SIKE is an IKKɛ/TBK1-associated suppressor of TLR3- and virus-triggered IRF-3 activation pathways (original) (raw)
Abstract
Viral infection or TLR3 engagement causes activation of the transcription factors IRF-3 and NF-κB, which collaborate to induce transcription of type I IFN genes. IKKɛ and TBK1 are two IKK-related kinases critically involved in virus- and TLR3-triggered activation of IRF-3. We identified a protein termed SIKE (for Suppressor of IKKɛ) that interacts with IKKɛ and TBK1. SIKE is associated with TBK1 under physiological condition and dissociated from TBK1 upon viral infection or TLR3 stimulation. Overexpression of SIKE disrupted the interactions of IKKɛ or TBK1 with TRIF, RIG-I and IRF-3, components in virus- and TLR3-triggered IRF-3 activation pathways, but did not disrupt the interactions of TRIF with TRAF6 and RIP, components in TLR3-triggered NF-κB activation pathway. Consistently, overexpression of SIKE inhibited virus- and TLR3-triggered interferon-stimulated response elements (ISRE) but not NF-κB activation. Knockdown of SIKE potentiated virus- and TLR3-triggered ISRE but not NF-κB activation. Moreover, overexpression of SIKE inhibited IKKɛ- and TBK1-mediated antiviral response. These findings suggest that SIKE is a physiological suppressor of IKKɛ and TBK1 and plays an inhibitory role in virus- and TLR3-triggered IRF-3 but not NF-κB activation pathways.
Keywords: antiviral response, IKKɛ, IRF-3, NF-κB, TBK1
Introduction
Viral infection results in induction of type I interferons (IFNs), including IFN-β and IFN-α family cytokines, which are critical mediators of antiviral responses (Durbin et al, 2000; Levy and Garcia-Sastre, 2001; Levy and Marie, 2004; Hengel et al, 2005). Transcriptional induction of type I IFN genes requires the coordinate activation of multiple transcription factors and their cooperative assembly into transcriptional enhancer complexes in vivo. For example, the enhancer of the IFN-β gene contains a κB site recognized by NF-κB, a site for ATF-2/c-Jun and two IFN-stimulated response elements (ISREs) recognized by phosphorylated IRF-3 and/or IRF-7 (Maniatis et al, 1998; Wathelet et al, 1998). It has been shown that transcriptional activation of the IFN-β gene requires coordinate and cooperative assembly of an enhanceosome that contains all of these transcription factors (Maniatis et al, 1998; Wathelet et al, 1998).
At least two distinct pathways for the activation of the innate antiviral responses by viral infection have been proposed (Levy and Garcia-Sastre, 2001; tenOever et al, 2002; Levy and Marie, 2004). First, viruses enter cells by membrane fusion at the plasma membrane or through an endocytic process, leading to the release of viral nucleocapsids (or ribonucleoproteins) into the cytoplasm. The viral dsRNA, accumulated during replication of the viral genome, triggers signaling cascades that lead to the activation of IRF-3 and NF-κB and subsequent production of type I IFNs (Levy and Garcia-Sastre, 2001; tenOever et al, 2002). Second, viral dsRNA, released upon lysis of infected cells, binds to TLR3 and triggers signaling pathways leading to IRF-3 and NF-κB activation (Alexopoulou et al, 2001; Yamamoto et al, 2002, 2003; Hoebe et al, 2003; Oshiumi et al, 2003; Han et al, 2004). It has been proposed that TLR3 is important for systemic responses to viral infection but is not required for the initial, cell-autonomous recognition of viral infection that induces the first wave of type I IFN production (Honda et al, 2003; Levy and Marie, 2004; Yoneyama et al, 2004).
The molecular components involved in virus-triggered TLR3-independent activation of IRF-3 and NF-κB have begun to emerge. It has been shown that two serine/threonine kinases, TBK1 (also known as NAK or T2K) and IKKɛ (also known as IKKi), phosphorylate IRF-3 and are involved in virus-triggered induction of type I IFNs in various cell types (Fitzgerald et al, 2003; Sharma et al, 2003; Hemmi et al, 2004; McWhirter et al, 2004; Perry et al, 2004). More recently, a CARD module-containing RNA helicase protein, RIG-I, has been identified as a sensor of cytoplasmic viral dsRNA (Levy and Marie, 2004; Yoneyama et al, 2004). Knockdown or knockout of RIG-I inhibits IFN-β production in response to Newcastle disease virus in different cell types, suggesting that RIG-I is required for virus-triggered signaling (Yoneyama et al, 2004; Kata et al, 2005). It has been further shown that RIG-I-mediated signaling is independent of TLR3 (Yoneyama et al, 2004; Kata et al, 2005).
In addition to virus-triggered TLR3-independent signaling pathways, TBK1 and IKKɛ are also involved in TLR3-mediated IRF-3 activation. It has been shown that TRIF, a TIR domain-containing adapter protein, is associated with TLR3 (Yamamoto et al, 2002, 2003; Hoebe et al, 2003; Oshiumi et al, 2003). TRIF mediates IRF-3 and NF-κB activation through two distinct pathways. TRIF interacts with TBK1 and/or IKKɛ, which phosphorylate IRF-3. TRIF interacts with TRAF6 and RIP, which activate NF-κB through TAK1 and IKK (Hoebe et al, 2003; Sato et al, 2003; Han et al, 2004; Jiang et al, 2004; Meylan et al, 2004).
Since IKKɛ and TBK1 are critically involved in virus-triggered TLR3-dependent and -independent type I IFN signaling, we investigated whether these kinases are regulated by additional proteins. In this report, we performed yeast two-hybrid screens for potential IKKɛ-interacting proteins. This effort identified a protein designated as SIKE (for Suppressor of IKKɛ). Our findings suggest that SIKE is a physiological inhibitor of IKKɛ and TBK1 and plays an inhibitory role in virus- and TLR3-triggered IRF-3 but not NF-κB activation pathways.
Results
Identification of SIKE as an IKK_ɛ_- and TBK1-interacting protein
To identify IKKɛ-interacting proteins, we used the yeast two-hybrid system to screen a human B-cell cDNA library with full-length IKKɛ as bait. We screened a total of ∼8 × 106 independent clones and obtained 123 β-galactosidase positive clones. Four of these clones encoded an uncharacterized protein of 207 amino acids (aa) that was designed as SIKE. Blast searches of the GenBank databases indicated that SIKE is an evolutionarily conserved protein whose orthologs could be found from Xenopus to human (data not shown). An alignment of human and mouse SIKE aa sequences is shown in Figure 1A. Blast searches of the GenBank databases also indicated that SIKE belongs to an uncharacterized protein family, which includes several uncharacterized proteins, such as FGF receptor 1 oncogene partner 2, hematopoietic stem/progenitor cells 123 (HSPC123), HSCP123-like protein and two invertebrate proteins, Schistosoma mansoni circulating cathodic antigen and Drosophila unknown protein CG10158-PA.
Figure 1.
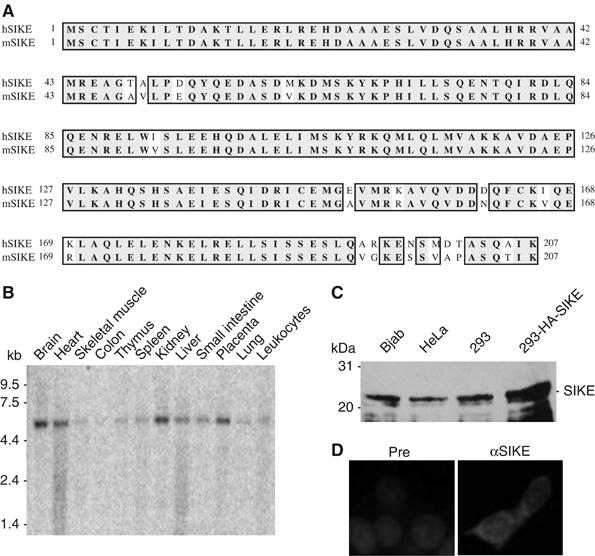
Sequence and expression analysis of SIKE. (A) Alignment of amino-acid sequences of human and mouse SIKE. (B) Tissue expression of SIKE mRNA. A human multiple tissue mRNA blot was hybridized with a probe corresponding to human SIKE cDNA coding sequence. (C) Expression of SIKE protein in mammalian cells. Lysates of the indicated cell types were analyzed by Western blot with a mouse polyclonal anti-SIKE antibody. The 293-HA-SIKE cells were 293 cells transfected with a mammalian expression plasmid for HA-tagged SIKE. (D) SIKE is a cytoplasmic protein. The 293 cells were stained with anti-SIKE antibody and DAPI. Pre, mouse preimmune serum; αSIKE, mouse anti-SIKE antiserum.
