decreased Cdk2 levels. Induced Cyclin E protein is related to increased Cdk2, which is further associated with Ki-67 staining. Thus, Cdk2 overexpression could facilitate lymph node metastasis and both Cyclin E and Cdk2 overexpression may trigger the progression of early cancer.
Cyclin E2 levels have also been shown to be elevated in some human primary tumors as compared to normal tissue [17, 50]. Elevated transcript levels of both cyclins E and E2 are found in breast, colorectal, lung, and ovary/uterus tumor samples as compared to levels in normal tissues. The results indicated that, although expression of Cyclin E is greater over the range of samples, Cyclin E2 expression is, on average, higher in breast primary tumors [50]. Also, it has been shown that the expression levels of Cyclins E and E2 are more likely to be elevated in breast tumors that lacked the estrogen receptor as compared to breast tumors with the receptor and normal breast tissue [50, 98, 108].
CONCLUSIONS AND FUTURE DIRECTIONS
Cyclin E represents a critical component required for regulation of the G1/S Restriction point, and it is the target of deregulation in many types of tumor cells. Although much has been learned in the last decade about cyclin E, many questions still remain. For example, are there other critical substrates that are modified by Cyclin E/Cdk2 phosphorylation? If there are, is this phosphorylation functionally significant? The discovery of cyclin E2, which has close structural homology with cyclin E, has revealed that this cyclin can also be aberrantly expressed in human cancers. This observation strengthens the possibility of future use of both cyclin E1 and/or E2 as therapeutic targets. Finally, our studies have revealed an important role for Cyclin E in apoptosis of hematopoietic cells, and perhaps of other malignancies. Increasing Cyclin E levels and the associated kinase activity, as well as the efficiency of its conversion to its p18-Cyclin E derivative, may be very important in cancer therapeutics.
Future studies will be directed to address the exact function of the proteolytic fragment of cyclin E during apoptosis. These studies may provide some significant insights into how the p18-cyclin E generation may be used as a practical goal of clinical therapy in hematopoietic and, perhaps, other malignancies. Its possible role in preventing DNA repair in cells with extensive DNA damage caused by activation of the apoptotic nucleases may shed light into how cells guard against oncogenic transformation. This may be particularly important in the hematopoietic cells known to have a high recombination potential and to undergo frequent chromosomal rearrangements. Cyclin E and its derivative p18-cyclin E are unique among most proteins, as it may have an essential role in as many as four fundamental biological processes: cell cycle control, DNA replication, apoptosis, and DNA repair. Augmenting apoptotic responses while minimizing DNA repair in tumor cells, and at the same time facilitating effective DNA repair in normal cells, is the ultimate goal of clinical therapy. Understanding how the p50- and p18-Cyclin E molecules are regulated may provide an important boost for many cancer therapeutic modalities.
Acknowledgments
We would like to thank Dr. K. Keyomarsi (UT-MD Anderson Cancer Center) for making available to us unpublished information. This work was supported by research grants from the National Institutes of Health (CA81504 and CA82858). We apologize to our colleagues whose work could not be cited due to space limitations.
References
- 1.Sherr CJ. The Pezcoller Lecture: Cancer Cell Cycles Revisited. Cancer Res. 2000;60:3689–3695. [PubMed] [Google Scholar]
- 2.Sherr CJ, Roberts JM. Inhibitors of Mammalian G1 Cyclin-Dependent Kinases. Genes Dev. 1995;9:1149–1163. doi: 10.1101/gad.9.10.1149. [DOI] [PubMed] [Google Scholar]
