Local translation of RhoA regulates growth cone collapse (original) (raw)
. Author manuscript; available in PMC: 2006 Feb 18.
Published in final edited form as: Nature. 2005 Aug 18;436(7053):1020–1024. doi: 10.1038/nature03885
Abstract
Neuronal development requires highly coordinated regulation of the cytoskeleton within the developing axon. This dynamic regulation manifests itself in axonal branching, turning, and pathfinding, presynaptic differentiation, and growth cone collapse and extension. Semaphorin 3A (Sema3A), a secreted guidance cue that primarily acts to repel axons from inappropriate targets, induces cytoskeletal rearrangements that results in growth cone collapse 1. These effects require intra-axonal mRNA translation. Here we show that transcripts for RhoA, a small GTPase that regulates the actin cytoskeleton, are localized to developing axons and growth cones, and this localization is mediated by an axonal targeting element located in the RhoA 3’UTR. Sema3A induces intra-axonal translation of RhoA mRNA and this local translation of RhoA is necessary and sufficient for Sema3A-mediated growth cone collapse. These studies indicate that local RhoA translation regulates the neuronal cytoskeleton and identify a novel mechanism for the regulation of RhoA signaling.
Studies using Xenopus retinal axons demonstrated that cytoskeletal regulation of the growth cone by Sema3A requires intra-axonal, or “local”, mRNA translation2. Sema3A treatment results in increased protein synthesis in growth cones as evidenced by metabolic labeling experiments and by phosphorylation of elongation factor 4E-BP12. These effects occur within minutes of Sema3A application2. Furthermore, Sema3A-mediated growth cone collapse is blocked by ribosomal inhibitors. The mRNA translation that is required for Sema3A-mediated growth cone collapse occurs in the axon, as both Sema3A-induced collapse and inhibition of this collapse by ribosomal inhibitors is preserved in axons that are severed from their cell bodies2.
To determine if intra-axonal mRNA translation is required for Sema3A signaling in mammalian neurons, we examined Sema3A-mediated growth cone collapse in embryonic rat dorsal root ganglia (DRG) explant cultures3-5. To eliminate the possibility that the effects of Sema3A were mediated through somatic translation, axons were severed from their cell bodies2 (Supplementary Fig. 1a). Treatment of severed axons with Sema3A for 60 min resulted in an increase in collapsed growth cones from 17 ± 1.3% to 75 ± 2.8 % (Supplementary Fig. 1b, c, g). This effect was blocked by pretreatment of axons with either cycloheximide or anisomycin (Supplementary Fig. 1d-f), both of which are ribosomal inhibitors. Pretreatment of these cultures with rapamycin, an inhibitor of cap-dependent translation6, also blocked Sema3A-mediated growth cone collapse. Together, these data indicate that the requirement for mRNA translation in mediating the cytoskeletal effects of Sema3A is a conserved feature among vertebrates.
Cytoskeletal alterations in axons are thought to involve members of the Rho family of small guanosine triphosphatases (GTPases) with roles for Cdc42 and Rac1 in regulating filopodia and lamellipodia formation in growth cones, and RhoA in triggering growth cone collapse and neurite retraction1. In DRG neurons, growth cone collapse in response to Sema3A requires the RhoA effector ROCK7 (Supplementary Fig. 1g), implying a role for RhoA activation in Sema3A signaling. Thus, local translation of RhoA or its effectors could contribute to Sema3A-mediated collapse. To identify axonal mRNAs that mediate the effects of Sema3A, we prepared DRG explant cultures and screened axons with a panel of probes by in situ hybridization. Transcripts for RhoA, as well as β-actin, an mRNA previously shown to be axonally-localized8, were present in axons at similar levels by fluorescent in situ hybridization (Fig. 1a-d, Supplementary Fig. 2a, b). RhoA mRNA localization was specific since transcripts for ROCK (Fig. 1e, f, Supplementary Fig. 2e, f), mDIA (Supplementary Fig. 2g, h), another RhoA effector, as well as other Rho family GTPases (Fig. 1g, Supplementary Fig. 2i-l) were not detectable in axons. We next examined RhoA mRNA localization in other types of developing neurons. In situ hybridization experiments with E18 rat hippocampal neurons and postnatal mouse basal pontine explant neurons also revealed axonal localization of RhoA transcripts (Supplementary Fig. 2m-p). To further confirm these findings, we performed RT-PCR on RNA obtained from mechanically-isolated DRG axons prepared in modified Boyden chambers (Supplementary Fig. 3a-c). RT-PCR of axonal RNA revealed the presence of RhoA and β-actin transcripts, but not transcripts for ROCK1 or Rac1 (Fig. 1i). The glial-specific transcript, glial fibrillary acidic protein (GFAP), and the neuronal soma-restricted transcript γ-actin9, were also absent, establishing the purity of the axonal preparations (Supplementary Fig. 3d). These data suggest that the axonal localization of RhoA mRNA is a general feature of developing axons.
Figure 1.
