Specificity of the STAR/GSG domain protein Qk1: Implications for the regulation of myelination (original) (raw)
Abstract
Inadequate formation and maintenance of myelin is the basis for several neurodegenerative disorders, including leukodystrophy and multiple sclerosis. In mice, oligodendrocyte differentiation and subsequent formation of myelin requires the Quaking gene. Mutation of this gene leads to embryonic lethality or to a trembling phenotype characteristic of dysmyelination. Quaking encodes Qk1, a member of the highly conserved STAR/GSG family of RNA-binding proteins that function as master developmental regulators in higher eukaryotes. Qk1 has been implicated in the regulation of alternative splicing, stability, and translation control of mRNAs that code for myelin structural components in glial cells. We have used quantitative gel mobility shift and fluorescence polarization assays to define the nucleotide sequence specificity of the Qk1 STAR/GSG domain, and to probe the interaction between Qk1 and the 3′-untranslated region (UTR) of myelin basic protein (MBP) mRNA. The results show that Qk1 recognizes a hexanucleotide consensus element that is similar although not identical to the specificity determinant recognized by the Caenorhabditis elegans STAR/GSG protein GLD-1. Several consensus sites are present in the 3′-UTR of MBP mRNA. The highest affinity site is located within the RNA localization region, suggesting a possible role for Qk1 in restricting MBP mRNA to the myelin compartment.
Keywords: RNA-binding protein, myelin, oligodendrocyte, STAR/GSG domain, fluorescence polarization
INTRODUCTION
Post-transcriptional regulation of gene expression plays a fundamental role in the temporal and spatial development of the central nervous system (CNS; Perrone-Bizzozero and Bolognani 2002; Richter and Lorenz 2002). The splicing pattern, subcellular localization, stability, and translation efficiency of a variety of mRNAs are controlled by a distinct set of CNS-specific RNA-binding proteins. Underscoring the importance of post-transcriptional control in the CNS, several well-characterized neurological disorders result from misregulation of RNA-binding protein activity. These include fragile X syndrome, a common form of inherited mental retardation that results from down-regulation of the KH domain protein FMRP (Ashley et al. 1993), and paraneoplastic encephalomyelitis (PEM), a small-cell lung carcinoma related autoimmune disorder in which antibodies cross the blood-brain barrier and inactivate a family of ELAV-like RRM domain proteins (Szabo et al. 1991).
RNA-binding proteins are also critical to the function of glial cells, which generate myelin around neuronal axons in the developing nervous system. The STAR/GSG domain protein Qk1 is expressed in neuronal progenitors and in mature glial cells, and may be a determinant of glial cell fate (Hardy et al. 1996; Hardy 1998b). Three major alternatively spliced isoforms are produced from the Quaking gene, one of which contains a C-terminal nuclear localization sequence (Fig. 1A; Wu et al. 1999). An ethylnitrosourea-induced mutation (_qk_kt4) in the Qk1 RNA-binding domain results in embryonic lethality (Justice and Bode 1988; Chen and Richard 1998). In contrast, a viable Quaking mutation (_qk_v), comprised of a deletion in an upstream regulatory sequence, yields a twitching phenotype similar to mutants in myelin structural components and displays severe dysmyelination of the CNS (Sidman et al. 1964; Ebersole et al. 1996). Because cytoplasmic Qk1 is not localized within the myelin compartment, the prevailing hypothesis is that Qk1 regulates the expression of myelin structural proteins (Hardy et al. 1996; Hardy 1998a).
FIGURE 1.

Qk1-STAR binds to TGE RNA. (A) Domain structure of the murine Quaking gene product. The STAR/GSG domain is comprised of the Qua1, KH, and Qua2 domains. Two proline-rich regions mediate interaction with SH3 domains. Several alternatively spliced isoforms yield proteins with variant C termini (denoted with a stippled pattern). The TGE RNA sequence is shown. The hexanucleotide consensus-binding element is highlighted in gray. (B) Trace of a typical fluorescence-polarization titration of Qk1-STAR into fluorescein-labeled TGE RNA. The error bars represent the standard deviation of 10 reads from the same plate. The equilibrium dissociation constant, hill coefficient, and the 95% confidence intervals of these parameters for this experiment are shown. (C) Electrophoretic mobility shift titration of Qk1-STAR into radiolabeled TGE RNA. Bound and free RNA species are annotated. The fit of the fraction of bound RNA vs. Qk1-STAR concentration, the equilibrium dissociation constant, hill coefficient, and 95% confidence intervals for this particular experiment are shown below.