- 3.Ekholm SV, Reed SI. Regulation of G(1) Cyclin-Dependent Kinases in the Mammalian Cell Cycle. Curr Opin Cell Biol. 2000;12:676–684. doi: 10.1016/s0955-0674(00)00151-4. [DOI] [PubMed] [Google Scholar]
- 4.Elledge SJ. Cell Cycle Checkpoints: Preventing an Identity Crisis. Science. 1996;274:1664–1672. doi: 10.1126/science.274.5293.1664. [DOI] [PubMed] [Google Scholar]
- 5.Ekholm SV, Zickert P, Reed SI, Zetterberg A. Accumulation of Cyclin E Is Not a Prerequisite for Passage through the Restriction Point. Mol Cell Biol. 2001;21:3256–3265. doi: 10.1128/MCB.21.9.3256-3265.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Serrano M, Hannon GJ, Beach D. A New Regulatory Motif in Cell-Cycle Control Causing Specific Inhibition of Cyclin D/CDK4. Nature. 1993;366:704–707. doi: 10.1038/366704a0. [DOI] [PubMed] [Google Scholar]
- 7.Toyoshima H, Hunter T. P27, A Novel Inhibitor of G1 Cyclin-Cdk Protein Kinase Activity, Is Related to P21. Cell. 1994;78:67–74. doi: 10.1016/0092-8674(94)90573-8. [DOI] [PubMed] [Google Scholar]
- 8.Koff A, Cross F, Fisher A, Schumacher J, Leguellec K, Philippe M, Roberts JM, Human Cyclin E. A New Cyclin That Interacts with Two Members of the CDC2 Gene Family. Cell. 1991;66:1217–1228. doi: 10.1016/0092-8674(91)90044-y. [DOI] [PubMed] [Google Scholar]
- 9.Lew DJ, Dulic V, Reed SI. Isolation of Three Novel Human Cyclins by Rescue of G1 Cyclin (Cln) Function in Yeast. Cell. 1991;66:1197–1206. doi: 10.1016/0092-8674(91)90042-w. [DOI] [PubMed] [Google Scholar]
- 10.Dulic V, Lees E, Reed SI. Association of Human Cyclin E with a Periodic G1-S Phase Protein Kinase. Science. 1992;257:1958–1961. doi: 10.1126/science.1329201. [DOI] [PubMed] [Google Scholar]
- 11.Koff A, Giordano A, Desai D, Yamashita K, Harper JW, Elledge S, Nishimoto T, Morgan DO, Franza BR, Roberts JM. Formation and Activation of a Cyclin E-Cdk2 Complex During the G1 Phase of the Human Cell Cycle. Science. 1992;257:1689–1694. doi: 10.1126/science.1388288. [DOI] [PubMed] [Google Scholar]
- 12.Reed SI. Control of the G1/S Transition. Cancer Surv. 1997;29:7–23. [PubMed] [Google Scholar]
- 13.Akli S, Keyomarsi K. Cyclin E and Its Low Molecular Weight Forms in Human Cancer and as Targets for Cancer Therapy. Cancer Biology & Therapy. 2003;2(4 Suppl 1):S38–47. [PubMed] [Google Scholar]
- 14.Gudas JM, Payton M, Thukral S, Chen E, Bass M, Robinson MO, Coats S. Cyclin E2, A Novel G1 Cyclin That Binds Cdk2 and Is Aberrantly Expressed in Human Cancers. Mol Cell Biol. 1999;19:612–622. doi: 10.1128/mcb.19.1.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lauper N, Beck AR, Cariou S, Richman L, Hofmann K, Reith W, Slingerland JM, Amati B. Cyclin E2: A Novel CDK2 Partner in the Late G1 and S Phases of the Mammalian Cell Cycle. Oncogene. 1998;17:2637–2643. doi: 10.1038/sj.onc.1202477. [DOI] [PubMed] [Google Scholar]
- 16.Zariwala M, Liu J, Xiong Y. Cyclin E2, A Novel Human G1 Cyclin and Activating Partner of CDK2 and CDK3, Is Induced by Viral Oncoproteins. Oncogene. 1998;17:2787–2798. doi: 10.1038/sj.onc.1202505. [DOI] [PubMed] [Google Scholar]
- 17.Payton M, Coats S. Cyclin E2, the Cycle Continues. Int J Biochem Cell Biol. 2002;34:315–320. doi: 10.1016/s1357-2725(01)00137-6. [DOI] [PubMed] [Google Scholar]
- 18.Ohtsubo M, Roberts JM. Cyclin-Dependent Regulation of G1 in Mammalian Fibroblasts. Science. 1993;259:1908–1912. doi: 10.1126/science.8384376. [DOI] [PubMed] [Google Scholar]
- 19.Ohtsubo M, Theodoras AM, Schumacher J, Roberts JM, Pagano M. Human Cyclin E, A Nuclear Protein Essential for the G1-to-S Phase Transition. Mol Cell Biol. 1995;15:2612–2624. doi: 10.1128/mcb.15.5.2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Resnitzky D, Gossen M, Bujard H, Reed SI. Acceleration of the G1/S Phase Transition By Expression of Cyclins D1 and E with an Inducible System. Mol Cell Biol. 1994;14:1669–1679. doi: 10.1128/mcb.14.3.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spruck CH, Won KA, Reed SI. Deregulated Cyclin E Induces Chromosome Instability. Nature. 1999;401:297–300. doi: 10.1038/45836. [DOI] [PubMed] [Google Scholar]