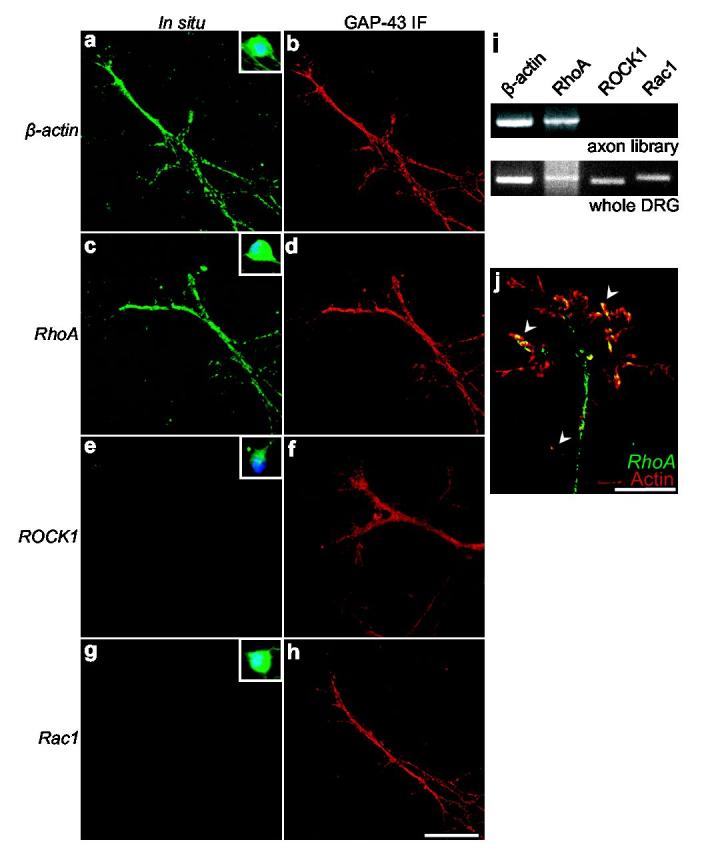
RhoA mRNA is localized in axons and growth cones. In situ hybridization experiments using digoxigenin-labeled riboprobes directed against a, β-actin, c, RhoA, e, ROCK1, and g, Rac1 transcripts. β-actin and RhoA transcripts are localized to axons while Rac1 and ROCK1 transcripts are not detectable. Insets (a,c,e,g), In situ hybridization of cell bodies of dissociated DRG neurons. DAPI staining is shown in blue. b,d,f,h, GAP-43 immunofluorescence of axons in (a,c,e,g), respectively. Scale bar, 20 μm. i, Detection of RhoA mRNA in axons by RT-PCR from purified axons. Only transcripts for RhoA and β-actin were detected in axonal preparations (upper panel), while all transcripts were detected when whole explant RNA was used as a template (lower panel). j, Growth cones were visualized by actin immunofluorescence (red). Arrowheads indicate RhoA mRNA (green) along the periphery of the growth cone. Scale bar, 10 μm.
In situ hybridization revealed that RhoA transcripts were localized in axons in punctate structures (Fig. 1c, Supplementary Fig. 2a). This localization may reflect the incorporation of RhoA mRNA in “RNA granules,” mobile macromolecular complexes of ribosomes, fragile X-mental retardation protein (FMRP), and mRNA10 that may function to transport mRNA or as sites of translation in neurites11. Indeed, Staufen, a marker for RNA granules12,13, partially colocalized with RhoA transcript-containing puncta (Supplementary Fig. 2q). We examined RhoA transcript localization in growth cones as detected by actin immunofluorescence (Fig. 1j). RhoA mRNA was found in the growth cone periphery indicating that RhoA mRNA may play a role in the regulation of the growth cone cytoskeleton.
Targeting elements in the 3’UTR of β-actin mRNA have been implicated in directing the localization of β-actin transcripts to axons14. We did not detect these elements in the RhoA 3’UTR. To determine if axonal localization of RhoA mRNA depends on other targeting elements in its 3’UTR, we expressed EGFP (EGFP) transcripts with various 3’UTRs in DRG explant cultures. Cultures were infected with Sindbis pseudovirus expressing EGFP mRNAs with the minimal viral 3’UTR (conserved sequence element, 3’CSE15, EGFP3’CSE), the 3’UTR of β-actin (EGFP3’β-actin), or the 3’UTR of RhoA (EGFP3’RhoA). In situ hybridization with an EGFP-specific probe revealed that only EGFP3’β-actin or EGFP3’RhoA were localized to axons and growth cones (Fig. 2a, Supplementary Fig. 4a). The absence of EGFP3’CSE in axons suggests that the observed axonal localization of the EGFP3’β-actin and EGFP3’RhoA is not a consequence of passive diffusion or nonspecific transport from the soma. In situ hybridization of infected cell bodies revealed that each EGFP transcript was expressed in the soma (Supplementary Fig. 4b), indicating that the absence of EGFP3’CSE in axons is not an effect of impaired transcript synthesis or stability. These studies indicate that the RhoA 3’UTR is sufficient to target heterologous transcripts to axons and growth cones.
Figure 2.
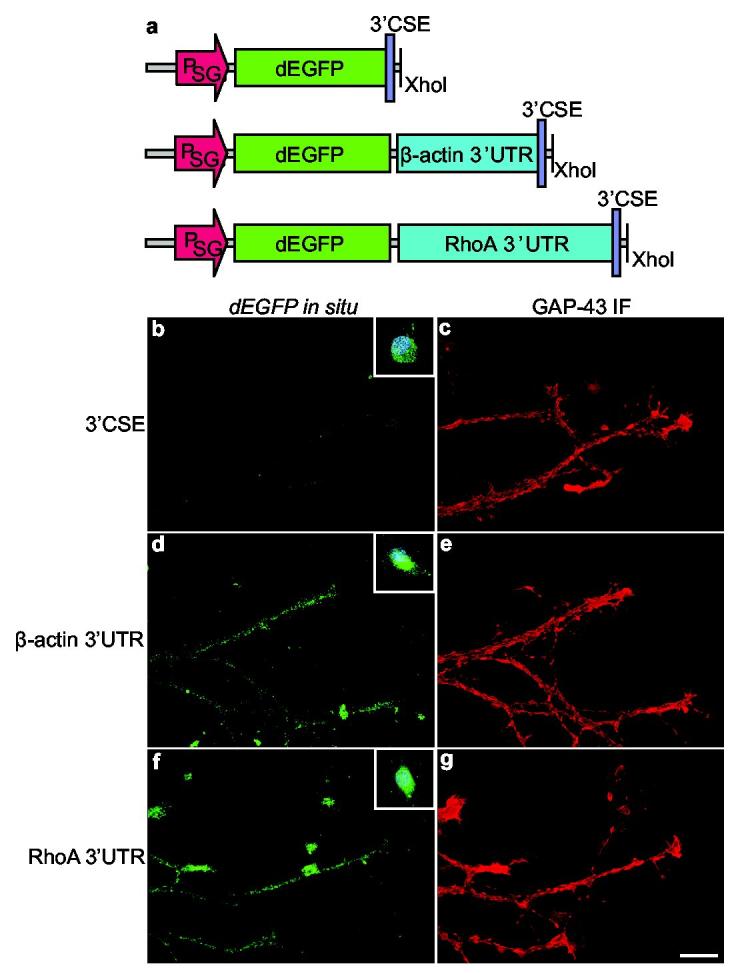
The RhoA 3’UTR contains an axonal targeting element. a, Schematic of Sindbis constructs. Transcripts encoding destabilized EGFP (dEGFP) contained either the conserved sequence element (3’CSE) of Sindbis15 as the 3’UTR of the transcript, the 3’UTR of RhoA, or the 3’UTR of β-actin. PSG, viral subgenomic promoter. b,d,f, In situ hybridization using riboprobes specific for dEGFP. dEGFP transcripts containing either the 3’UTR of β-actin or RhoA were targeted to axons. dEGFP transcripts containing only the viral 3’CSE sequence were not detected in axons. Insets (b,d,f), In situ hybridization of cell bodies. c,e,g, GAP-43 immunofluorescence of axons in (b,d,f).