Northern blot analysis indicated that human SIKE is ubiquitously expressed as a ∼5.5 kDa band in all examined tissues, including brain, heart, skeletal muscle, colon, thymus, spleen, kidney, liver, small intestine, placenta, lung and leukocytes (Figure 1B). Consistently, blast searches of the GenBank databases identified a human SIKE mRNA sequence of ∼5.5 kb with an open reading frame of 207 aa.
To determine whether SIKE is expressed in mammalian cells at the protein level, we raised mouse polyclonal antiserum against recombinant human SIKE. Western blot analysis indicated that SIKE was expressed as a ∼26 kDa protein in all examined cells, including Bjab, HeLa and 293 cells (Figure 1C). The size of endogenous SIKE was slightly smaller than overexpressed HA-tagged SIKE, suggesting that the identified SIKE cDNA encodes a full-length protein. Immunofluorescent staining experiments indicated that SIKE is a cytoplasmic protein (Figure 1D).
SIKE is associated with IKK_ɛ_ and TBK1
To determine whether SIKE interacts with IKKɛ in mammalian cells, we transfected 293 cells with expression plasmids for HA-tagged SIKE and Flag-tagged IKKɛ. Co-immunoprecipitation experiments indicated that SIKE interacted with IKKɛ (Figure 2A). In these experiments, SIKE also interacted with TBK1 but not RIP (Figure 2A), a kinase that is involved in TNF receptor and TLR3-induced NF-κB activation (Hsu et al, 1996; Meylan et al, 2004).
Figure 2.
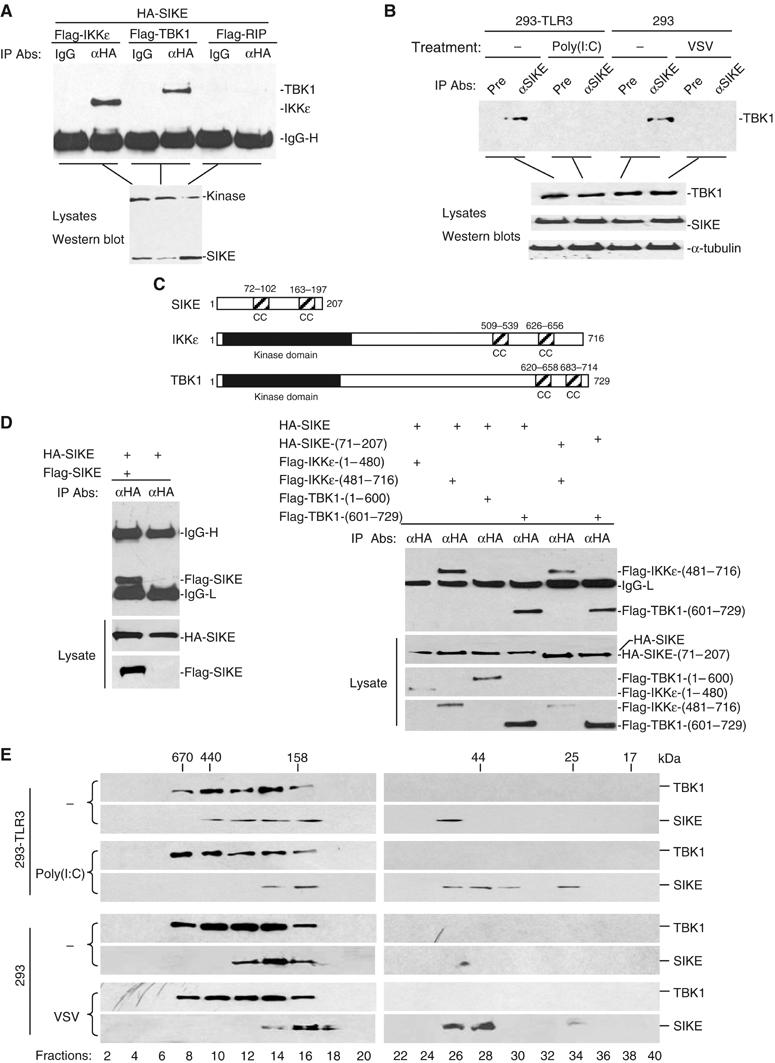
SIKE is associated with IKKɛ and TBK1. (A) SIKE interacts with IKKɛ and TBK1 but not RIP in mammalian overexpression system. The 293 cells (1 × 106) were transfected with the indicated plasmids (5 μg each). Co-immunoprecipitations were performed with anti-HA antibody (αHA) or control mouse IgG (IgG). Western blot was performed with anti-Flag antibody (upper panel). Expression of the transfected plasmids was confirmed by Western blot analysis of the lysates with anti-HA and anti-Flag antibodies (lower panel). (B) Association of SIKE with TBK1 and effects of stimulation on the association in untransfected cells. The 293-TLR3 or 293 cells (5 × 107) were left untreated or treated with poly(I:C) (50 μg/ml) for 10 min or infected with VSV for 4 h. The cells were lysed and the lysates were immunoprecipitated with anti-SIKE antiserum or preimmune control serum. The immunoprecipitates were analyzed by Western blots with anti-TBK1 antibody (upper panel). The expression levels of the endogenous proteins were detected by Western blot analysis with anti-TBK1, anti-SIKE and anti-α-tubulin antibodies (lower panels). (C) A schematic presentation of the coiled-coil motifs identified in SIKE, IKKɛ and TBK1. CC, coiled coil. (D) SIKE interacts with itself, IKKɛ and TBK1 through their respective coiled-coil motifs. The 293 cells (1 × 106) were transfected with the indicated plasmids (5 μg each) and co-immunoprecipitations were performed as in (A). (E) Analysis of protein complexes containing SIKE and TBK1 by size-exclusion chromatography. The 293-TLR3 (2 × 108) cells were treated with poly(I:C) (50 μg/ml) for 10 min or left untreated. The 293 cells were infected with VSV for 4 h or left uninfected before lysis. Cell lysates were analyzed by size-exclusion chromatography on Superdex 200 column. The individual fractions were analyzed by Western blots with anti-TBK1 and anti-SIKE antibodies, respectively.
Previously, it has been demonstrated that TBK1 is constitutively expressed in most cell types, while IKKɛ is undetectable under physiological conditions but inducible in lymphoid and other cell types (Pomerantz and Baltimore, 1999; Shimada et al, 1999; Peters et al, 2000). Therefore, we examined whether SIKE is associated with TBK1 in untransfected cells. In co-immunoprecipitation experiments, endogenous SIKE interacted with endogenous TBK1 under physiological condition. This interaction was significantly decreased following virus infection or poly(I:C) treatment of TLR3-expressing 293 cells (Figure 2B). These data suggest that SIKE is constitutively associated with TBK1 under physiological condition, but dissociated from TBK1 upon virus infection or TLR3 activation.
Since SIKE interacts with IKKɛ and TBK1, we performed structural analysis to determine whether they are structurally related to each other. Using the paircoil program, we found that SIKE contains two coiled-coil motifs at aa72–102 and aa163–197, respectively (Figure 2C). Interestingly, IKKɛ and TBK1 also contain two potential coiled-coil motifs at their C-terminuses (Figure 2C). Since coiled-coil motifs mediate homophilic multimerization, we determined whether SIKE could dimerize or oligomerize. In co-immunoprecipitation experiments, Flag-tagged SIKE could interact with HA-tagged SIKE (Figure 2D), suggesting that SIKE could form homodimmers or -oligomers.
We also made series of mutants of SIKE, IKKɛ and TBK1 and performed domain mapping experiments. We found that SIKE interacted with IKKɛ and TBK1 through their respective coiled-coil domains (Figure 2D).