- 22.Gray-Bablin J, Zalvide J, Fox MP, Knickerbocker CJ, Decaprio JA, Keyomarsi K. Cyclin E, A Redundant Cyclin in Breast Cancer. Proc Natl Acad Sci USA. 1996;93:15215–15220. doi: 10.1073/pnas.93.26.15215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Geng Y, Whoriskey W, Park MY, Bronson RT, Medema RH, Li T, Weinberg RA, Sicinski P. Rescue of Cyclin D1 Deficiency by Knockin Cyclin E. Cell. 1999;97:767–777. doi: 10.1016/s0092-8674(00)80788-6. [DOI] [PubMed] [Google Scholar]
- 24.Dowdy SF, Hinds PW, Louie K, Reed SI, Arnold A, Weinberg RA. Physical Interaction of the Retinoblastoma Protein with Human D Cyclins. Cell. 1993;73:499–511. doi: 10.1016/0092-8674(93)90137-f. [DOI] [PubMed] [Google Scholar]
- 25.Resnitzky D, Reed SI. Different Roles for Cyclins D1 and E in Regulation of the G1- to-S Transition. Mol Cell Biol. 1995;15:3463–3469. doi: 10.1128/mcb.15.7.3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alevizopoulos K, Vlach J, Hennecke S, Amati B. Cyclin E and C-Myc Promote Cell Proliferation in the Presence of p16Ink4a and of Hypophosphorylated Retinoblastoma Family Proteins. EMBO J. 1997;16:5322–5333. doi: 10.1093/emboj/16.17.5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lukas J, Herzinger T, Hansen K, Moroni MC, Resnitzky D, Helin K, Reed SI, Bartek J. Cyclin E-Induced S Phase Without Activation of the pRb/E2F Pathway. Genes Dev. 1997;11:1479–1492. doi: 10.1101/gad.11.11.1479. [DOI] [PubMed] [Google Scholar]
- 28.Duronio RJ, Brook A, Dyson N, O’Farrell PH. E2F-Induced S Phase Requires Cyclin E. Genes Dev. 1996;10:2505–2513. doi: 10.1101/gad.10.19.2505. [DOI] [PubMed] [Google Scholar]
- 29.Jackson PK, Chevalier S, Philippe M, Kirschner MW. Early Events in DNA Replication Require Cyclin E and are Blocked By p21cip1. J Cell Biol. 1995;130:755–769. doi: 10.1083/jcb.130.4.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rao H, Stillman B. The Origin Recognition Complex Interacts with a Bipartite DNA Binding Site Within Yeast Replicators. Proc Natl Acad Sci USA. 1995;92:2224–2228. doi: 10.1073/pnas.92.6.2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lei M, Tye BK. Initiating DNA Synthesis: From Recruiting to Activating the MCM Complex. J Cell Sci. 2001;114:1447–1454. doi: 10.1242/jcs.114.8.1447. [DOI] [PubMed] [Google Scholar]
- 32.Takisawa H, Mimura S, Kubota Y. Eukaryotic DNA Replication: from Prereplication Complex to Initiation Complex. Curr Opin Cell Biol. 2000;12:690–696. doi: 10.1016/s0955-0674(00)00153-8. [DOI] [PubMed] [Google Scholar]
- 33.Furstenthal L, Kaiser BK, Swanson C, Jackson PK. Cyclin E Uses Cdc6 as a Chromatin-Associated Receptor Required for DNA Replication. J Cell Biol. 2001;152:1267–1278. doi: 10.1083/jcb.152.6.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Furstenthal L, Swanson C, Kaiser BK, Eldridge AG, Jackson PK. Triggering Ubiquitination of a CDK Inhibitor at Origins of DNA Replication. Nat Cell Biol. 2001;3:715–722. doi: 10.1038/35087026. [DOI] [PubMed] [Google Scholar]
- 35.Zhao J, Kennedy BK, Lawrence BD, Barbie DA, Matera AG, Fletcher JA, Harlow E. NPAT Links Cyclin E-Cdk2 to the Regulation of Replication-Dependent Histone Gene Transcription. Genes Dev. 2000;14:2283–2297. [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao J, Dynlacht B, Imai T, Hori T, Harlow E. Expression of NPAT, A Novel Substrate of Cyclin E-CDK2, Promotes S-Phase Entry. Genes Dev. 1998;12:456–461. doi: 10.1101/gad.12.4.456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Okuda M, Horn HF, Tarapore P, Tokuyama Y, Smulian AG, Chan PK, Knudsen ES, Hofmann IA, Snyder JD, Bove KE, Fukasawa K. Nucleophosmin/B23 Is a Target of CDK2/Cyclin E in Centrosome Duplication. Cell. 2000;103:127–140. doi: 10.1016/s0092-8674(00)00093-3. [DOI] [PubMed] [Google Scholar]