The enrichment of RhoA mRNA in axons and growth cones suggests that its local translation could regulate the actin cytoskeleton. The finding that polyadenylated mRNA and large numbers of polyribosomes are present in developing vertebrate axons and growth cones8,9,16, as well as evidence that axons and axonal preparations have the capacity to synthesize proteins17-19, suggests that axonal translation has a functional role in neuronal development. To determine if RhoA translation is induced in response to Sema3A, we monitored RhoA levels after Sema3A treatment. Total RhoA levels were determined by summing RhoA immunofluorescence in image Z-stacks collected in 1 μm steps and normalizing to the volume of the growth cone, using GAP-43 immunofluorescence to define the cellular borders. DRG explants were cultured in medium containing 125 ng/ml NGF rather than 75 ng/ml to slow the rate of growth cone collapse7 an additional 2 h, thus allowing images of not yet fully collapsed growth cones (Supplementary Fig. 5a, b) to be taken after 1 h. In severed axons, Sema3A treatment resulted in a 2.3 ± 0.2 fold increase in RhoA immunofluorescence (Fig. 3a-f). This increase was blocked by pretreatment with either anisomycin or rapamycin, indicating that protein translation is required for Sema3A-induced increases in RhoA immunofluorescence. This increase was specific to RhoA, as total GAP-43 immunofluorescence levels were unchanged in response to Sema3A treatment (Fig. 3g).
Figure 3.
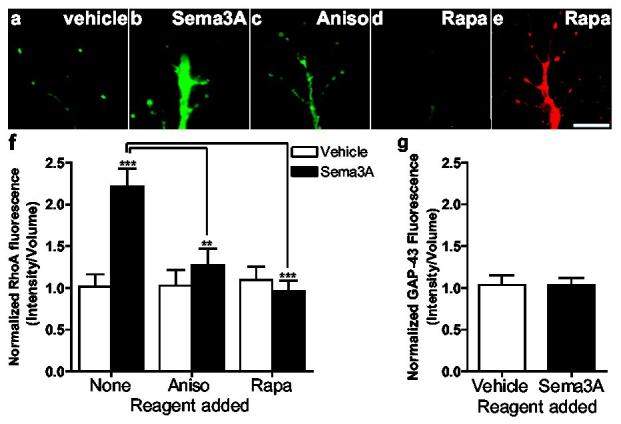
Sema3A induces RhoA translation. Severed axons were preincubated with vehicle, 10 nM rapamycin, or 40 μM anisomycin, treated with Sema3A for 60 min, and RhoA was detected by immunofluorescence and normalized to axonal volume using GAP-43-defined cellular borders. a,b, Sema3A treatment increases RhoA levels. c, Sema3A-induced increases in RhoA are blocked by anisomycin. d, Sema3A-induced increases in RhoA are blocked by rapamycin. e, Example of GAP-43 immunofluorescence staining of the axon terminal seen in (d). Scale bar, 10 μm. f, Summary of results from a-d. n=50 growth cones per condition **p < 0.01. g, Normalized GAP-43 immunofluorescence is unaltered by Sema3A treatment. n=50 growth cones per condition.
To monitor the spatial distribution of protein synthesis, we utilized myr-dEGFP, a destabilized EGFP containing an N-terminal myristoylation consensus sequence that limits the diffusion of the fluorescent protein from the site of translation20. The myr-dEGFP reporter has a half-life of one hour, thus fluorescent signals represent newly synthesized protein. Infection of DRG explants with a Sindbis viral construct comprising myr-dEGFP and the RhoA 3’UTR (Fig. 4a) resulted in the appearance of fluorescent puncta distributed throughout axons. Treatment with Sema3A for 60 min resulted in increased intensity of pre-existing puncta as well as the appearance of new puncta in infected axons (Fig. 4b-d). New or increased fluorescence of pre-existing myr-dEGFP puncta were infrequent in the absence of Sema3A treatment (Supplementary Fig. 6a, b). To test the role of protein translation in Sema3A-mediated increases in reporter expression, we pretreated axons with anisomycin 30 min prior to addition of Sema3A. Anisomycin pretreatment resulted in a complete loss of Sema3A-mediated increases in myr-dEGFP throughout the axon (Supplementary Fig. 6c, d). The absence of fluorescence in anisomycin-treated axons after 1 h of Sema3A treatment reflects the lability of myr-dEGFP and indicates that signals at this time point must derive from newly synthesized protein. Thus, the increase in myr-dEGFP levels upon Sema3A-treatement is due to increased translation. To assess the effects of Sema3A more comprehensively, 580 puncta were analyzed from both the Sema3A-treated condition and the vehicle-treated condition, and the fluorescence signal at 0 min and 60 min were plotted (Fig. 4d). Analysis of puncta presented in this manner showed distinct populations of puncta after 60 min of Sema3A treatment, comprising both newly-formed puncta, as well as puncta that displayed increased intensity after Sema3A treatment. Most puncta from vehicle-treated axons displayed minimal changes in fluorescence intensity over the 60 min period. The punctate localization may reflect translation at the sites of RNA granules. Furthermore, these experiments indicate that the RNA elements that direct Sema3A-induced translation are located in the 3’UTR. The 3’UTR of RhoA contains several elements implicated in translational regulation, including several binding sites for microRNAs21, and a binding site for FMRP, an RNA-binding protein that regulates translation22 (Supplementary Fig. 7).