To further characterize the association of SIKE with TBK1, we examined the biochemical nature of SIKE and TBK1 in untransfected cells. In these experiments, 293-TLR3 cells were treated with poly(I:C) for 10 min or left untreated, or 293 cells were infected with vesicular stomatitis virus (VSV) for 4 h or left uninfected before lysis. Cell lysates were separated by size-exclusion chromatography and individual fractions were analyzed by Western blots with antibodies against SIKE and TBK1, respectively. As shown in Figure 2E, SIKE and TBK1 exist in overlapping fractions that eluted in the 158–670 kDa range in uninfected cells. However, following poly(I:C) treatment or VSV infection, a significant part of SIKE was shifted to fractions peaking at about 25 and 44 kDa. Based on these data, we propose that SIKE is associated with TBK1, as well as unidentified proteins, as complexes of 158–670 kDa under physiological conditions. Upon poly(I:C) stimulation or viral infection, SIKE is dissociated from the TBK1-containing complexes to become free monomer (in 25 kDa fraction) or dimer (in 44 kDa fraction) (Figure 2E).
In the size-exclusion chromatography experiments, the molecular weights of TBK1-containing complexes were not decreased following virus infection or poly(I:C) treatment. It is possible that the molecular weight of dissociated SIKE is too small to have apparent effect on the molecular weights of TBK1-containing complexes. Alternatively, virus infection or poly(I:C) treatment may cause both dissociation of SIKE from the TBK1-containing complexes and recruitment of other proteins to the TBK1-containing complexes.
SIKE inhibits IKK_ɛ_- and TBK1-mediated activation of ISRE and the IFN-β promoter
Overexpression of IKKɛ and TBK1 activates both ISRE and NF-κB (Pomerantz and Baltimore, 1999; Peters et al, 2000; Fitzgerald et al, 2003; Han et al, 2004). Since SIKE interacts with IKKɛ and TBK1, we determined the effects of SIKE on IKKɛ- and TBK1-mediated signaling. In reporter gene assays, SIKE inhibited both IKKɛ- and TBK1-mediated activation of ISRE and the IFN-β promoter (Figure 3A). In similar experiments, SIKE did not inhibit IKKɛ- and TBK1-mediated NF-κB activation (Figure 3A). These results suggest that SIKE specifically inhibits IKKɛ- and TBK1-mediated IRF-3 pathways.
Figure 3.
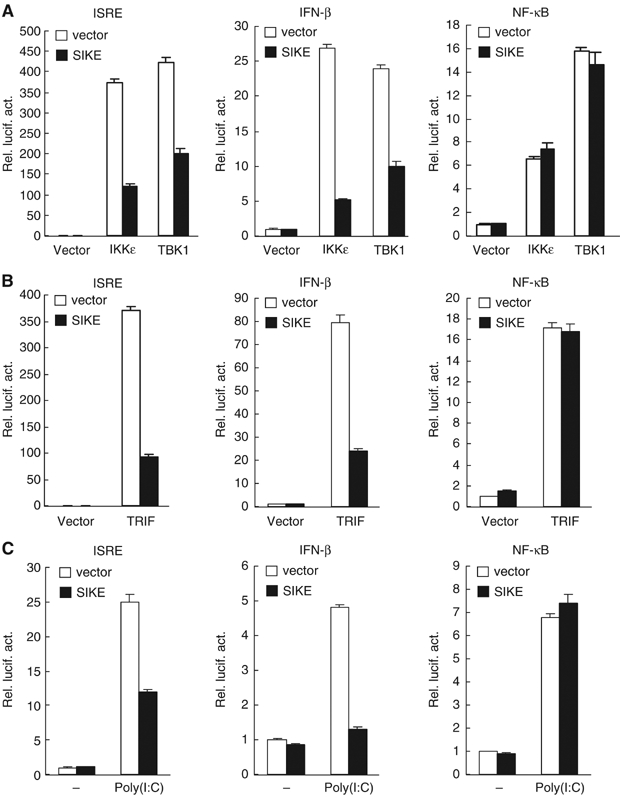
SIKE inhibits IKKɛ-, TBK1-, TRIF- and TLR3-mediated activation of ISRE and the IFN-β promoter but not NF-κB. (A) SIKE inhibits IKKɛ- and TBK1-mediated activation of ISRE and the IFN-β promoter but not NF-κB. The 293 cells (1 × 105) were transfected with the indicated luciferase reporter plasmid (0.1 μg), an expression plasmid for IKKɛ or TBK1 (0.1 μg) and an empty control plasmid (open bars) or an expression plasmid for SIKE (filled bars) (1 μg). Luciferase assays were performed 16 h after transfection. (B) SIKE inhibits TRIF-mediated activation of ISRE and the IFN-β promoter but not NF-κB. The experiments were similarly performed as in (A), except that a TRIF expression plasmid (0.1 μg) was used to replace the IKKɛ or TBK1 plasmid. (C) SIKE inhibits poly(I:C)-induced activation of ISRE and the IFN-β promoter but not NF-κB. The 293-TLR3 cells (1 × 105) were transfected with the indicated luciferase reporter plasmid (0.1 μg) and an empty control vector (empty bars) or a SIKE expression plasmid (filled bars) (1 μg). At 14 h after transfection, cells were treated with poly(I:C) (50 μg/ml) or left untreated for 6 h before luciferase assays were performed.
SIKE inhibits TLR3-mediated activation of ISRE and the IFN-β promoter
Since IKKɛ and TBK1 are critically involved in TLR3-triggered ISRE but not NF-κB activation pathways (Fitzgerald et al, 2003; Sharma et al, 2003; Hemmi et al, 2004; McWhirter et al, 2004; Perry et al, 2004), we determined the effects of SIKE on these pathways. TRIF is a signaling component upstream of IKKɛ and TBK1 in TLR3-mediated signaling. In reporter gene assays, SIKE inhibited TRIF-mediated activation of ISRE and the IFN-β promoter (Figure 3B), but had no effect on TRIF-mediated NF-κB activation (Figure 3B). In a poly(I:C)-responsive 293 cell line that stably expresses TLR3, SIKE inhibited poly(I:C)-induced activation of ISRE and the IFN-β promoter but not NF-κB (Figure 3C). In similar experiments, SIKE did not inhibit IRF3-mediated activation of ISRE and the IFN-β promoter (data not shown). These data suggest that SIKE specifically inhibits TLR3-mediated ISRE but not NF-κB activation pathways at TBK1 and/or IKKɛ level.
SIKE disrupts the interactions of IKK_ɛ_ or TBK1 with TRIF and IRF-3
One of the simplest explanations for the inhibitory role of SIKE on TLR3-mediated ISRE activation is that SIKE sequesters IKKɛ and/or TBK1 in an inactive complex and blocks their interactions with TRIF and/or IRF-3, two signaling components that are associated with IKKɛ and TBK1 in the TLR3-mediated ISRE activation pathway. To test this, we performed transient transfection and co-immunoprecipitation experiments. In these experiments, either IKKɛ or TBK1 interacted with TRIF and IRF-3 as expected. The interactions were completely disrupted with the addition of SIKE (Figure 4A and B). In similar experiments, SIKE did not disrupt the interactions of TRIF with TRAF6 and RIP, two components that are involved in TLR3-mediated NF-κB activation pathway (Figure 4C). Since SIKE did not interact with TRIF and IRF-3 in co-immunoprecipitation experiments (data not shown), these results suggest that SIKE specifically blocks the interactions of IKKɛ or TBK1 with TRIF or IRF-3 by its association with IKKɛ or TBK1.
Figure 4.