- 38.Fisk HA, Winey M. The Mouse Mps1p-Like Kinase Regulates Centrosome Duplication. Cell. 2001;106:95–104. doi: 10.1016/s0092-8674(01)00411-1. [DOI] [PubMed] [Google Scholar]
- 39.Mussman JG, Horn HF, Carroll PE, Okuda M, Tarapore P, Donehower LA, Fukasawa K. Synergistic Induction of Centrosome Hyperamplification by Loss of p53 and Cyclin E Overexpression. Oncogene. 2000;19:1635–1646. doi: 10.1038/sj.onc.1203460. [DOI] [PubMed] [Google Scholar]
- 40.Lingle WL, Salisbury JL. Altered Centrosome Structure Is Associated with Abnormal Mitoses in Human Breast Tumors. Am J Pathol. 1999;155:1941–1951. doi: 10.1016/S0002-9440(10)65513-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Le Cam L, Polanowska J, Fabbrizio E, Olivier M, Philips A, Ng Eaton E, Classon M, Geng Y, Sardet C. Timing of Cyclin E Gene Expression Depends On the Regulated Association of a Bipartite Repressor Element with a Novel E2F Complex. EMBO J. 1999;18:1878–1890. doi: 10.1093/emboj/18.7.1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Polanowska J, Fabbrizio E, Le Cam L, Trouche D, Emiliani S, Herrera R, Sardet C. The Periodic Down Regulation of Cyclin E Gene Expression from Exit of Mitosis to End of G(1) Is Controlled by a Deacetylase- and E2F- Associated Bipartite Repressor Element. Oncogene. 2001;20:4115–4127. doi: 10.1038/sj.onc.1204514. [DOI] [PubMed] [Google Scholar]
- 43.Zhang HS, Gavin M, Dahiya A, Postigo AA, Ma D, Luo RX, Harbour JW, Dean DC. Exit from G1 and S Phase of the Cell Cycle Is Regulated by Repressor Complexes Containing HDAC-Rb-Hswi/SNF and Rb-Hswi/SNF. Cell. 2000;101:79–89. doi: 10.1016/S0092-8674(00)80625-X. [DOI] [PubMed] [Google Scholar]
- 44.Shanahan F, Seghezzi W, Parry D, Mahony D, Lees E. Cyclin E Associates with BAF155 and BRG1, Components of the Mammalian SWI-SNF Complex, and Alters the Ability of BRG1 to Induce Growth Arrest. Mol Cell Biol. 1999;19:1460–1469. doi: 10.1128/mcb.19.2.1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Almasan A, Yin Y, Kelly RE, Lee EY, Bradley A, Li WW, Bertino JR, Wahl GM. Deficiency of Retinoblastoma Protein Leads to Inappropriate S-Phase Entry, Activation of E2F-Responsive Genes, and Apoptosis. Proc Natl Acad Sci USA. 1995;92:5436–5440. doi: 10.1073/pnas.92.12.5436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martin LG, Demers GW, Galloway DA. Disruption of the G1/S Transition in Human Papillomavirus Type 16 E7-Expressing Human Cells Is Associated with Altered Regulation of Cyclin E. J Virol. 1998;72:975–985. doi: 10.1128/jvi.72.2.975-985.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lawrence TS, Davis MA, Loney TL. Fluoropyrimidine-Mediated Radiosensitization Depends On Cyclin E-Dependent Kinase Activation. Cancer Res. 1996;56:3203–3206. [PubMed] [Google Scholar]
- 48.Jones DL, Thompson DA, Munger K. Destabilization of the RB Tumor Suppressor Protein and Stabilization of P53 Contribute to HPV Type 16 E7-Induced Apoptosis. Virology. 1997;239:97–107. doi: 10.1006/viro.1997.8851. [DOI] [PubMed] [Google Scholar]
- 49.Geng Y, Yu Q, Whoriskey W, Dick F, Tsai KY, Ford HL, Biswas DK, Pardee AB, Amati B, Jacks T, Richardson A, Dyson N, Sicinski P. Expression of Cyclins E1 and E2 During Mouse Development and in Neoplasia. Proc Natl Acad Sci USA. 2001;98:13138–13143. doi: 10.1073/pnas.231487798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Payton M, Scully S, Chung G, Coats S. Deregulation of Cyclin E2 Expression and Associated Kinase Activity in Primary Breast Tumors. Oncogene. 2002;21:8529–8534. doi: 10.1038/sj.onc.1206035. [DOI] [PubMed] [Google Scholar]