Figure 4.
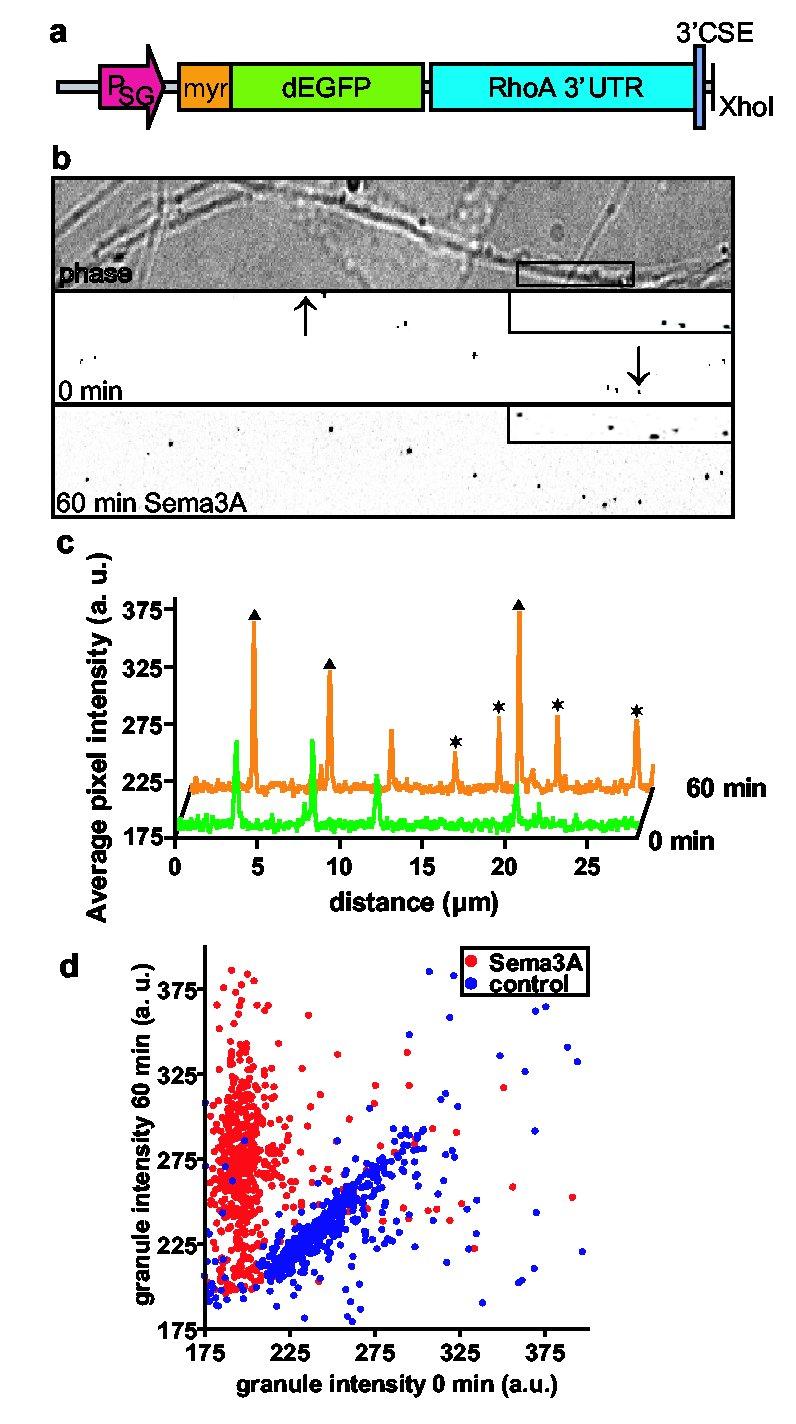
Sema3A activates translation of a RhoA reporter. a, Schematic of Sindbis reporter construct. The reporter contains a myristoylated, destabilized EGFP (d1EGFP) with the 3’UTR of RhoA. b, Sema3A induces translation of the reporter. Phase (top) and EGFP fluorescence (middle) images of axons. Fluorescence images are shown with inverted contrast to facilitate visualization. Lower panel, Newly formed puncta and puncta with increased signal intensity are seen following Sema3A treatment. Inset, a region of the axon in the top panel is magnified to show changes in puncta number and intensity. Scale bar, 10 μm. c, A line scan performed at 0 (green) and 60 min (orange) in the region demarcated by arrows. Newly formed puncta (asterisks) and puncta with increased signal intensity (triangles) are seen following Sema3A treatment. d, Scatter plot of puncta intensities at 0 and 60 min. 580 puncta from experiments monitoring Sema3A- (red) and vehicle-treated (blue) axons were plotted. The majority of points from Sema3A-treated axons remained above the diagonal, indicating newly formed puncta or pre-existing puncta that increased in intensity upon Sema3A treatment. Most points from vehicle-treated axons remained along the diagonal, indicating that puncta were largely unaffected by vehicle treatment. The background levels in these images averaged approximately 185.