Several lines of evidence support this proposed role. First, the level of myelin basic protein (MBP) mRNA is reduced in qk_v/qk_v mice (Lu et al. 2003). Second, the subcellular localization pattern of the remaining mRNA is altered such that less is present in the myelin compartment and more RNA is retained in the nucleus (Li et al. 2000; Larocque et al. 2002). Third, the splicing pattern of MBP, proteolipid protein (PLP), and myelin-associated glycoprotein (MAG) is affected in Quaking mice (Wu et al. 2002). Finally, Qk1 has been shown to interact with the 3′-untranslated region (UTR) of MBP mRNA by RNase protection assay, coimmunoprecipitation, and electrophoretic mobility shift assay (Li et al. 2000; Larocque et al. 2002). Two large fragments of the 3′-UTR were shown to bind to Qk1 (Larocque et al. 2002). However, the specific functional sites of Qk1 interaction with RNA and its nucleotide binding specificity remain unclear.
In a previous effort, we characterized the assembly of the Caenorhabditis elegans STAR/GSG protein GLD-1 with its RNA target (Ryder et al. 2004). The STAR/GSG domain is composed of a canonical KH domain flanked by two conserved extensions termed Qua1 and Qua2 (Vernet and Artzt 1997). The results demonstrated that the KH and Qua2 regions are necessary for RNA binding, whereas the Qua1 domain is both necessary and sufficient to mediate dimerization in the absence of RNA or other cellular components. Furthermore, GLD-1 binds with highest affinity to RNA sequences containing a hexanucleotide consensus sequence termed SBE (for STAR Binding Element; Fig. 1A). Finally, each protomer of the GLD-1 dimer is capable of individually recognizing this consensus, defining a mechanism for specific GLD-1 recognition of RNA.
Because Qk1 shares 56% sequence identity with GLD-1 across the STAR/GSG domain, it seems plausible that Qk1 binds to a similar specificity determinant. To test this hypothesis, two avenues of analysis were pursued. First, we determined the ability of Qk1 to bind to a library of GLD-1 target RNAs using quantitative gel mobility shift and fluorescence-polarization. Second, we measured the binding of Qk1 to fragments of the 3′-UTR of MBP mRNA previously implicated in mediating regulation of this gene. The results show that Qk1 binds to the same hexanucleotide consensus as does GLD-1. However, the rank order of binding within the consensus is different, with Qk1 displaying a strong preference for an adenosine residue at the fifth position. Five consensus sites are present in the 3′-UTR of MBP, and one of these located within a previously defined RNA localization region binds to Qk1 with very high affinity. This result suggests that Qk1 mediates localization of MBP mRNA in the myelin compartment.
RESULTS
Analysis of Qk1 interaction with TGE RNA
The C. elegans STAR/GSG domain protein GLD-1 represses the translation of tra-2 by specifically binding to a 28-nt sequence element (termed TGE) present in the 3′-UTR of the mRNA transcript (Jan et al. 1999). In a published report, Qk1-6 (6-kb isoform) was shown to functionally substitute for GLD-1 in the repression of a reporter gene containing the tra-2 3′-UTR in transgenic worms (Saccomanno et al. 1999). Given this result and the strong similarity between GLD-1 and Qk1, it seemed likely that Qk1 might also bind the TGE with high affinity. To test this hypothesis, the equilibrium dissociation constant (Kd) between recombinant purified Qk1-STAR domain and this RNA was determined by two independent quantitative methods: electrophoretic gel mobility shift assay and fluorescence-polarization (Fig. 1B, C; Table 1). The affinity of Qk1 for TGE RNA by each method is 44 ± 6 nM and 52 ± 4 nM, respectively, which is equivalent within error. Therefore, both assays can be effectively utilized to monitor protein–RNA interactions in this system.
TABLE 1.
Analysis of Qk1-STAR binding to RNA
| RNA | Kd (nM) EMSA | Kd (nM) FPA | Kc (nM) FPA |
|---|---|---|---|
| TGE | 44 ± 6 | 52 ± 4 | 41 ± 6 |
| QSBE | 25 ± 4 | 9.7 ± 1 | 12 ± 5 |
| QRE | 110 ± 5 | — | 10 ± 5 |
In comparison, the affinity of GLD-1-STAR for TGE RNA is 11.4 ± 2 by gel mobility shift and 22 ± 5 nM by fluorescence-polarization, which is three- to fourfold tighter than Qk1. This difference is quite modest, corresponding to a change in the standard free energy change is <0.5 kcal/mole. These data are consistent with the hypothesis that Qk1 binds to TGE RNA in a manner similar to GLD-1, but it remained to be determined if the binding specificity was also similar.