- 51.Deed RW, Hara E, Atherton GT, Peters G, Norton JD. Regulation of Id3 Cell Cycle Function by Cdk-2-Dependent Phosphorylation. Mol Cell Biol. 1997;17:6815–6821. doi: 10.1128/mcb.17.12.6815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hara E, Hall M, Peters G. Cdk2-Dependent Phosphorylation of Id2 Modulates Activity of E2A-Related Transcription Factors. EMBO J. 1997;16:332–342. doi: 10.1093/emboj/16.2.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Seghezzi W, Chua K, Shanahan F, Gozani O, Reed R, Lees E. Cyclin E Associates with Components of the Pre-Mrna Splicing Machinery in Mammalian Cells. Mol Cell Biol. 1998;18:4526–4536. doi: 10.1128/mcb.18.8.4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang C, Chua K, Seghezzi W, Lees E, Gozani O, Reed R. Phosphorylation of Spliceosomal Protein SAP 155 Coupled with Splicing Catalysis. Genes Dev. 1998;12:1409–1414. doi: 10.1101/gad.12.10.1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Blattner C, Sparks A, Lane D. Transcription Factor E2F-1 Is Upregulated in Response to DNA Damage in a Manner Analogous to that of p53. Mol Cell Biol. 1999;19:3704–3713. doi: 10.1128/mcb.19.5.3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mazumder S, Gong B, Almasan A. Cyclin E Induction By Genotoxic Stress Leads to Apoptosis of Hematopoietic Cells. Oncogene. 2000;19:2828–2835. doi: 10.1038/sj.onc.1203623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Won KA, Reed SI. Activation of Cyclin E/CDK2 Is Coupled to Site-Specific Autophosphorylation and Ubiquitin-Dependent Degradation of Cyclin E. EMBO J. 1996;15:4182–4193. [PMC free article] [PubMed] [Google Scholar]
- 58.Clurman BE, Sheaff RJ, Thress K, Groudine M, Roberts JM. Turnover of Cyclin E by the Ubiquitin-Proteasome Pathway Is Regulated by Cdk2 Binding and Cyclin Phosphorylation. Genes Dev. 1996;10:1979–1990. doi: 10.1101/gad.10.16.1979. [DOI] [PubMed] [Google Scholar]
- 59.Bai C, Sen P, Hofmann K, Ma L, Goebl M, Harper JW, Elledge SJ. SKP1 Connects Cell Cycle Regulators to the Ubiquitin Proteolysis Machinery through a Novel Motif, the F-Box. Cell. 1996;86:263–274. doi: 10.1016/s0092-8674(00)80098-7. [DOI] [PubMed] [Google Scholar]
- 60.Winston JT, Koepp DM, Zhu C, Elledge SJ, Harper JW. A Family of Mammalian F-Box Proteins. Curr Biol. 1999;9:1180–1182. doi: 10.1016/S0960-9822(00)80021-4. [DOI] [PubMed] [Google Scholar]
- 61.Strohmaier H, Spruck CH, Kaiser P, Won KA, Sangfelt O, Reed SI. Human F-Box Protein Hcdc4 Targets Cyclin E for Proteolysis and Is Mutated in a Breast Cancer Cell Line. Nature. 2001;413:316–322. doi: 10.1038/35095076. [DOI] [PubMed] [Google Scholar]
- 62.Koepp DM, Schaefer LK, Ye X, Keyomarsi K, Chu C, Harper JW, Elledge SJ. Phosphorylation-Dependent Ubiquitination of Cyclin E by the Scffbw7 Ubiquitin Ligase. Science. 2001;294:173–177. doi: 10.1126/science.1065203. [DOI] [PubMed] [Google Scholar]
- 63.Hoffmann I, Draetta G, Karsenti E. Activation of the Phosphatase Activity of Human Cdc25a by a Cdk2-Cyclin E Dependent Phosphorylation at the G1/S Transition. EMBO J. 1994;13:4302–4310. doi: 10.1002/j.1460-2075.1994.tb06750.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Falck J, Mailand N, Syljuasen RG, Bartek J, Lukas J. The ATM-Chk2-Cdc25A Checkpoint Pathway Guards Against Radioresistant DNA Synthesis. Nature. 2001;410:842–847. doi: 10.1038/35071124. [DOI] [PubMed] [Google Scholar]
- 65.Raff MC. Social Controls On Cell Survival and Cell Death. Nature. 1992;356:397–400. doi: 10.1038/356397a0. [DOI] [PubMed] [Google Scholar]
- 66.Thompson CB. Apoptosis in the Pathogenesis and Treatment of Disease. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 67.Evan GI, Vousden KH. Proliferation, Cell Cycle and Apoptosis in Cancer. Nature. 2001;411:342–348. doi: 10.1038/35077213. [DOI] [PubMed] [Google Scholar]
- 68.Sherr CJ. Cancer Cell Cycles. Science. 1996;274:1672–1677. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- 69.Harbour JW, Dean DC. The Rb/E2F Pathway: Expanding Roles and Emerging Paradigms. Genes Dev. 2000;14:2393–2409. doi: 10.1101/gad.813200. [DOI] [PubMed] [Google Scholar]