We next examined the role of RhoA in Sema3A-mediated growth cone collapse. To assess the role of RhoA in Sema3A-mediated collapse, we examined the effects of Clostridium botulinum C3 exoenzyme, an ADP-ribosyltransferase that inactivates RhoA23. Treatment of DRG explant cultures with C3 exoenzyme significantly reduced Sema3A-induced collapse (Supplementary Fig. 8), indicating a requirement for RhoA in the cytoskeletal remodeling effects of Sema3A.
We next examined the requirement for axonal RhoA mRNA in Sema3A-mediated growth cone collapse. Treatment of cultured dissociated DRG neurons with siRNA directed against the 5'UTR of RhoA mRNA essentially abolished RhoA mRNA in axons (Fig. 5a-d). In these neurons, we “Greplaced” endogenous RhoA transcripts with heterologous RhoA mRNAs that exhibit selective localizations. Transcripts that contain the RhoA 3’UTR exhibit axonal localization, while transcripts that contain the viral 3’CSE are restricted to the soma (Fig. 2). These viral-encoded mRNAs lack the RhoA 5'UTR, rendering them resistant to the siRNA. Thus, we infected siRNA-treated neurons with Sindbis pseudovirus expressing EGFP-RhoA transcripts containing either the viral 3’CSE (EGFP-RhoA3’CSE) or the RhoA 3’UTR (EGFP-RhoA3’RhoA). In DRG neurons, siRNA directed against the 5'UTR of RhoA markedly reduced Sema3A-mediated growth cone collapse (Fig. 5e). Infection of these neurons with Sindbis pseudovirus that express EGFP-RhoA3’CSE failed to restore Sema3A-mediated collapse. Infection with Sindbis pseudovirus that express EGFP-RhoA3’RhoA restored Sema3A-mediated growth cone collapse (Fig. 5e).
Figure 5.
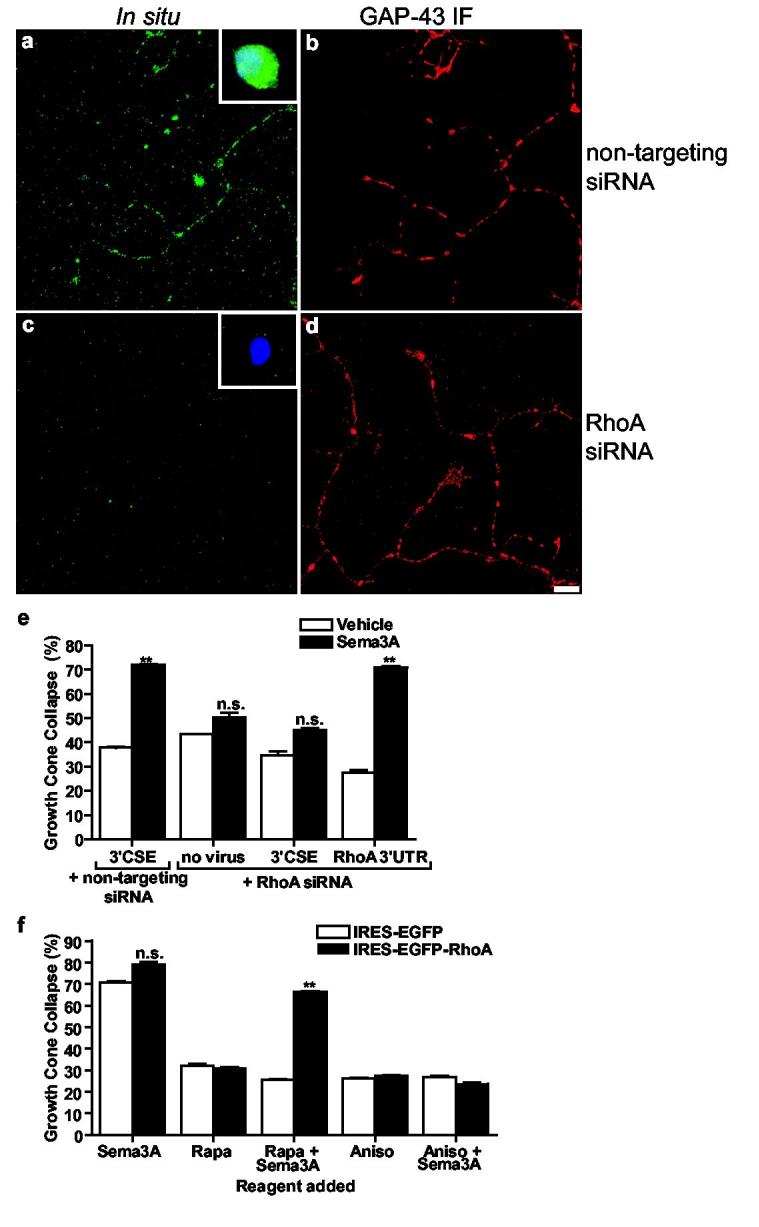
Axonal RhoA translation mediates Sema3A signaling. a-d, siRNA-mediated knockdown of RhoA transcripts in axons. In situ hybridization of RhoA transcripts in DRG axons was unaffected by control siRNA (a), and abolished in neurons transfected RhoA 5'UTR-directed siRNA (c). GAP-43 immunofluorescence (b,d) of axons in a,c. Insets, cell body staining. Scale bar, 10 μm. e, Axonal RhoA transcripts are required for Sema3A-induced collapse. Endogenous RhoA was knocked down with RhoA 5'UTR-directed siRNA, and RhoA was restored via Sindbis pseudoviruses expressing EGFP-RhoA3’CSE or EGFP-RhoA3’RhoA. The domain targeted by the siRNA is absent from the viral constructs. Sema3A-mediated collapse was significantly reduced in RhoA siRNA-transfected neurons infected with pseudovirus expressing EGFP-RhoA3’CSE, but was restored in RhoA siRNA-transfected neurons infected with pseudovirus expressing EGFP-RhoA3’RhoA. n=50 growth cones per condition, **p <0.01. f, Sema3A-mediated growth cone collapse was blocked by rapamycin (10 nM) in axons expressing IRES-EGFP and restored in axons expressing IRES-EGFP-RhoA. n=50 growth cones per condition, **p <0.01.