Determination of Qk1 binding specificity
In a previous effort, the smallest fragment of the TGE capable of binding to GLD-1-STAR was identified as a half-site RNA with the sequence 5′-AUCUACUCAUAU-3′ (Ryder et al. 2004). This RNA interacts with GLD-1-STAR almost as well as full-length TGE and binds with 1:1 protein/RNA stoichiometry. This simple, minimal system was used to probe the nucleotide sequence specificity of GLD-STAR via extensive mutagenesis. The relative affinity of 18 point mutations was determined by competition gel mobility shift. By this approach, GLD-1 was shown to bind with highest affinity to RNA sequences containing a hexanucleotide 5′-UACU(C/A)A-3′ consensus (termed SBE), and less well to RNA containing a relaxed consensus of (U>G>C/A)A(C>A)U(C/A>U)A.
Because Qk1 binds reasonably well to TGE RNA, we compared the nucleotide sequence specificity of Qk1 to GLD-1 by using the same library of 18 mutant RNA sequences. The relative ability of each mutant to compete with the Qk1-TGE interaction was determined by competition fluorescence-polarization (Fig. 2A). Surprisingly, the 12-nt fragment competes poorly for Qk1-STAR binding, 30-fold weaker than self-competition with the full length TGE sequence (relative IC50 TGE/12-mer = 0.03). This is quite different from GLD-1, in which the 12-mer RNA binds just twofold weaker than the full 28-nt TGE sequence.
FIGURE 2.

Determination of Qk1 binding specificity. (A) Plot of the normalized polarization as a function of competitor RNA concentration for TGE self-competition and for several representative mutant 12-mer RNAs. The error bars represent the standard deviation of 10 reads from the same plate. (B) Table of the relative IC50 value for each mutant 12-mer compared with the wild-type 12-mer sequence. The relative IC50 is listed as <0.3 if the apparent IC50 is >15 μM.
However, two individual point mutations of the hexanucleotide consensus restore high-affinity binding to the shorter RNA. One of these, C21A, binds with slightly higher affinity than the full-length TGE and 40-fold tighter than the wild-type 12-mer fragment (relative IC50 12-mer/C21A = 41). The other (C19A) is just threefold weaker than the TGE and ninefold tighter than the wild-type fragment (relative IC50 12-mer/C19A 12-mer = 0.3). In comparison, the same mutations (C19A and C21A) have limited affect on the interaction with GLD-1-STAR. C19A actually binds fourfold weaker than the wild-type 12-mer, whereas C21A binds only twofold tighter.
The consensus-binding site for Qk1-STAR can be derived from comparison of the relative affinity of each individual point mutation (Fig. 2B). The specificity determinant for this protein is 5′-NA(A>C)U(A≫C)A-3′, where N equals any of the four bases. All of the mutations that are permissible within the relaxed GLD-1-STAR consensus are also consistent with the Qk-1-STAR consensus. However, there are a few notable differences in the individual nucleotide rank order between the Qk1-STAR and GLD-1-STAR consensus sequences. First, the preference of GLD-1-STAR for a uridine at the first position appears to be absent in Qk1. Furthermore, there is a slight preference for an adenosine over a cytidine at the third position for Qk1, whereas for GLD-1 this preference is reversed. Last, Qk1 binds with much higher affinity to the mutant with an adenosine at the fifth position, where GLD-1 will tolerate an adenosine or cytidine equally well. Together, these results show that Qk1 can recognize RNA sequences in a manner very similar to GLD-1 but with a few specific differences. However, the structural basis for these sequence preferences is not immediately clear.
Identification of Qk1 binding sites in the 3′-UTR of MBP mRNA
With the Qk1 high-affinity consensus in hand, we searched for putative binding sites within the 3′-UTR of one if its cognate targets, MBP mRNA. Five sites matching the consensus sequence were identified (Fig. 3A). Two sites are located proximal to each other within a previously defined functional region of the UTR necessary for MBP mRNA localization (Ainger et al. 1997), and three more are located toward the 3′-end of the UTR. No consensus binding sites are found within the first 900 nt of the 3′-UTR.
FIGURE 3.

Identification of Qk1 binding sites in the 3′-UTR of MBP mRNA. (A) Structure of the 3′-UTR of MBP mRNA. The numbering scheme begins at the 5′-end of the 3′-UTR. Consensus Qk1 binding sites are labeled with white boxes. Previously identified elements in the UTR are labeled with gray boxes. (B) Plot of normalized polarization vs. competitor RNA concentration for each of the five consensus sites and for the TGE half-site. (C) Nucleotide sequence and position of Qk1 binding sites in the 3′-UTR of MBP mRNA. (D) Typical electrophoretic mobility shift of Qk1-STAR binding to QSBE RNA. Bound and free RNA species are labeled. A plot of fraction bound QSBE RNA vs. Qk1-STAR concentration is shown. The equilibrium dissociation constant, Hill coefficient, and 95% confidence intervals for this experiment are given. (E) Typical fluorescence-polarization titration of Qk1-STAR into fluorescein-labeled QSBE RNA. The error bars are derived as in Figure 1B.