- 70.Gong B, Almasan A. Apo2 Ligand/TNF-Related Apoptosis-Inducing Ligand and Death Receptor 5 Mediate the Apoptotic Signaling Induced by Ionizing Radiation in Leukemic Cells. Cancer Res. 2000;60:5754–5760. [PubMed] [Google Scholar]
- 71.Chen Q, Gong B, Almasan A. Distinct Stages of Cytochrome C Release from Mitochondria: Evidence for a Feedback Amplification Loop Linking Caspase Activation to Mitochondrial Dysfunction in Genotoxic Stress Induced Apoptosis. Cell Death Differ. 2000;7:227–233. doi: 10.1038/sj.cdd.4400629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gong B, Chen Q, Endlich B, Mazumder S, Almasan A. Ionizing Radiation-induced, Bax-Mediated Cell Death Is Dependent On Activation of Serine and Cysteine Proteases. Cell Growth Diff. 1999;10:491–502. [PubMed] [Google Scholar]
- 73.Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of Apoptotic Program in Cell-Free Extracts: Requirement for DAtp and Cytochrome C. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 74.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome C and DAtp-Dependent Formation of Apaf-1/Caspase-9 Complex Initiates an Apoptotic Protease Cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 75.Hengartner MO. The Biochemistry of Apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 76.Janicke RU, Walker PA, Lin XY, Porter AG. Specific Cleavage of the Retinoblastoma Protein by an ICE-Like Protease in Apoptosis. EMBO J. 1996;15:6969–6978. [PMC free article] [PubMed] [Google Scholar]
- 77.Tan X, Martin SJ, Green DR, Wang JYJ. Degradation of Retinoblastoma Protein in Tumor Necrosis Factor- and CD95-Induced Cell Death. J Biol Chem. 1997;272:9613–9616. doi: 10.1074/jbc.272.15.9613. [DOI] [PubMed] [Google Scholar]
- 78.Chen L, Marechal V, Moreau J, Levine AJ, Chen J. Proteolytic Cleavage of the MDM2 Oncoprotein During Apoptosis. J Biol Chem. 1997;272:22966–22973. doi: 10.1074/jbc.272.36.22966. [DOI] [PubMed] [Google Scholar]
- 79.Beyaert R, Kidd VJ, Cornelis S, Van De Craen M, Denecker G, Lahti JM, Gururajan R, Vandenabeele P, Fiers W. Cleavage of PITSLRE Kinases by ICE/CASP-1 and CPP32/CASP-3 During Apoptosis Induced by Tumor Necrosis Factor. J Biol Chem. 1997;272:11694–11697. doi: 10.1074/jbc.272.18.11694. [DOI] [PubMed] [Google Scholar]
- 80.Levkau B, Koyoma H, Raines EW, Clurman BE, Herren B, Orth K, Roberts JM. Cleavage of P21/Cip1/Waf1 and P27Kip1 Mediates Apoptosis of Endothelial Cells through Activation of Cdk2: Role of a Caspase Cascade. Molec Cell. 1997;1:553–563. doi: 10.1016/s1097-2765(00)80055-6. [DOI] [PubMed] [Google Scholar]
- 81.Meikrantz W, Schlegel R. Apoptosis and the Cell Cycle. J Cell Biochem. 1995;58:160–174. doi: 10.1002/jcb.240580205. [DOI] [PubMed] [Google Scholar]
- 82.Meikrantz W, Schlegel R. Suppression of Apoptosis by Dominant Negative Mutants of Cyclin-Dependent Protein Kinases. J Biol Chem. 1996;271:10205–10209. doi: 10.1074/jbc.271.17.10205. [DOI] [PubMed] [Google Scholar]
- 83.Brady HJ, Gil-Gomez G, Kirberg J, Berns AJ. Bax Alpha Perturbs T Cell Development and Affects Cell Cycle Entry of T Cells. EMBO J. 1996;15:6991–7001. [PMC free article] [PubMed] [Google Scholar]
- 84.Mazel S, Burtrum D, Petrie HT. Regulation of Cell Division Cycle Progression by Bcl-2 Expression: A Potential Mechanism for Inhibition of Programmed Cell Death. J Exp Med. 1996;183:2219–2226. doi: 10.1084/jem.183.5.2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Linette GP, Li Y, Roth K, Korsmeyer SJ. Cross Talk Between Cell Death and Cell Cycle Progression: BCL-2 Regulates NFAT-Mediated Activation. Proc Natl Acad Sci USA. 1996;93:9545–9552. doi: 10.1073/pnas.93.18.9545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.O’Reilly LA, Huang DC, Strasser A. The Cell Death Inhibitor Bcl-2 and Its Homologues Influence Control of Cell Cycle Entry. EMBO J. 1996;15:6979–6990. [PMC free article] [PubMed] [Google Scholar]