We next examined the sufficiency of RhoA translation in Sema3A-mediated growth cone collapse. Rapamycin, which blocks Sema3A-mediated growth cone collapse, blocks cap-dependent translation. However, rapamycin does not affect cap-independent translation, such as translation initiated at an internal ribosome entry site (IRES)24. Thus, we generated Sindbis viral constructs that expressed RhoA in axons via an encephalomyocarditis virus IRES (Supplementary Fig. 9). We monitored growth cone collapse in severed axons from DRG explant cultures infected with either IRES-EGFP or IRES-EGFPRhoA (Fig. 5f). Sema3A-mediated growth cone collapse in neurons infected with IRES-EGFP (Fig. 5f) was similar to that observed in uninfected neurons (Supplementary Fig. 1g) and was blocked by rapamycin. We next monitored growth cone collapse in DRG neurons infected with IRES-EGFP-RhoA, which permits EGFP-RhoA translation in the presence of rapamycin. We found that rapamycin treatment failed to block Sema3A-mediated growth cone collapse in neurons that express IRES-EGFP-RhoA (Fig. 5f), indicating that expression of EGFP-RhoA is sufficient to restore growth cone collapse in the presence of rapamycin. Additionally, in neurons that express IRES-EGFP-RhoA, but are not treated with Sema3A, baseline levels of growth cone collapse are not significantly different than in IRES-EGFP-infected neurons, indicating that EGFP-RhoA expression alone is not sufficient for collapse. Sema3A signaling likely involves both translation of RhoA and activation of the newly synthesized protein, perhaps by a Sema3A-regulated guanine nucleotide-exchange factor (GEF). The ability of EGFP-RhoA translation alone to overcome rapamycin blockade of Sema3A suggests that translation of RhoA is required for Sema3A-mediated collapse.
In summary, we have found that RhoA mRNA is enriched in developing axons, and Sema3A-regulated local translation of this transcript mediates cytoskeletal rearrangements in growth cones. Thus, regulation of mRNA translation is an effector pathway of Sema3A, most likely through its receptor Plexin-A25. Given the prominent axonal localization of RhoA mRNA in diverse neuronal types, regulated local RhoA translation is likely to be a widespread mechanism involved in many aspects of neuronal morphogenesis. Local translation permits epistatic regulation of RhoA signaling distinct from regulation achieved by canonical GEF pathways by restricting activation of RhoA and its downstream effectors to sites of RhoA translation. GEFs for Rho family GTPases have been identified that mediate cytoskeletal remodeling in axons but do not display high selectivity for specific Rho family members26. Activation of these GEFs may result in different outcomes that depend on whether RhoA translation pathways have been activated.
Methods
Dorsal root ganglia cultures and collapse assays
E15-16 dissociated DRG neurons27 and explants were plated on glass pre-coated with 33 μg/ml poly-Dlysine and 1 μg/ml fibronectin. DRGs were cultured in B27/F-12/MEM (Invitrogen) supplemented with 75 ng/ml NGF, 40 mM glucose, 10 μM 1-(β-D-arabinofuranosyl)cytosine, and 20 μM 5-fluorodeoxyuridine. RhoA 5'UTR-directed siRNA (target sequence: 5'-AAUGAGCCUUGCAUCUAAGAA-3’) was transfected using GeneSilencer (Genlantis) according to the manufacturer's protocol. siRNA transfection was performed at 2 DIV, 1 day following viral infection. For RhoA rescue, EGFP-RhoA viruses were infected at titers giving equivalent EGFP cell-body fluorescence.
Cycloheximide, anisomycin, rapamycin, or C. botulinum C3 exoenzyme was bath-applied 30 min prior to the application of 450 ng/ml Sema3A (R&D) or vehicle (PBS/0.1% BSA) for 60 min in collapse assays. This concentration of Sema3A was empirically determined to cause the maximal degree of collapse within 60 min. Only axons without varicosities or blebbing and whose growth cones remained healthy for the duration of the experiment were evaluated. Phase-contrast images of the same growth cones were taken at 0 and 60 min. A growth cone was considered collapsed if it had less than two filopodia, each shorter than 10 μm2. Values are presented as percent collapsed growth cones ± s.e.m. and p values were determined using the student _t_-test from experiments repeated a minimum of three times.
For measurement of rapamycin-resistant collapse, DIV2 DRGs were infected with IRES viruses and collapse assays were performed 48 h after infection. Boyden chambers were prepared as described28 with modifications as described in Supplementary Fig. 3.
In situ hybridization
Antisense riboprobes were in vitro transcribed from sense oligonucleotides (Supplementary Table) using MEGAscript (Ambion) with digoxigenin-conjugated UTP.