To determine the relative affinity of each site, competition-binding experiments were performed with 12-nt RNAs containing each putative site, using Qk1-STAR bound to TGE as a probe (Fig 3B, C; Table 2). Each of the consensus sites binds to Qk1 as well or better than the TGE half-site. The first site (906–918; numbered from the 5′-end of the 3′-UTR) binds with the highest affinity, equaling that of the C21A point mutant. The third site (1060–1072) binds with the second highest affinity, sevenfold tighter than the TGE half-site. The second and fourth sites (923–935, 1273–1285) bind equivalently well, three- to fourfold tighter than the TGE half-site. The last site (1457–1469) binds with an affinity identical to the TGE half-site.
TABLE 2.
Consensus site competitions
| RNA | Kc (nM) FPA | Relative IC50 (half-site/n) |
|---|---|---|
| TGE half-site | 1300 ± 230 | 1 |
| TGE half-site C21A | 32 ± 3 | 42 |
| MBP 906–918 | 41 ± 9 | 37 |
| MBP 923–935 | 390 ± 85 | 3 |
| MBP 1060–1072 | 180 ± 35 | 7 |
| MBP 1273–1285 | 330 ± 64 | 4 |
| MBP 1457–1469 | 1300 ± 460 | 1 |
Because site (906–918) binds with the highest affinity, we prepared a longer MBP mRNA variant that contains this site (Fig. 3C). This RNA, termed QSBE (Qk1 STAR-Binding Element), is identical in size to the full-length TGE and positions the consensus hexanucleotide in a similar location relative to the 5′ and 3′ termini. The affinity of this RNA for Qk1-STAR was determined by quantitative gel mobility shift, direct titration fluorescence-polarization, and competition fluorescence-polarization (Fig. 3D, E; Table 1). The Kd by each method is 25 ± 4 nM, 9.7 ± 1 nM, and 12 ± 5 nM, respectively. In comparison, GLD-1 binds to the QSBE with an equilibrium dissociation constant of 50 ± 15 nM (direct titration fluorescence-polarization). The reduced affinity of GLD-1 for QSBE is likely due to the C-for-U substitution in the consensus site and is consistent with its affinity for a U17C mutant of full-length TGE RNA. Thus, Qk1 binds to QSBE RNA with an affinity similar to that of GLD-1 for TGE RNA.
Analysis of Qk1 binding with QRE RNA
Previously, two fragments of MBP mRNA were identified that interact with Qk1 (Larocque et al. 2002). By using immunoprecipitated Qk1 from transfected HeLa cells, the relative capacity of radiolabeled fragments of the MBP 3′-UTR to interact with Qk1 was determined using a bead-binding assay. One fragment (815–1144) contains the first three consensus sites, including the high-affinity QSBE. The other site (54–164: termed QRE for Quaking Response Element), however, is located toward the 5′-end of the UTR in a region devoid of obvious consensus binding sites. To verify the interaction with this region and to define the affinity of this fragment for Qk1, we prepared QRE RNA by in vitro transcription and performed a series of quantitative binding experiments.
First, the ability of QRE RNA to compete with a Qk1-STAR/QSBE complex was determined by fluorescence-polarization (Fig, 4B; Table 1), and the relative IC50 QSBE/QRE is equal to 0.85. The fact that the QRE competes efficiently for Qk1 binding is in excellent agreement with the previous report but is also somewhat surprising as consensus-binding sites are not apparent in this region. The efficient competition of the QRE sequence that lacks a consensus-binding site can be rationalized by at least two models. First, the QRE sequence may contain a single high-affinity site that does not correlate to the hexanucleotide consensus. Second, because QRE RNA is four times longer than QSBE RNA, several weak partial Qk1 binding sites may collaborate in order to yield efficient competition.
FIGURE 4.

Analysis of Qk1 binding to QRE RNA. (A) Plot of competition fluorescence-polarization experiments for QSBE, QRE, and a control poly-U RNA. (B) Electrophoretic mobility shift of Qk1-STAR binding to QRE RNA. Bound and free RNA species are labeled. The fraction of bound RNA was determined by grouping all of the shifted species, and plotted vs. the concentration of Qk1-STAR. The equilibrium dissociation constant, Hill coefficient, and 95% confidence intervals from this fit are shown.
To distinguish between these models, the interaction between Qk1 and 5′-end–labeled QRE RNA was monitored by gel mobility shift. If one high-affinity site is present, then a single shifted species should be apparent, similar to what is observed for the QSBE (Fig. 3D). If there are several weak binding sites, then it may be possible to observe several shifted species, each with a weaker apparent equilibrium dissociation constant. We observe the latter (Fig. 4B). At least three individual shifts are present. In addition, the apparent equilibrium dissociation constant for this interaction is 10-fold weaker than binding to the QSBE (110 ± 5 nM, n = 1.8 ± 0.2). The 10-fold weaker apparent binding by direct titration is consistent with the multiple weak sites model, and the higher Hill coefficient suggests that Qk1 interaction with this larger RNA is cooperative. Together, these data show that Qk1 binding to the QRE is driven by cooperative recognition of multiple sites, rather than one high-affinity consensus element, as is observed for QSBE RNA. However, these experiments do not address the functional relevance of Qk1 binding to either QSBE or QRE RNA.