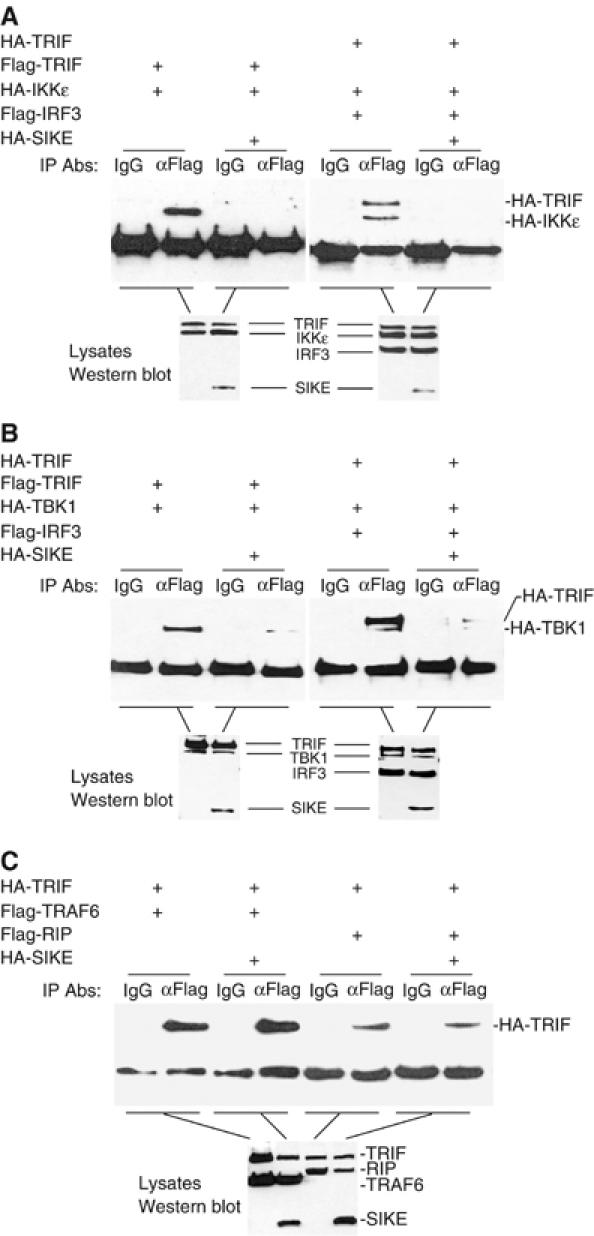
Effects of SIKE on assembly of the TRIF-IKKɛ-IRF-3, TRIF-TBK1-IRF3 and TRIF-TRAF6-RIP complexes. (A) SIKE disrupts the TRIF-IKKɛ-IRF-3 complex. The 293 cells (1 × 106) were transfected with 5 μg of the indicated plasmids. Cell lysates were immunoprecipitated with control mouse IgG (IgG) or anti-Flag (αFlag) antibody. The immunoprecipitates were analyzed by Western blot with anti-HA antibody (upper panel). The expression levels of the transfected proteins were detected by Western blot analysis with anti-HA and anti-Flag antibodies (low panel). (B) SIKE disrupts the TRIF-TBK1-IRF-3 complex. The experiments were carried out similarly as in (A), except that TBK1 plasmid was used to replace IKKɛ plasmid. (C) SIKE does not disrupt the interactions of TRIF with TRAF6 or RIP. The 293 cells were transfected with the indicated plasmids and the co-immunoprecipitation and Western blot analysis were carried out as in (A).
SIKE inhibits virus-triggered TLR3-independent activation of ISRE and the IFN-β promoter
In addition to TLR3-mediated ISRE activation, IKKɛ and TBK1 are also involved in virus-triggered TLR3-independent activation of ISRE and the IFN-β promoter. Recent studies have indicated that RIG-I is a sensor of viral dsRNA in the cytoplasm and is critically involved in virus-triggered TLR3-indepenent signaling (Levy and Marie, 2004; Yoneyama et al, 2004). Overexpression of the CARD modules of RIG-I activates ISRE, NF-κB and the IFN-β promoter (Yoneyama et al, 2004). Therefore, we examined the effects of SIKE on virus-induced and RIG-I-mediated TLR3-independent signaling. The parent 293 cells do not express TLR3 and are not responsive to poly(I:C) (Yoneyama et al, 2004 and data not shown). In these cells, SIKE inhibited VSV-induced and RIG-I-mediated activation of ISRE and the IFN-β promoter (Figure 5A and B). In similar experiments, SIKE did not inhibit VSV-induced and RIG-I-mediated activation of NF-κB (Figure 5C).
Figure 5.
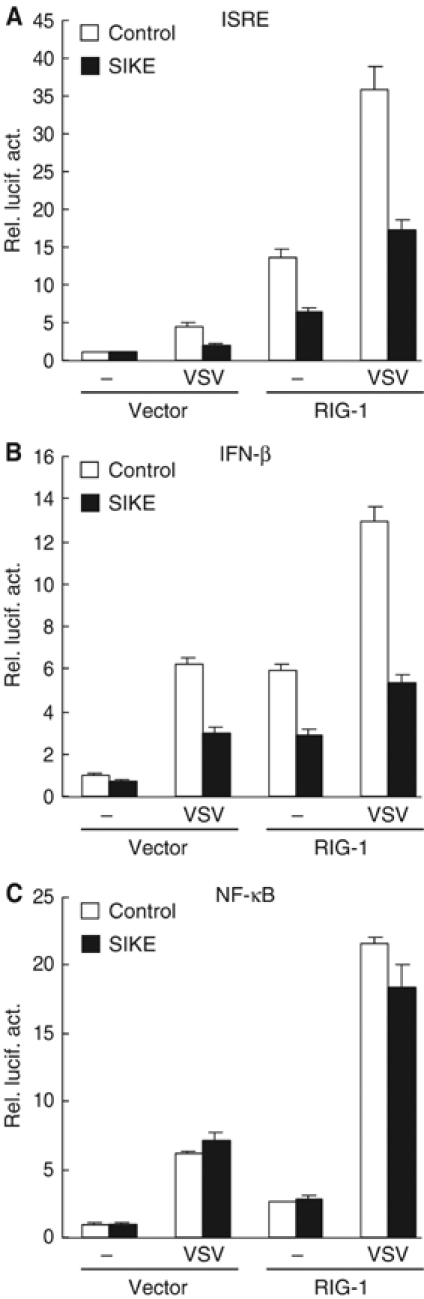
SIKE inhibits VSV-induced and RIG-I-mediated activation of ISRE (A) and the IFN-β promoter (B) but not NF-κB (C). The 293 cells (1 × 105) were transfected with the indicated luciferase reporter plasmid (0.1 μg), an empty control vector or an expression plasmid for RIG-I (1 μg) as indicated, and an empty control vector (empty bars) or an expression plasmid for SIKE (filled bars). At 14 h after transfection, cells were infected with VSV or left infected for 18 h before luciferase assays were performed.
SIKE blocks the interaction of IKK_ɛ_ and TBK1 with RIG-I
It has been demonstrated that both RIG-I and IKKɛ or TBK1 are involved in virus-triggered TLR3-independent signaling (Hemmi et al, 2004; Levy and Marie, 2004; Perry et al, 2004; tenOever et al, 2004). However, the relationship between RIG-I and IKKɛ or TBK1 has not been revealed. We found that RIG-I could interact with IKKɛ and TBK1 in co-immunoprecipitation experiments (Figure 6A and B). The CARD modules but not the helicase domain of RIG-I is responsible for its interaction with IKKɛ and TBK1 (Figure 6A and B). In these experiments, the interactions of IKKɛ and TBK1 with RIG-I and its CARD modules were completely disrupted by the addition of SIKE (Figure 6A and B).
Figure 6.