- 87.Schlegel J, Peters I, Orrenius S, Miller DK, Thornberry NA, Yamin TT, Nicholson DW. CPP32/Apopain Is a Key Interleukin 1 Beta Converting Enzyme-Like Protease Involved in Fas-Mediated Apoptosis. J Biol Chem. 1996;271:1841–1844. doi: 10.1074/jbc.271.4.1841. [DOI] [PubMed] [Google Scholar]
- 88.Zhou BB, Li H, Yuan J, Kirschner MW. Caspase-Dependent Activation of Cyclin-Dependent Kinases During Fas-Induced Apoptosis in Jurkat Cells. Proc Natl Acad Sci USA. 1998;95:6785–6790. doi: 10.1073/pnas.95.12.6785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Park DS, Morris EJ, Padmanabhan J, Shelanski ML, Geller HM, Greene LA. Cyclin-Dependent Kinases Participate in Death of Neurons Evoked by Dna damaging Agents. J Cell Biol. 1998;143:457–467. doi: 10.1083/jcb.143.2.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang J, Walsh K. Resistance to Apoptosis Conferred by Cdk Inhibitors During Myocyte Differentiation. Science. 1996;273:359–361. doi: 10.1126/science.273.5273.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hakem A, Sasaki T, Kozieradzki I, Penninger JM. The Cyclin-Dependent Kinase Cdk2 Regulates Thymocyte Apoptosis. J Exp Med. 1999;189:957–968. doi: 10.1084/jem.189.6.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sofer-Levi Y, Resnitzky D. Apoptosis Induced by Ectopic Expression of Cyclin D1 but Not Cyclin E. Oncogene. 1996;13:2431–2437. [PubMed] [Google Scholar]
- 93.Han EK, Begemann M, Sgambato A, Soh JW, Doki Y, Xing WQ, Liu W, Weinstein IB. Increased Expression of Cyclin D1 in a Murine Mammary Epithelial Cell Line Induces P27kip1, Inhibits Growth, and Enhances Apoptosis. Cell Growth Differ. 1996;7:699–710. [PubMed] [Google Scholar]
- 94.Mazumder S, Chen Q, Gong B, Drazba JA, Buchsbaum JC, Almasan A. Proteolytic Cleavage of Cyclin E Leads to Inactivation of Associated Kinase Activity and Amplification of Apoptosis in Hematopoietic Cells. Mol Cell Biol. 2002;22:2398–2409. doi: 10.1128/MCB.22.7.2398-2409.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pardee AB. G1 Events and Regulation of Cell Proliferation. Science. 1989;246:603–608. doi: 10.1126/science.2683075. [DOI] [PubMed] [Google Scholar]
- 96.Keyomarsi K, Samet J, Molnar G, Pardee AB. The Thymidylate Synthase Inhibitor, ICI D1694, Overcomes Translational Detainment of the Enzyme. J Biol Chem. 1993;268:15142–15149. [PubMed] [Google Scholar]
- 97.Hunter T. 1001 Protein Kinases Redux--Towards 2000. Semin Cell Biol. 1994;5:367–376. doi: 10.1006/scel.1994.1044. [DOI] [PubMed] [Google Scholar]
- 98.Keyomarsi K, O’Leary N, Molnar G, Lees E, Fingert HJ, Pardee AB. Cyclin E, A Potential Prognostic Marker for Breast Cancer. Cancer Res. 1994;54:380–385. [PubMed] [Google Scholar]
- 99.Keyomarsi K, Conte D, Jr, Toyofuku W, Fox MP. Deregulation of Cyclin E in Breast Cancer. Oncogene. 1995;11:941–950. [PubMed] [Google Scholar]
- 100.Keyomarsi K, Tucker SL, Buchholz TA, Callister M, Ding Y, Hortobagyi GN, Bedrosian I, Knickerbocker C, Toyofuku W, Lowe M, Herliczek TW, Bacus SS. Cyclin E and Survival in Patients with Breast Cancer. N Engl J Med. 2002;347:1566–1575. doi: 10.1056/NEJMoa021153. [DOI] [PubMed] [Google Scholar]
- 101.Semenov I, Akyuz C, Roginskaya V, Chauhan D, Corey SJ. Growth Inhibition and Apoptosis of Myeloma Cells by the CDK Inhibitor Flavopiridol. Leuk Res. 2002;26:271–280. doi: 10.1016/s0145-2126(01)00103-5. [DOI] [PubMed] [Google Scholar]
- 102.Senderowicz AM. Small Molecule Modulators of Cyclin-Dependent Kinases for Cancer Therapy. Oncogene. 2000;19:6600–6606. doi: 10.1038/sj.onc.1204085. [DOI] [PubMed] [Google Scholar]
- 103.Senderowicz AM. Development of Cyclin-Dependent Kinase Modulators As Novel Therapeutic Approaches for Hematological Malignancies. Leukemia. 2001;15:1–9. doi: 10.1038/sj.leu.2401994. [DOI] [PubMed] [Google Scholar]