DISCUSSION
Qk1 and GLD-1 recognize similar sequence specificity determinants
STAR/GSG domain proteins function in the regulation of developmental processes in higher eukaryotes. Members of this family have been implicated in the regulation of mRNA translation, mRNA stability, and alternative splicing of pre-mRNA (Baehrecke 1997; Jan et al. 1999; Wu et al. 2002; Coyle et al. 2003; Di Fruscio et al. 2003). The STAR/GSG protein GLD-1 is a master regulator of C. elegans germline development; it represses the translation of several key determinants during the spatial and temporal development of the hermaphrodite gonad (Francis et al. 1995; Jan et al. 1999; Lee and Schedl 2001; Marin and Evans 2003). Similarly, a Mus musculus STAR/GSG protein (Qk1) is involved in the regulation of development at the post-transcriptional level (Ebersole et al. 1996; Larocque et al. 2002; Wu et al. 2002; Lu et al. 2003). Qk1 is necessary for the proper formation of myelin in the CNS. It regulates the splicing pattern of several of the structural components of myelin and is required for the proper subcellular localization of MBP mRNA in oligodendrocytes.
In a previous effort, the minimal RNA sequence and the nucleotide specificity determinant for GLD-1 was defined (Ryder et al. 2004). This protein recognizes a hexanucleotide consensus 5′-UACU(C/A)A-3′ with high affinity, and with lower affinity, a relaxed consensus of 5′-(U>G>C/A)A(C>A)U(C/A>U)A-3′. To determine if STAR/GSG domain proteins have conserved sequence specificity, the ability of Qk1 to cross-react with a GLD-1 target (TGE RNA) was measured. Qk1 binds to this RNA just threefold weaker than GLD-1, demonstrating that Qk1 is capable of binding to a similar RNA sequence. However, this analysis does not define the consensus sequence required for Qk1 binding.
To address this question, the ability of Qk1 to bind to a library of mutant TGE sequences was determined by competition fluorescence-polarization. Via this analysis, the consensus element necessary for Qk1 binding was determined. Qk1 recognizes a 5′-NA(A>C)U(A>>C/U)A-3′ consensus, which correlates very well to the relaxed consensus necessary for GLD-1 binding. Therefore, the specificity of these two STAR/GSG domain proteins is conserved.
Although Qk1 can recognize all of the same nucleotides present in the relaxed GLD-1 consensus, there are a few notable differences in the sequences that comprise the highest affinity sites. First, Qk1 lacks the preference for uridine at the first position. Every mutation at this position binds as well as wild-type RNA. Second, Qk1 binds better to sequences with an adenosine at the third position, whereas GLD-1 binds better to cytidine. Last, Qk1 displays a very strong preference for an adenosine at the fifth position, whereas GLD-1 binds to an adenosine or cytidine equally well.
Previously, homology models of GLD-1 and Qk1 were prepared based upon the NMR structure of the related KH/Qua2 protein SF-1 (Liu et al. 2001; Ryder et al. 2004). In these models, the RNA contact amino acids in GLD-1 and Qk1 are 100% conserved. This is consistent with the conservation of binding specificity between these proteins but does not explain the differences between highest affinity binding sites. It is likely that additional residues not identified in the models contribute to the binding specificity. In the absence of high-resolution structures for either of these proteins in complex with RNA, it is difficult to identify additional amino acids that specify the binding consensus. Identification of these residues will require further experiments that characterize the binding specificity of GLD-1 and Qk1 mutants.
Identification of Qk1 binding sites in MBP mRNA
MBP is a primary structural component of the myelin sheath (Mikoshiba et al. 1991). This protein is highly positively charged and functions in the compaction of membrane layers in developing myelin. As such, expression of this protein is localized to the myelin compartment. Expression of this protein outside of the compartment is cytotoxic and leads to premature cell death.
Accordingly, several pathways of post-transcriptional regulation control the localized expression of this gene product (Fig. 5; de Ferra et al. 1985; Ainger et al. 1993). First, MBP mRNA is spliced into one of several isoforms and then transported from the nucleus. Once in the cytoplasm, this RNA is packaged into granules with 80S ribosomes and translationally silenced. The granules are then shuttled from the perikaryon through the processes to the myelin compartment. Last, the mRNA is localized into the compartment, where translation of MBP commences.
FIGURE 5.