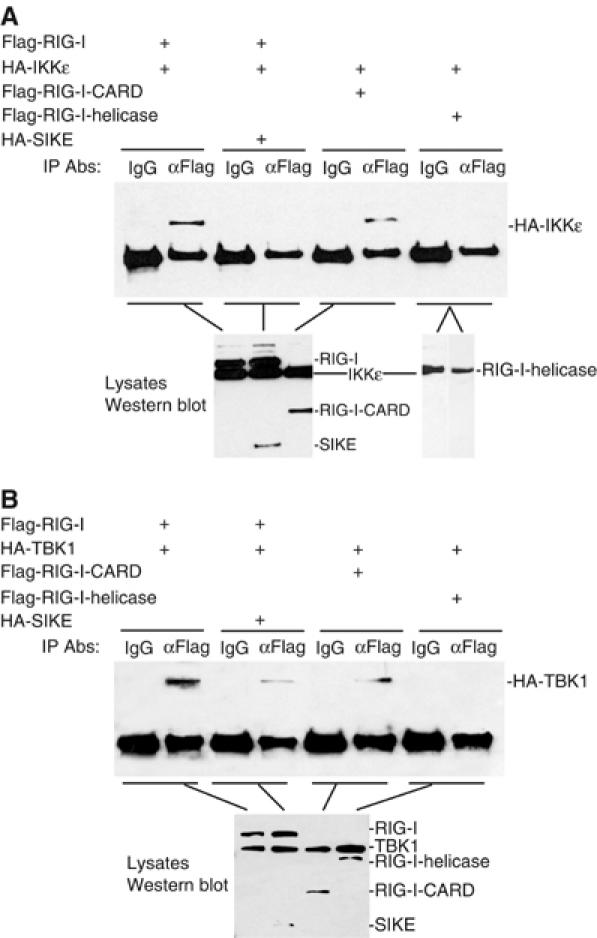
SIKE disrupts the interactions of RIG-I with IKKɛ or TBK1. (A) SIKE disrupts the interaction of IKKɛ with RIG-I. The 293 cells (1 × 106) were transfected with 5 μg of the indicated plasmids. Cell lysates were immunoprecipitated with control mouse IgG (IgG) or anti-Flag (αFlag) antibody. The immunoprecipitates were analyzed by Western blot with anti-HA antibody (upper panel). The expression levels of the transfected proteins were detected by Western blot analysis with anti-HA and anti-Flag antibodies (low panels). (B) SIKE disrupts the interaction of TBK1 with RIG-I. The experiments were carried out as in (A), except that the TBK1 plasmid was used.
Knockdown of SIKE expression potentiates poly(I:C)- and VSV-induced activation of ISRE and the IFN-β promoter
To further determine whether SIKE is involved in the regulation of the ISRE activation pathways under physiological conditions, we constructed five human SIKE RNAi plasmids. One of these RNAi plasmids (#4) could significantly inhibit the expression of transfected and endogenous SIKE in 293 cells as suggested by Western blot analysis (Figure 7A). In reporter gene assays, this SIKE RNAi plasmid potentiated poly(I:C)- and VSV-triggered activation of ISRE and the IFN-β promoter but not NF-κB (Figure 7B–G). These data suggest that SIKE is a physiological inhibitor of TLR3- and virus-triggered ISRE but not NF-κB activation pathways.
Figure 7.
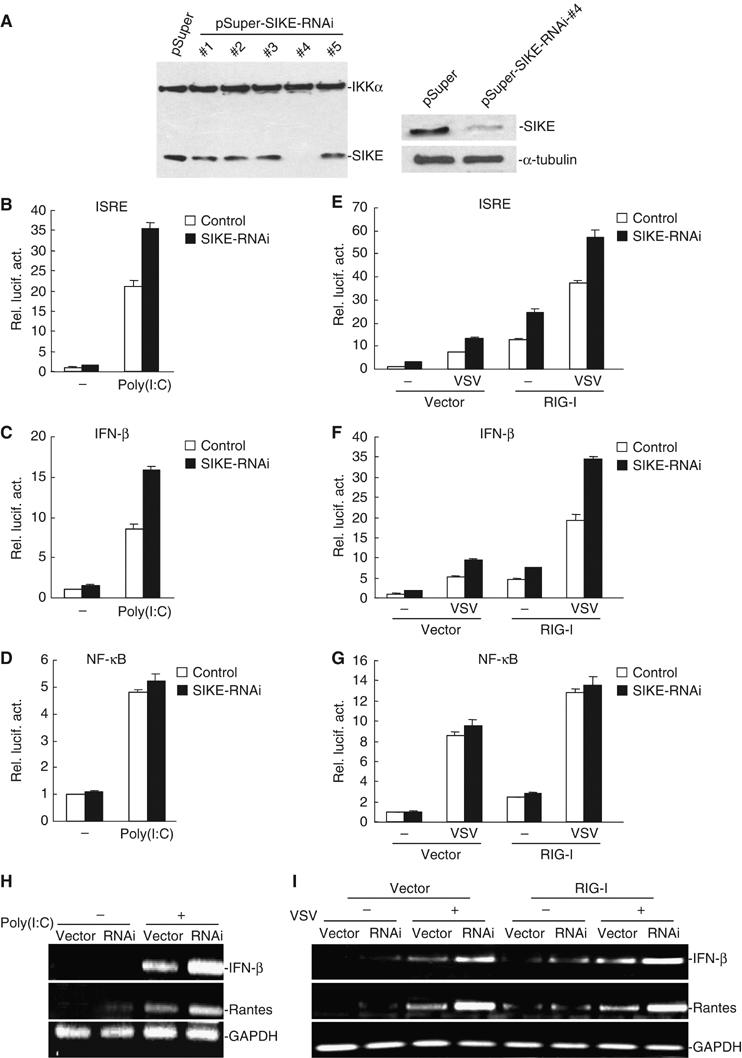
Effects of SIKE RNAi on poly(I:C)- and VSV-induced activation of ISRE, NF-κB and the IFN-β promoter. (A) Effects of SIKE RNAi plasmids on the expression of transfected and endogenous SIKE. In the left panel, 293 cells (1 × 105) were transfected with expression plasmids for Flag-SIKE and Flag-IKKα (0.5 μg each), and the indicated RNAi plasmids (1 μg). At 24 h after transfection, cell lysates were analyzed by Western blot with anti-Flag antibody. In the right panels, 293 cells (1 × 105) were transfected with 1 μg of pSuper.Retro or the #4 SIKE RNAi plasmid for 24 h. Cell lysates were then analyzed by Western blots with a mouse anti-SIKE (upper panel) or a monoclonal anti-α-tubulin (lower panel) antibody. (B–D) SIKE RNAi potentiates poly(I:C)-induced activation of ISRE and the IFN-β promoter but not NF-κB in 293-TLR3 cells. The 293-TLR3 cells (1 × 105) were transfected with 0.1 μg of the indicated reporter plasmid, 0.1 μg of pRL-TK Renilla luciferase plasmid, 1 μg of the #4 SIKE RNAi or pSuper.Retro control plasmid as indicated. At 36 h after transfection, cells were treated with poly(I:C) (50 μg/ml) (filled bars) or left untreated (open bars) for 6 h before luciferase assays were performed. (E–G) SIKE RNAi potentiates VSV-induced RIG-I-mediated activation of ISRE and the IFN-β promoter but not NF-κB in 293 cells. The 293 cells (1 × 105) were transfected with 0.1 μg of the indicated reporter plasmid, 0.1 μg of pRL-TK Renilla luciferase plasmid, 1 μg of an expression plasmid for RIG-I, 1 μg of the #4 SIKE RNAi (filled bars) or pSuper.Retro control plasmid (open bars) as indicated. At 36 h after transfection, cells were infected with VSV or left uninfected for 18 h before luciferase assays were performed. (H) SIKE RNAi potentiated basal as well as poly(I:C)-induced expression of endogenous IFN-β and Rantes mRNA. The 293-TLR3 cells were transfected with control or SIKE RNAi plasmid. At 36 h after transfection, cells were treated with poly(I:C) (50 μg/ml) for 6 h before RT–PCR was performed. (I) SIKE RNAi potentiated virus-triggered RIG-I-mediated expression of endogenous IFN-β and Rantes mRNA. The 293 cells were transfected with control or SIKE RNAi plasmid, together with empty control or RIG-I expression plasmid as indicated. At 36 h after transfection, cells were infected with VSV or left uninfected for 18 h before RT–PCR was performed.
Since SIKE inhibits TLR3- and virus-triggered ISRE activation, we further examined whether SIKE inhibits TLR3-mediated and virus-triggered expression of endogenous IFN-β and Rantes. In reverse-transcription polymerase chain reaction (RT–PCR) experiments, we found that SIKE RNAi potentiated basal as well as poly(I:C)- and virus-triggered expression of endogenous IFN-β and Rantes mRNA (Figure 7H and I). These data suggest that SIKE is a physiological inhibitor of TLR3-mediated and virus-triggered gene expression.