For fluorescent in situ hybridizations (FISH), DRGs were fixed overnight at 4°C in 4% paraformaldehyde in cytoskeleton buffer (CSB: 10 mM MES pH 6.1, 138 mM KCl, 3 mM MgCl2, 2 mM EGTA, 0.4 M sucrose). Unless indicated, washes were performed in TBST (20 mM Tris pH 8.0, 150 mM NaCl, 0.1% Triton X-100) for 3 × 5 min. DRGs were permeabilized (0.5% Triton X-100/TBS) for 10 min, then post-fixed (4% PFA/TBS) for 5 min, followed by acetylation (0.25% acetic anhydride, 0.1 M HEPES) for 10 min before equilibration with 4X SSC/50% formamide for 20 min. For hybridization, coverslips were incubated with 15 ng riboprobes in 15 μl hybridization buffer (10% dextran sulfate, 4X SSC, 1X Denhardt's Solution, 40% formamide, 20 mM ribonucleoside vanadyl complex, 10 mM DTT, 1 mg/ml yeast tRNA, and 1 mg/ml salmon sperm DNA) at 37°C overnight. The coverslips were washed with 40% formamide/1X SSC at 37°C for 20 min, three times each with 1X SSC and 0.1X SSC. DRGs were blocked (100 mM Tris-HCl pH 8.0, 150 mM NaCl, 8% formamide, 5% BSA, 2.5% normal horse serum, and 2.5% normal goat serum) for 30 min. Anti-digoxin antibody (1:500, DI-22, Sigma) was precleared with rat embryo powder for 2 h at 25°C. Antibodies were as follows: GAP-43, GFAP, Staufen (Chemicon); tau, (Sigma); actin, RhoA (Santa Cruz Biotechnology), and Alexa Fluor 488 and 546 secondary antibodies (Molecular Probes).
Image analysis and quantification of immunofluorescence
DRGs were fixed with 4% PFA in CBS overnight at 4°C, permeabilized with 0.5% Triton X-100/TBS, and blocked in 2% BSA/20 mM Tris-HCl pH 8.0 for 30 min. DRGs were labeled with anti-RhoA (26C4, 1:1000). Similar results were obtained with a RhoA antibody against a separate epitope (sc-179, 1:1000).
For image acquisition of RhoA mRNA (in situ hybridization), RhoA immunofluorescence, or GAP-43 immunofluorescence, exposure times were kept constant and below grey scale saturation. Three-dimensional deconvolution was done with AutoDeblur (AutoQuant).
For RhoA immunofluorescence normalization, the signal in the GAP-43 immunofluorescence stack was thresholded and a volume mask was created around fluorescent objects. This mask was then applied onto the RhoA Z series and the total pixel intensity within this volume was measured. The summed immunofluorescence intensity was normalized to the volume of the growth cone. Quantification of in situ hybridization was performed similarly, using the GAP-43 labeling to demarcate cellular boundaries.
For quantification of myr-dEGFP puncta a stack of images was three-dimensionally deconvoluted. The stack of images was collapsed to a single image using the maximal projection. The fluorescence signal was thresholded, a top-hat filter was applied and regions around the fluorescent puncta were automatically traced. To exclude debris, a size exclusion limit was defined. The regions were transferred to the corresponding picture (0 or 60 minutes, respectively) and the average pixel intensities in the regions were plotted. Image analyses were performed with MetaMorph (Universal Imaging).
Generation and infection of Sindbis virus
We used a Sindbis vector, pSinRep5, containing a point mutation in nsP2 (P726S) that reduces neuronal cytotoxicity30, and the helper plasmid DH-BB (S. Schlesinger, Washington University, St. Louis). The 3’ nontranslated region (3’NTR) of pSinRep5 was replaced with 3’UTRs of interest, preserving the 29-nucleotide CSE required for replication and polyadenylation15. Pseudoviruses were prepared according to the manufacturer's instructions (Invitrogen), purified via sucrose gradient, concentrated, and titered using BHK-21 cells.
Supplementary Material
SI fig legend
Sfig9
Supp table
Sfig1
Sfig2
Sfig3
Sfig4
Sfig5
Sfig6
Sfig7
Sfig8
Acknowledgements
We thank S. Schlesinger (Washington Univ. at St. Louis) for Sindbis plasmids and N. O'Connor (Universal Imaging) for advice on 3-D deconvolution and axonal volume calculation. Supported by the National Institute of Mental Health (S.R.J.), the National Alliance for Autism Research (S.R.J.), the Charles E. Dana foundation, and the Medical Scientist Training Program (E.Z.M.). A.J. is supported by D. Johnston (Baylor Medical College).
Footnotes
Supplementary Information accompanies the paper on Nature's website (http://www.nature.com).
Competing interests statement The authors declare that they have no competing financial interests.
References
- 1.Gallo G, Letourneau PC. Regulation of growth cone actin filaments by guidance cues. Journal of Neurobiology. 2004;58:92–102. doi: 10.1002/neu.10282. [DOI] [PubMed] [Google Scholar]
- 2.Campbell DS, Holt CE. Chemotropic responses of retinal growth cones mediated by rapid local protein synthesis and degradation. Neuron. 2001;32:1013–26. doi: 10.1016/s0896-6273(01)00551-7. [DOI] [PubMed] [Google Scholar]
- 3.Luo Y, Raible D, Raper JA. Collapsin: a protein in brain that induces the collapse and paralysis of neuronal growth cones. Cell. 1993;75:217–227. doi: 10.1016/0092-8674(93)80064-l. [DOI] [PubMed] [Google Scholar]
- 4.Kolodkin AL, et al. Neuropilin is a semaphorin III receptor. Cell. 1997;90:753–762. doi: 10.1016/s0092-8674(00)80535-8. [DOI] [PubMed] [Google Scholar]
- 5.He Z, Tessier-Lavigne M. Neuropilin is a receptor for the axonal chemorepellent Semaphorin III. Cell. 1997;90:739–751. doi: 10.1016/s0092-8674(00)80534-6. [DOI] [PubMed] [Google Scholar]
- 6.Beretta L, Gingras AC, Svitkin YV, Hall MN, Sonenberg N. Rapamycin blocks the phosphorylation of 4E-BP1 and inhibits cap-dependent initiation of translation. EMBO Journal. 1996;15:658–664. [PMC free article] [PubMed] [Google Scholar]