Stages of the post-transcriptional regulation of MBP mRNA in oligodendrocyte glial cells. Transcripts are spliced into one of several isoforms (I), exported from the nucleus to the perikaryal space where it is packaged into granules (II), transported along the processes (III), and then localized to the myelin compartment (IV).
By using an RNA microinjection assay of primary rat oligodendrocytes, two functionally separable regulatory elements were defined within the 3′-UTR of MBP mRNA (Ainger et al. 1997). One region, termed the RTS for RNA transport signal, is required for the movement of the granules along the processes (Fig. 3A). Subsequent efforts defined a high-affinity hnRNP A2 binding site within this region that is required for transport activity (Kwon et al. 1999). The second region, termed the RLR for RNA localization region, is necessary for MBP mRNA to enter the myelin compartment. Unfortunately, the protein cofactors that recognize this region have not been identified.
Several lines of evidence suggest that Qk1 plays a role in the post-transcriptional regulation of MBP mRNA. First, MBP mRNA colocalizes with Qk1 in mouse and rat oligodendrocytes and when cotransfected into COS cells (Larocque et al. 2002). Second, overexpression of the nucleus specific isoform of Qk1 (Qk1-5) leads to aberrant accumulation of MBP mRNA in the nucleus (Li et al. 2000; Li et al. 2003; Larocque et al. 2002). Third, the abundance of MBP mRNA is reduced in the myelin compartment of qk_v/qk_v mice. Finally, recombinant Qk1 has been shown to interact with the 3′-UTR of MBP mRNA in vitro by several methods (Li et al. 2000; Larocque et al. 2002).
To define binding sites for Qk1 in the 3′-UTR of this transcript, we searched for the presence of the Qk1 consensus binding sites in this RNA. Five consensus sites are present within this sequence, all of which lie downstream of the RTS. Interestingly, two sites are present in the RLR (sites 906–918 and 923–935). Three additional sites are located further downstream (1060–1072, 1273–1285, 1457–1469). The first three sites are found within a fragment of the 3′-UTR shown by Larocque and coworkers (2002) to interact with Qk1 in a bead-binding analysis (1–3). None of the consensus sites are present within a second element identified by these investigators near the 5′-end of the UTR previously termed QRE.
The ability of Qk1 to interact with the consensus sites was determined by competition fluorescence-polarization assay. Consistent with the consensus identity, all of the sites bind as well or better than the site from TGE RNA. Site (906–918), which is present within the localization element, binds to Qk1 with the highest affinity. Three of the five sites (906–918, 1273–1285, 1457–1469) are conserved between the mouse and rat 3′-UTR. Therefore, these sites are the best candidates for functional Qk1 interaction sites within this RNA.
Because site (906–918) is located within a previously defined functional element, competes most efficiently, and is conserved between the mouse and rat genes, we chose to further characterize this position. A longer RNA variant (termed QSBE) containing this site was prepared that is equivalent in length to the TGE and positions the consensus in an equivalent position relative to the 5′ and 3′ termini. The ability of this RNA to bind to recombinant Qk1-STAR was determined by fluorescence polarization and gel mobility shift assay. Qk1-STAR binds to this RNA with an equilibrium dissociation constant of 10 nM, which is equivalent to the Kd of GLD-1-STAR for TGE RNA. In addition, only one shifted species is observed, consistent with a single high-affinity binding site. In contrast, several shifted species are observed for the interaction between Qk1-STAR and QRE RNA. This suggests that binding to this sequence is mediated by multiple weak binding sites, rather than a single high-affinity site.
At least two factors influence the ability of a particular RNA sequence to form a functional Qk1 binding element. First, the relative affinity of Qk1 for an individual sequence is defined by its nucleotide-binding consensus. Second, the affinity of Qk1 for a particular sequence may be modulated by cooperative interactions with other factors that bind to nearby sequence elements or by its oligomeric state. Here, we show that Qk1 binds with high affinity to a single site within the localization element of the UTR, and with weaker affinity to four additional consensus sites further downstream. Furthermore, consistent with previous work, we observe cooperative binding of Qk1 to several weaker sites within the QRE sequence (Larocque et al. 2002). It remains to be seen which, if any, of these sites functions individually to mediate the post-transcriptional regulation of this transcript. It is also possible that these sites are functionally redundant or that Qk1 binding to each site may play a distinct role in the regulation of MBP mRNA, including regulating splicing, nuclear export, and localization to the myelin compartment.
The role of Qk1 in the regulation of myelination
The presence of a Qk1 binding site within the RLR immediately suggests a functional role for Qk1 in restricting MBP mRNA to the myelin compartment. This hypothesis is consistent with three previous observations. First, the amount of MBP mRNA present in purified myelin from qk_v/qk_v mice is significantly reduced (Li et al. 2000). Second, MBP mRNA is localized aberrantly in cultured _qk_v oligodendrocytes (Barbarese 1991). Third, MBP mRNA is predominantly found in the nucleus in oligodendrocytes sectioned from brains of qk_v/qk_v mice (Larocque et al. 2002). Together with the identification of a high-affinity binding site, these results suggest that Qk1 is the cofactor that mediates localization via the RLR. However, because binding does not unequivocally correlate to biological function, more experiments will be necessary to validate this hypothesis.