- 104.Stadler WM, Vogelzang NJ, Amato R, Sosman J, Taber D, Liebowitz D, Vokes EE. Flavopiridol, a Novel Cyclin-Dependent Kinase Inhibitor, in Metastatic Renal Cancer: A University of Chicago Phase II Consortium Study. J Clin Oncol. 2000;18:371–375. doi: 10.1200/JCO.2000.18.2.371. [DOI] [PubMed] [Google Scholar]
- 105.D’Urso G, Marraccino RL, Marshak DR, Roberts JM. Cell Cycle Control of DNA Replication by a Homologue from Human Cells of the p34cdc2 Protein Kinase. Science. 1990;250:786–791. doi: 10.1126/science.2173140. [DOI] [PubMed] [Google Scholar]
- 106.Porter PL, Malone KE, Heagerty PJ, Alexander GM, Gatti LA, Firpo EJ, Daling JR, Roberts JM. Expression of Cell-Cycle Regulators p27Kip1 and Cyclin E, Alone and in Combination, Correlate with Survival in Young Breast Cancer Patients. Nat Med. 1997;3:222–225. doi: 10.1038/nm0297-222. [DOI] [PubMed] [Google Scholar]
- 107.Dutta A, Chandra R, Leiter LM, Lester S. Cyclins as Markers of Tumor Proliferation: Immunocytochemical Studies in Breast Cancer. Proc Natl Acad Sci USA. 1995;92:5386–5390. doi: 10.1073/pnas.92.12.5386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nielsen NH, Arnerlov C, Emdin SO, Landberg G. Cyclin E Overexpression, a Negative Prognostic Factor in Breast Cancer with Strong Correlation to Estrogen Receptor Status. Br J Cancer. 1996;74:874–880. doi: 10.1038/bjc.1996.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Keyomarsi K, Pardee AB. Redundant Cyclin Overexpression and Gene Amplification in Breast Cancer Cells. Proc Natl Acad Sci USA. 1993;90:1112–1116. doi: 10.1073/pnas.90.3.1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Porter DC, Zhang N, Danes C, Mcgahren MJ, Harwell RM, Faruki S, Keyomarsi K. Tumor-Specific Proteolytic Processing of Cyclin E Generates Hyperactive Lower Molecular-Weight Forms. Mol Cell Biol. 2001;21:6254–6269. doi: 10.1128/MCB.21.18.6254-6269.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Porter DC, Keyomarsi K. Novel Splice Variants of Cyclin E with Altered Substrate Specificity. Nucleic Acids Res. 2000;28:E101. doi: 10.1093/nar/28.23.e101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Harwell RM, Porter DC, Danes C, Keyomarsi K. Processing of Cyclin E Differs Between Normal and Tumor Breast Cells. Cancer Res. 2000;60:481–489. [PubMed] [Google Scholar]
- 113.Li JQ, Miki H, Ohmori M, Wu F, Funamoto Y. Expression of Cyclin E and Cyclin-Dependent Kinase 2 Correlates with Metastasis and Prognosis in Colorectal Carcinoma. Hum Pathol. 2001;32:945–953. doi: 10.1053/hupa.2001.27116. [DOI] [PubMed] [Google Scholar]
- 114.Chen IT, Akamatsu M, Smith ML, Lung FD, Duba D, Roller PP, Fornace AJ, Jr, O’Connor PM. Characterization of p21Cip1/Waf1 Peptide Domains Required for Cyclin E/Cdk2 and PCNA Interaction. Oncogene. 1996;12:595–607. [PubMed] [Google Scholar]
- 115.Singer JD, Gurian-West M, Clurman B, Roberts JM. Cullin-3 Targets Cyclin E for Ubiquitination and Controls S Phase in Mammalian Cells. Genes Dev. 1999;13:2375–2387. doi: 10.1101/gad.13.18.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chen J, Jackson PK, Kirschner MW, Dutta A. Separate Domains of p21 Involved in the Inhibition of Cdk Kinase and PCNA. Nature. 1995;374:386–388. doi: 10.1038/374386a0. [DOI] [PubMed] [Google Scholar]
- 117.El-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. WAF1, a Potential Mediator of p53 Tumor Suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 118.Almasan A, Linke S, Paulson T, Huang LC, Wahl GM. Genetic Instability as a Consequence of Inappropriate Entry and Progression through S-Phase. Cancer Metast Rev. 1995;14:59–73. doi: 10.1007/BF00690212. [DOI] [PubMed] [Google Scholar]
- 119.Wang X. The Expanding Role of Mitochondria in Apoptosis. Genes Dev. 2001;15:2922–2933. [PubMed] [Google Scholar]