SIKE inhibits IKK_ɛ_- and TBK1-mediated antiviral response
IKKɛ and TBK1 have been demonstrated to be required for establishing the antiviral state and this activity is IRF3-dependent. Since SIKE could inhibit IKKɛ- and TBK1-mediated activation of ISRE and the IFN-β promoter, we determined whether SIKE has effects on IKKɛ- and TBK-mediated antiviral response. We transfected 293 cells with IKKɛ/TBK1, together with a control plasmid or a SIKE expression or RNAi plasmid. The transfected cells were infected with VSV and replicated viruses released into the medium were quantitated by standard plaque assays. The results indicated that VSV production in the presence of IKKɛ/TBK1 decreased by ∼4 logs to 104 plaque-forming units (PFU)/ml at 12 h postinfection (Figure 8), which is consistent with the data reported previously (Sharma et al, 2003). Overexpression of SIKE restored the VSV production to more than 106 PFU/ml (Figure 8A), while SIKE RNAi potentiated IKKɛ- and TBK1-mediated inhibition of VSV production (Figure 8B). These data indicate that SIKE is a physiological inhibitor of IKKɛ- and TBK1-mediated antiviral response.
Figure 8.
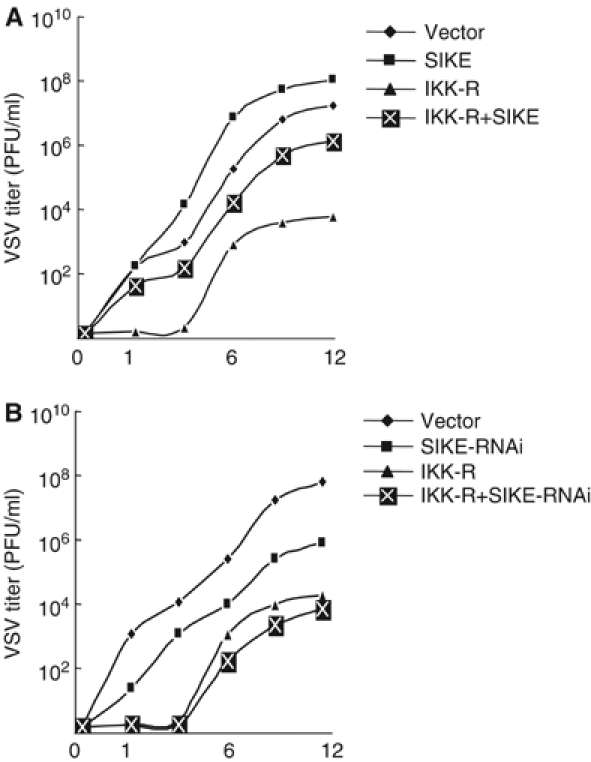
Effects of SIKE on IKKɛ- and TBK1-mediated antiviral response. (A) SIKE inhibits IKKɛ- and TBK1-mediated antiviral response. The 293 cells (1 × 105) were transfected with IRF-3 expression plasmid (2 μg) together with an empty control plasmid (2 μg), SIKE (2 μg), IKK-R (IKKɛ+TBK1, each 0.5 μg) or IKK-R+SIKE (0.5 μg of IKKɛ, 0.5 μg of TBK1 and 2 μg of SIKE). At 24 h after transfection, cells were infected with VSV (MOI 10) and supernatants were harvested at 0, 1, 3, 6, 9 and 12 h postinfection. Supernatants were analyzed for VSV production using standard plaque assays. Plaques were counted and titers calculated as PFU/ml. (B) Knockdown of SIKE potentiates IKKɛ- and TBK1-mediated antiviral response. The experiments were carried out similarly as in (A), except that SIKE-RNAi plasmid was used to replace SIKE expression plasmid and the 293 cells were infected with VSV at 36 h after transfection.
Discussion
The immune system has evolved distinct virus recognition strategies. Viruses can enter cells by membrane fusion or through endocytosis. The viral dsRNA accumulated during replication of the viral genome is detected by the RNA helicase protein RIG-I or MDA5 in the cytoplasm (Andrejeva et al, 2004; Yoneyama et al, 2004). During late stage of infection, viral dsRNA is released upon lysis of infected cells and recognized by TLR3 (Alexopoulou et al, 2001; Levy and Marie, 2004). These distinct virus recognition strategies trigger signaling pathways that converge on activation of the transcription factors NF-κB and IRF-3, leading to transcription of the antiviral type I IFNs. In these converged pathways, two noncanonical IKK family members, IKKɛ and TBK1, phosphorylate IRF-3 and are critically involved in virus-triggered TLR3-independent and TLR3-mediated induction of type I IFNs (Fitzgerald et al, 2003; Hoebe et al, 2003; Oshiumi et al, 2003; Sharma et al, 2003; Yamamoto et al, 2003; Han et al, 2004; Hemmi et al, 2004; McWhirter et al, 2004; Perry et al, 2004).
Although it is clear that IKKɛ and TBK1 play essential roles in antiviral responses in various cell types, how their activities are regulated is unknown. To get insight into this, we searched for additional IKKɛ-interacting proteins by the yeast two-hybrid system. This effort identified SIKE, an evolutionarily conserved protein.
In mammalian overexpression systems, SIKE interacts with both IKKɛ and TBK1 (Figure 2A). These interactions are mediated through their respective coiled-coil motifs (Figure 2D). We further demonstrated that SIKE is associated with TBK1 in untransfected cells. Upon viral infection or TLR3 engagement, SIKE is dissociated from the TBK1 complex as suggested by co-immunoprecipitation and gel filtration experiments (Figure 2B and E). Since IKKɛ is undetectable under physiological conditions in most cell types (Shimada et al, 1999; Peters et al, 2000), an endogenous interaction between SIKE and IKKɛ is not confirmed. It is possible that SIKE is associated with IKKɛ when IKKɛ is induced by certain stimuli.
In mammalian overexpression system, SIKE blocked the interactions of IKKɛ or TBK1 with TRIF, RIG-I and IRF-3 (Figures 4 and 6), signaling components required for TLR3-mediatd or virus-triggered IRF-3 activation (Yamamoto et al, 2002; Honda et al, 2003; Levy and Marie, 2004; Yoneyama et al, 2004). In reporter gene assays, SIKE inhibited activation of ISRE and the IFN-β promoter mediated by IKKɛ, TBK1, TRIF and RIG-I, as well as triggered by poly(I:C) and VSV (Figures 3 and 5), while knockdown of SIKE potentiated activation of ISRE and the IFN-β promoter triggered by poly(I:C) and VSV infection (Figure 7). In VSV replication experiments, SIKE inhibited IKKɛ- and TBK1-mediated antiviral response, while knockdown of SIKE potentiated IKKɛ- and TBK1-mediated antiviral state (Figure 8).
SIKE does not disrupt the interactions of TRIF with TRAF6 and RIP (Figure 4C), components required for TLR3-mediated NF-κB activation (Hoebe et al, 2003; Sato et al, 2003; Han et al, 2004; Jiang et al, 2004; Meylan et al, 2004). Consistently, SIKE does not inhibit NF-κB activation triggered by TNF, poly(I:C) and VSV. These results suggest that SIKE specifically inhibits IKKɛ- and TBK1-mediated ISRE activation pathways.
Since SIKE is associated with TBK1 under physiological conditions and dissociated with TBK1 upon TLR3 engagement or viral infection (Figure 2), the simplest explanation for our observations is that SIKE functions as a physiological suppressor of IKKɛ and TBK1 by sequestering IKKɛ and TBK1 in inactive complexes. SIKE-mediated regulation of signaling is not caused by change of its protein level, because SIKE protein level was not significantly changed following TLR3 engagement or VSV infection (Figure 2B and data not shown). Although the experiments carried out in this study are not enough to offer a precise explanation on the regulatory mechanisms of SIKE in TLR3-mediated or virus-triggered signaling, some plausible scenarios can be envisioned. For example, upon TLR3 engagement or viral infection, the recruitment of TRIF to TLR3, or binding of RIG-I to dsRNA, may cause their conformational changes and thus provide a high-affinity binding site for IKKɛ/TBK1, causing dissociation of SIKE with IKKɛ/TBK1 and subsequent activation of IKKɛ/TBK1. Alternatively, poly(I:C) engagement or viral infection may directly signal certain post-translational modifications or conformational changes of IKKɛ/TBK1 or SIKE, causing their dissociation and enabling the association of IKKɛ/TBK1 with TRIF or RIG-I.