In addition to the regulation of MBP mRNA, nuclear Qk1 has been shown to affect the splicing pattern of several mRNAs that code for several myelin structural components, including MAG, PLP, and MBP (Wu et al. 2002). As mentioned above, Qk1 is related to the splicing factor SF-1, necessary for branch-site selection (Berglund et al. 1997). SF-1 recognizes RNA containing a 5′-YNCURAY-3′ consensus and cooperatively associates with U2AF65/35 in the splicing commitment complex (Berglund et al. 1998; Peled-Zehavi et al. 2001). Because the STAR/GSG binding element partially overlaps with the branch-site consensus, we previously postulated that these proteins could affect splicing by competing with SF-1 for branch-site RNA (Ryder et al. 2004). If this is true for Qk1, then consensus Qk1 binding sites should be present within introns that flank the alternatively spliced exons.
Consistent with a role for Qk1 in the regulation of these processes, at least one consensus Qk1 binding site is located in the intronic sequence adjacent to each of the regulated exons. However, the form of alternative splicing that occurs for each gene is quite diverse, suggesting that the role of Qk1 in regulating these processes may be variable (Wu et al. 2002). In the PLP gene, the selection the 5′-splice site within exon 3 is altered. For MAG, inclusion of exon 12 is enhanced. For MBP, the inclusion of exon 2 and the exclusion of exons 5 and 6 are enhanced. Because the splicing defect for each of these genes is widely variable, the mechanism of Qk1 in regulating alternative splicing remains unclear.
In summary, this work defines the nucleotide binding specificity of the STAR/GSG protein Qk1. The consensus sequence recognized by this protein is very similar to that recognized by the C. elegans STAR/GSG protein GLD-1, showing that binding specificity is conserved for a subset of this family of RNA-binding proteins. However, there are a few differences between the highest affinity sites, indicating that amino acids not conserved between Qk1 and GLD-1 can modulate specificity. Based on this result, several Qk1 binding sites were identified and characterized within one of its cognate targets, MPB mRNA. The highest-affinity site is present within a previously characterized functional region required to localize MBP mRNA to the myelin compartment. This result is consistent with a role for Qk1 in restricting MBP expression to the developing myelin compartment.
MATERIALS AND METHODS
Protein expression constructs and purification
The STAR/GSG domain of Qk1 was PCR amplified from pRO-EXHT-Qk1-STAR by using the following primers: 5′-CGG GAT CCA TGG TCG GGG AAA TGG AA-3′ and 5′-CCC AAG CTT TCA TTA TCT GTA GGT GCC ATT CAG-3′. The resultant product was digested with BamH1 and HindIII and subcloned into pMal-c (NEB) to generate pMal-Qk1-STAR.
Qk1-STAR was expressed from this plasmid in Escherichia coli strain JM109 by inducing mid-log phase cultures for three hours with 1 mM IPTG at 37°C. The protein was purified from 1 L of culture as fusion to maltose binding protein via three steps. Initial purification was achieved from soluble cellular lysate via affinity chromatography using amylose resin (NEB) in lysis buffer (50 mM Tris-Cl at pH 8.0, 200 mM NaCl, and 2 mM DTT). Protein was eluted from the column in lysis buffer supplemented with 10 mM maltose (Sigma). Fractions containing Qk1-STAR were pooled, diluted fivefold into HS buffer (50 mM MOPS at pH 6.0, 2 mM DTT), and further purified with a POROS-HS cation exchange column over a gradient from 0 to 2 M NaCl by using a SPRINT Bio-CAD (PerSeptive Biosystems). Fractions containing protein were pooled, diluted into HQ buffer (75 mM Tris at pH 8.8, 2 mM DTT), and purified to homogeneity with a POROS-HQ anion exchange column over a gradient from 0 to 2 M NaCl. Pure fractions were combined, dialyzed into storage buffer (20 mM Tris at pH 8.0, 25 mM NaCl, 2 mM DTT), and stored at 4°C. The concentration of the purified protein was determined in 6 M guanidinium-HCl by UV spectroscopy at 280 nm using a calculated molar extinction coefficient of 76,810 M−1. Typical yields for this preparation are ~15 mg/L culture, and purity is >95% as judged by a coommassie-stained SDS-polyacrylamide gel and by MALDI-TOF (matrix-assisted laser desorption ionization time of flight) mass spectrometry. GLD-1-STAR was purified as previously described (Ryder et al. 2004).