- 7.Dontchev VD, Letourneau PC. Nerve growth factor and semaphorin 3A signaling pathways interact in regulating sensory neuronal growth cone motility. J.Neurosci. 2002;22:6659–6669. doi: 10.1523/JNEUROSCI.22-15-06659.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Olink-Coux M, Hollenbeck PJ. Localization and active transport of mRNA in axons of sympathetic neurons in culture. Journal of Neuroscience. 1996;16:1346–1358. doi: 10.1523/JNEUROSCI.16-04-01346.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bassell GJ, et al. Sorting of β-actin mRNA and protein to neurites and growth cones in culture. Journal of Neuroscience. 1998;18:251–265. doi: 10.1523/JNEUROSCI.18-01-00251.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kanai Y, Dohmae N, Hirokawa N. Kinesin transports RNA: isolation and characterization of an RNA-transporting granule. Neuron. 2004;43:513–525. doi: 10.1016/j.neuron.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 11.Krichevsky AM, Kosik KS. Neuronal RNA granules: a link between RNA localization and stimulation-dependent translation. Neuron. 2001;32:683–696. doi: 10.1016/s0896-6273(01)00508-6. [DOI] [PubMed] [Google Scholar]
- 12.Tang SJ, Meulemans D, Vazquez L, Colaco N, Schuman E. A role for a rat homolog of staufen in the transport of RNA to neuronal dendrites. Neuron. 2001;32:463–475. doi: 10.1016/s0896-6273(01)00493-7. [DOI] [PubMed] [Google Scholar]
- 13.Kohrmann M, et al. Microtubule-dependent recruitment of Staufen-green fluorescent protein into large RNA-containing granules and subsequent dendritic transport in living hippocampal neurons. Molecular Biology of the Cell. 1999;10:2945–53. doi: 10.1091/mbc.10.9.2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kislauskis EH, Zhu X, Singer RH. Sequences responsible for intracellular localization of beta-actin messenger RNA also affect cell phenotype. Journal of Cell Biology. 1994;127:441–451. doi: 10.1083/jcb.127.2.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Raju R, Hajjou M, Hill KR, Botta V, Botta S. In vivo addition of poly(A) tail and AU-rich sequences to the 3’ terminus of the Sindbis virus RNA genome: a novel 3’-end repair pathway. Journal of Virology. 1999;73:2410–2419. doi: 10.1128/jvi.73.3.2410-2419.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tennyson VM. The fine structure of the axon and growth cone of the dorsal root neuroblast of the rabbit embryo. Journal of Cell Biology. 1970;44:62–79. doi: 10.1083/jcb.44.1.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brittis PA, Lu Q, Flanagan JG. Axonal protein synthesis provides a mechanism for localized regulation at an intermediate target. Cell. 2002;110:223–235. doi: 10.1016/s0092-8674(02)00813-9. [DOI] [PubMed] [Google Scholar]
- 18.Eng H, Lund K, Campenot RB. Synthesis of β-tubulin, actin, and other proteins in axons of sympathetic neurons in compartmented cultures. Journal of Neuroscience. 1999;19:1–9. doi: 10.1523/JNEUROSCI.19-01-00001.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Steward O, Schuman EM. Compartmentalized synthesis and degradation of proteins in neurons. Neuron. 2003;40:347–59. doi: 10.1016/s0896-6273(03)00635-4. [DOI] [PubMed] [Google Scholar]
- 20.Aakalu G, Smith WB, Nguyen N, Jiang C, Schuman EM. Dynamic visualization of local protein synthesis in hippocampal neurons. Neuron. 2001;30:489–502. doi: 10.1016/s0896-6273(01)00295-1. [DOI] [PubMed] [Google Scholar]
- 21.John B, et al. Human MicroRNA targets. PLoS Biology. 2004;2:e363. doi: 10.1371/journal.pbio.0020363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen L, Yun SW, Seto J, Liu W, Toth M. The fragile X mental retardation protein binds and regulates a novel class of mRNAs containing U rich target sequences. Neuroscience. 2003;120:1005–1017. doi: 10.1016/s0306-4522(03)00406-8. [DOI] [PubMed] [Google Scholar]
- 23.Aktories K. Rho proteins: targets for bacterial toxins. Trends in Microbiology. 1997;5:282–8. doi: 10.1016/S0966-842X(97)01067-6. [DOI] [PubMed] [Google Scholar]
- 24.Svitkin YV, Hahn H, Gingras AC, Palmenberg AC, Sonenberg N. Rapamycin and wortmannin enhance replication of a defective encephalomyocarditis virus. Journal of Virology. 1998;72:5811–5819. doi: 10.1128/jvi.72.7.5811-5819.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fiore R, Puschel AW. The function of semaphorins during nervous system development. Frontiers in Bioscience. 2003;8:s484–99. doi: 10.2741/1080. [DOI] [PubMed] [Google Scholar]
- 26.Shamah SM, et al. EphA receptors regulate growth cone dynamics through the novel guanine nucleotide exchange factor ephexin. Cell. 2001;105:233–44. doi: 10.1016/s0092-8674(01)00314-2. [DOI] [PubMed] [Google Scholar]
- 27.Svenningsen AF, Shan WS, Colman DR, Pedraza L. Rapid method for culturing embryonic neuron-glial cell cocultures. Journal of Neuroscience Research. 2003;72:565–73. doi: 10.1002/jnr.10610. [DOI] [PubMed] [Google Scholar]
- 28.Zheng JQ, et al. A functional role for intra-axonal protein synthesis during axonal regeneration from adult sensory neurons. Journal of Neuroscience. 2001;21:9291–303. doi: 10.1523/JNEUROSCI.21-23-09291.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tietjen I, et al. Single-cell transcriptional analysis of neuronal progenitors. Neuron. 38:161–75. doi: 10.1016/s0896-6273(03)00229-0. [DOI] [PubMed] [Google Scholar]
- 30.Jeromin A, Yuan LL, Frick A, Pfaffinger P, Johnston D. A modified Sindbis vector for prolonged gene expression in neurons. Journal of Neurophysiology. 2003;90:2741–2745. doi: 10.1152/jn.00464.2003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SI fig legend
Sfig9
Supp table
Sfig1
Sfig2
Sfig3
Sfig4
Sfig5
Sfig6
Sfig7
Sfig8