Since type I IFNs play a vital role in innate antiviral response and provide potent regulatory interactions with the adaptive immune system for efficient virus elimination, they are tightly regulated at transcription level. Expression of type I IFNs is nearly undetectable in uninfected individuals, but rapidly induced to high concentrations following infection. The identification of a physiological suppressor of IKKɛ and TBK1 provides an explanation on the inability of these kinases to activate the IFN pathways under physiological conditions.
Materials and methods
Reagents
Mouse monoclonal and rabbit polyclonal antibodies against Flag and HA epitopes (Sigma, St Louis, MO), and rabbit polyclonal antibodies against NAK (TBK1) (eBioscience 14-6351) were purchased from the indicated manufacturers. VSV was provided by Dr Hong-Kui Deng. Mouse anti-SIKE and anti-IKKɛ antisera were raised against recombinant human SIKE(aa26–207) and IKKɛ proteins.
Yeast two-hybrid screening
To construct an IKKɛ bait vector, a cDNA fragment encoding full-length human IKKɛ was inserted in-frame into the Gal4 DNA-binding domain vector pGBT (Clontech, Palo Alto, CA). A human B-cell cDNA library (ATCC, Manassas, VA) was screened following protocols recommended by Clontech.
Immunofluorescent staining
The 293 cells cultured on glass coverslips were plunged sequentially into methanol and acetone at −20°C, each for 10 min. The cells were rehydrated in PBS, blocked with 1% bovine serum albumin in PBS for 15 min and stained with primary antibody in blocking buffer for 1 h at room temperature. The cells were rinsed with PBS and stained with Texas Red-conjugated Affinipure goat anti-mouse IgG (1:200 dilution) for 45 min at room temperature. The cells were then rinsed with PBS containing DAPI and mounted in Prolong Antifade (Molecular Probes). The cells were observed with a Leica DMR/XA immunofluorescent microscope using a × 100 plan objective.
Northern blot analysis
Human multiple tissue mRNA blot was purchased from Clontech. The blot was hybridized with 32P-dCTP-labeled cDNA probe corresponding to human SIKE coding sequence. Hybridization was performed in the Rapid Hybridization Buffer (Clontech) under high stringency conditions.
Constructs
Mammalian expression plasmid pcDNA3-Flag-IKKɛ was provided by Dr Tom Maniatis. Mammalian expression plasmids for SIKE and its deletion mutants were constructed by PCR amplification of SIKE cDNA from human B-cell library and subsequently cloning into CMV promoter-based vectors containing an HA or Flag tag. HA-IKKɛ and its deletion mutants were constructed by PCR amplification of the IKKɛ cDNA from the pcDNA3-Flag-IKKɛ plasmid and subsequently cloning into a CMV promoter-based vector containing a tag. TBK1 deletion mutants were constructed by PCR amplification of the TBK1 cDNA from the PRK-Flag-TBK1 plasmid and subsequently cloning into a CMV promoter-based vector containing a tag. Mammalian expression plasmids for Flag-IKKα, Flag-TBK1, HA-TBK1, Flag-RIP, Flag- and HA-TRIF, Flag-TRAF6, Flag-IRF-3, Flag-RIG-1 and its deletion mutants were described previously (Hu et al, 1999; Bin et al, 2003; Han et al, 2004). ISRE-luciferase reporter plasmid was purchased from Stratagene (West Cedar Creek, TX). NF-κB luciferase reporter plasmid was provided by Dr Gary Johnson. Human IFN-β promoter luciferase reporter plasmid was described previously (Han et al, 2004).
Transfection and reporter gene assays
The 293 cells (∼1 × 105) were seeded on 12-well dishes and transfected the following day by standard calcium phosphate precipitation. In the same experiment, where necessary, empty control plasmid was added to ensure that each transfection receives the same amount of total DNA. To normalize for transfection efficiency, 0.1 μg of pRL-TK Renilla luciferase reporter plasmid was added to each transfection. Approximately 16 h after transfection, luciferase assays were performed using a dual-specific luciferase assay kit (Promega, Madison, WI). Firefly luciferase activities were normalized based on Renilla luciferase activities. All reporter gene assays were repeated for at least three times. Data shown were average values±s.d. from one representative experiment.
Co-immunoprecipitation and Western blot analysis
For transient transfection and co-immunoprecipitation experiments, 293 cells (∼1 × 106) were transfected for 24 h. The transfected cells were lysed in 1 ml of lysis buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 1% Triton, 1 mM EDTA, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 1 mM phenylmethylsulfonyl fluoride). For each immunoprecipitation, 0.4 ml aliquot of lysate was incubated with 0.5 μg of the indicated antibody or control IgG and 25 μl of a 1:1 slurry of GammaBind G Plus-Sepharose (Amersham Biosciences) for 2 h. The Sepharose beads were washed three times with 1 ml of lysis buffer containing 500 mM NaCl. The precipitates were analyzed by Western blots as described (Huang et al, 2004).
For endogenous co-immunoprecipitation experiments, cells (5 × 107) were treated with poly(I:C) (50 μg/ml) for 10 min or infected with VSV virus for 4 h or left untreated. Cells were then lysed in 5 ml lysis buffer and the lysate was incubated with 1 μl of the indicated antiserum or preimmune control serum. The subsequent procedures were carried out as described above.
Size-exclusion chromatography
Cells (2 × 108) were lysed in 1.5 ml lysis buffer. The lysate was centrifuged for 1 h at 15 000 r.p.m. The supernatant was recovered and loaded onto a Superdex 200 gel filtration chromatography column pre-equilibrated with lysis buffer. The samples were eluted from the column in lysis buffer at a flow rate of 0.5 ml/min and collected in fractions of 1 ml. The fractions were precipitated with 10% trichloroacetic acid and analyzed by Western blots with antibodies against IKKɛ, TBK1 and SIKE, respectively.
RNAi experiments
Double-strand oligonucleotides corresponding to the target sequences were cloned into the pSuper.Retro RNAi plasmid (Oligoengine Inc.). The following sequences were targeted for human SIKE cDNA: (1) GAGGATGCATCCGATATGA; (2) AGCTCACCAGTCTCACTCT; (3) GGAACTTCGAGAATTATTG; (4) GGACATGTCCAAATACAAA; and (5) ATACAAACCTCACATTCTG.
RT–PCR
Total RNA was isolated from 293 cells using Trizol reagent (Tianwei, China) and subjected to RT–PCR analysis to measure expression of IFN-β, Rantes and GAPDH according to the manufacturer's instructions. Gene-specific primer sequences were as follows: IFN-β, 5′-CAGCAATTTTCAGTGTCAGAAGCT-3′ and 5′-TCATCCTGTCCTTGAGGCAGTAT-3′; Rantes, 5′-ATGAAGGTCTCCGCGGCACGCCT-3′ and 5′-CTAGCTCATCTCCAAAGAGTTG-3′; and GAPDH, 5′-AAAATCAAGTGGGGCGATGCT-3′ and 5′-GGGCAGAGATGATGACCCTTT-3′.
VSV plaque assay
The 293 cells (1 × 105) were transfected with the indicated plasmids for 24 or 36 h prior to VSV infection (MOI of 10.0). At 1 h after infection, cells were washed with PBS for three times and then medium was added. The supernatants were harvested at 0, 1, 3, 6, 9 and 12 h after washing. The supernatants were diluted 1:106 and then used to infect confluent BHK21 cells cultured on 24-well plates. At 1 h postinfection, supernatant was removed and 3% methylcellulose was overplayed. At 3 days postinfection, overlay was removed, cells were fixed with 4% formaldehyde for 1 h and stained with 0.2% Crystal violet in 20% methanol. Plaques were counted, averaged and multiplied by the dilution factor to determine viral titer as PFU/ml.
Acknowledgments
We thank Tom Maniatis and Hong-Kui Deng for reagents, Qiang Chen, Fei Yuan and members of our laboratories for technical help and discussion. This work was supported by grants from the National Institute of Health (R01 AI062739) and the Chinese High-Technology Program (#2003AA221030).
References
- Alexopoulou L, Holt AC, Medzhitov R, Flavell RA (2001) Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 413: 732–738 [DOI] [PubMed] [Google Scholar]
- Andrejeva J, Childs KS, Young DF, Carlos TS, Stock N, Goodbourn S, Randall RE (2004) The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc Natl Acad Sci USA 101: 17264–17269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bin LH, Xu LG, Shu HB (2003) TIRP, a novel Toll/interleukin-1 receptor (TIR) domain-containing adapter protein involved in TIR signaling. J Biol Chem 278: 24526–24532 [DOI] [PubMed] [Google Scholar]
- Durbin JE, Fernandez-Sesma A, Lee CK, Rao TD, Frey AB, Moran TM, Vukmanovic S, Garcia-Sastre A, Levy DE (2000) Type I IFN modulates innate and specific antiviral immunity. J Immunol 164: 4220–4228 [DOI] [PubMed] [Google Scholar]
- Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, Maniatis T (2003) IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol 4: 491–496 [DOI] [PubMed] [Google Scholar]
- Han KJ, Su X, Xu LG, Bin LH, Zhang J, Shu HB (2004) Mechanisms of the TRIF-induced interferon-stimulated response element and NF-kappaB activation and apoptosis pathways. J Biol Chem 279: 15652–15661 [DOI] [PubMed] [Google Scholar]
- Hemmi H, Takeuchi O, Sato S, Yamamoto M, Kaisho T, Sanjo H, Kawai T, Hoshino K, Takeda K, Akira S (2004) The roles of two IkappaB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J Exp Med 199: 1641–1650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengel H, Koszinowski UH, Conzelmann KK (2005) Viruses know it all: new insights into IFN networks. Trends Immunol 26: 396–401 [DOI] [PubMed] [Google Scholar]
- Hoebe K, Du X, Georgel P, Janssen E, Tabeta K, Kim SO, Goode J, Lin P, Mann N, Mudd S, Crozat K, Sovath S, Han J, Beutler B (2003) Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature 424: 743–748 [DOI] [PubMed] [Google Scholar]
- Honda K, Sakaguchi S, Nakajima C, Watanabe A, Yanai H, Matsumoto M, Ohteki T, Kaisho T, Takaoka A, Akira S, Seya T, Taniguchi T (2003) Selective contribution of IFN-alpha/beta signaling to the maturation of dendritic cells induced by double-stranded RNA or viral infection. Proc Natl Acad Sci USA 100: 10872–10877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu H, Huang J, Shu HB, Baichwal V, Goeddel DV (1996) TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity 4: 387–396 [DOI] [PubMed] [Google Scholar]
- Hu WH, Johnson H, Shu HB (1999) Tumor necrosis factor-related apoptosis-inducing ligand receptors signal NF-kappaB and JNK activation and apoptosis through distinct pathways. J Biol Chem 274: 30603–30610 [DOI] [PubMed] [Google Scholar]
- Huang J, Teng L, Li L, Liu T, Chen D, Xu LG, Zhai Z, Shu HB (2004) ZNF216 is an A20-like and IkappaB kinase gamma-interacting inhibitor of NFkappaB activation. J Biol Chem 279: 16847–16853 [DOI] [PubMed] [Google Scholar]
- Jiang Z, Mak TW, Sen G, Li X (2004) Toll-like receptor 3-mediated activation of NF-kappaB and IRF3 diverges at Toll-IL-1 receptor domain-containing adapter inducing IFN-beta. Proc Natl Acad Sci USA 101: 3533–3538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kata H, Sato S, Yoneyama M, Yamamoto M, Uematsu S, Matsui K, Tsujimura T, Takeda K, Fujita T, Takeuchi O, Akira S (2005) Cell type-specific involvement of RIG-I in antiviral response. Immunity 23: 19–28 [DOI] [PubMed] [Google Scholar]
- Levy DE, Garcia-Sastre A (2001) The virus battles: IFN induction of the antiviral state and mechanisms of viral evasion. Cytokine Growth Factor Rev 12: 143–156 [DOI] [PubMed] [Google Scholar]
- Levy DE, Marie IJ (2004) RIGging an antiviral defense—it's in the CARDs. Nat Immunol 5: 699–701 [DOI] [PubMed] [Google Scholar]
- Maniatis T, Falvo JV, Kim TH, Kim TK, Lin CH, Parekh BS, Wathelet MG (1998) Structure and function of the interferon-beta enhanceosome. Cold Spring Harb Symp Quant Biol 63: 609–620 [DOI] [PubMed] [Google Scholar]
- McWhirter SM, Fitzgerald KA, Rosains J, Rowe DC, Golenbock DT, Maniatis T (2004) IFN-regulatory factor 3-dependent gene expression is defective in Tbk1-deficient mouse embryonic fibroblasts. Proc Natl Acad Sci USA 101: 233–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meylan E, Burns K, Hofmann K, Blancheteau V, Martinon F, Kelliher M, Tschopp J (2004) RIP1 is an essential mediator of Toll-like receptor 3-induced NF-kappaB activation. Nat Immunol 5: 503–507 [DOI] [PubMed] [Google Scholar]
- Oshiumi H, Matsumoto M, Funami K, Akazawa T, Seya T (2003) TICAM-1, an adaptor molecule that participates in Toll-like receptor 3-mediated interferon-beta induction. Nat Immunol 4: 161–167 [DOI] [PubMed] [Google Scholar]
- Perry AK, Chow EK, Goodnough JB, Yeh WC, Cheng G (2004) Differential requirement for TANK-binding kinase-1 in type I interferon responses to toll-like receptor activation and viral infection. J Exp Med 199: 1651–1658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters RT, Liao SM, Maniatis T (2000) IKKepsilon is part of a novel PMA-inducible IkappaB kinase complex. Mol Cell 5: 513–522 [DOI] [PubMed] [Google Scholar]
- Pomerantz JL, Baltimore D (1999) NF-kappaB activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. EMBO J 18: 6694–6704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S, Sugiyama M, Yamamoto M, Watanabe Y, Kawai T, Takeda K, Akira S (2003) Toll/IL-1 receptor domain-containing adaptor inducing IFN-beta (TRIF) associates with TNF receptor-associated factor 6 and TANK-binding kinase 1, and activates two distinct transcription factors, NF-kappa B and IFN-regulatory factor-3, in the Toll-like receptor signaling. J Immunol 171: 4304–4310 [DOI] [PubMed] [Google Scholar]
- Sharma S, tenOever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J (2003) Triggering the interferon antiviral response through an IKK-related pathway. Science 300: 1148–1151 [DOI] [PubMed] [Google Scholar]
- Shimada T, Kawai T, Takeda K, Matsumoto M, Inoue J, Tatsumi Y, Kanamaru A, Akira S (1999) IKK-i, a novel lipopolysaccharide-inducible kinase that is related to IkappaB kinases. Int Immunol 11: 1357–1362 [DOI] [PubMed] [Google Scholar]
- tenOever BR, Servant MJ, Grandvaux N, Lin R, Hiscott J (2002) Recognition of the measles virus nucleocapsid as a mechanism of IRF-3 activation. J Virol 76: 3659–3669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- tenOever BR, Sharma S, Zou W, Sun Q, Grandvaux N, Julkunen I, Hemmi H, Yamamoto M, Akira S, Yeh WC, Lin R, Hiscott J (2004) Activation of TBK1 and IKKvarepsilon kinases by vesicular stomatitis virus infection and the role of viral ribonucleoprotein in the development of interferon antiviral immunity. J Virol 78: 10636–10649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wathelet MG, Lin CH, Parekh BS, Ronco LV, Howley PM, Maniatis T (1998) Virus infection induces the assembly of coordinately activated transcription factors on the IFN-beta enhancer in vivo. Mol Cell 1: 507–518 [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S (2003) Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 301: 640–643 [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Sato S, Mori K, Hoshino K, Takeuchi O, Takeda K, Akira S (2002) Cutting edge: a novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-beta promoter in the Toll-like receptor signaling. J Immunol 169: 6668–6672 [DOI] [PubMed] [Google Scholar]
- Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T (2004) The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol 5: 730–737 [DOI] [PubMed] [Google Scholar]