RNA construct preparation
All of the RNA oligonucleotides, with the exception of the QRE RNA, were purchased from Dharmacon. RNA oligonucleotides were deprotected, purified, lyophilized, and stored as per the manufacturer’s protocol. RNA constructs used in gel mobility shift experiments were radiolabeled at the 5′-end and purified using γ-32P-ATP (Perkin Elmer) and T4 polynucleotide kinase (NEB) as previously described (Ryder et al. 2004). 5′-Fluorescein–labeled RNAs were purchased from Dharmacon and handled in the dark.
QRE RNA was prepared by in vitro transcription from pUC-18-QRE. This plasmid was generated from the following six DNA oligonucleotides: QRE-1, GCGCGAATTCTAATACGACTCAC TATAGGCCTTAAACTTTTAAT; QRE-2, CTAGCTAATCGGTG CAAGTAGAATTAAAAGTTTAAGGCCTATAG; QRE-3, TCTAC TTGCACCGATTAGCTAGTTAGAGCAGACCCTCTCTTAAT; QRE-4, ACCGCGATCACGGCTCCACGGGATTAAGAGAGGGTCT GCTCTAA; QRE-5, CCCGTGGAGCCGTGATCGCGGTGGGG CCAGGCCCACGGCACCCC; and QRE-6, GCGCGGATCCCTCTT CGCAGTCGGGGTGCCGTGGGCCTGGCCCC. Each oligonucleotide was individually phosphorylated by using T4 polynucleotide kinase, then mixed in an annealing reaction, and treated with T4 DNA ligase. The resultant duplex product was used as a template for PCR with QRE-1 and QRE-2. The product was digested with EcoR1 and HindIII and cloned into pUC-18. Transcription reactions were performed in transcription buffer (40 mM Tris at pH 8.0, 5 mM DTT, 2 mM Spermidine, 12.5 mM MgCl2, 1 Mm each NTP) using 40 ng/mL of Ear1-linearized plasmid template and 0.1 mg/mL T7 RNA polymerase for 3 h. The RNA product was purified on a denaturing gel (8% 39:1 polyacrylamide, 7 M urea, 1× TBE), visualized by UV shadowing, eluted into TEN by the crush/soak method, ethanol precipitated, resuspended into TE, and stored at −20°C. The concentration of the QRE RNA was measured by UV spectroscopy at 260 nm by using a calculated extinction coefficient of 1183.7 mM−1.
Electrophoretic mobility shift assay
The complex between recombinant STAR/GSG domain proteins and TGE, QSBE, or QRE RNA was visualized by electrophoretic gel mobility shift assay. A constant concentration of radiolabeled RNA (100 pM) was equilibrated with varying concentrations fusion protein in equilibration buffer (10 mM Tris at pH 8.0, 25 mM NaCl, 0.1 mM EDTA, 0.1 mg/mL tRNA, 5 μg/mL heparin, and 0.01% IGEPAL CA630). Radiolabeled RNA was heated to 90°C for 2 min and then allowed to slow-cool over 30 min to room temperature prior to equilibration with protein. Typically, 20 μL reactions were allowed to incubate at room temperature for at least 3 h. Prior to loading, 4 μL of type III loading dye (30% v/v glycerol, 0.05% w/v xylene cyanol) was added to each sample. A 5 μL portion of each reaction was loaded onto a prerun native polyacrylamide gel (6% 29:1 acrylamide/bis-acrylamide, 0.5× TBE) with the voltage set to 100 V. Gels were run at 600 V for 30 min before they were dried and exposed to a PhosphorImager screen overnight (Molecular Dynamics). The fraction of bound RNA was determined by using ImageQuant software (Molecular Dynamics).
Fluorescence polarization assay
Quantitative analysis of protein/nucleic acid interactions by fluorescence-polarization has been described (LeTilly and Royer 1993). The experimental setup for the fluorescence-polarization titration assay was identical to the electrophoretic mobility shift assay, except 1 nM fluorescein-labeled TGE or QSBE RNA was used in place of the radiolabeled RNA and reactions were performed in a total volume of 100 μL. Samples were equilibrated in 96-well opaque fluotrak 200 microtiter plates (Grenier) and the polarization determined using a Fusion α-FP plate reader (Packard). The average and standard deviation of the mP value for each protein concentration was calculated from 10 consecutive readings of each plate.
Competition fluorescence-polarization experiments were performed by using identical conditions, except that a constant concentration of Qk1-STAR was included in each equilibration. The protein concentration was chosen in order to yield a 70%–90% bound complex. In TGE competition experiments, a constant concentration of 200 nM of Qk1-STAR was used. In the QSBE competition experiments, 80 nM of protein was used. Unlabeled competitor RNA was titrated into each reaction at varying concentrations and allowed to equilibrate for 3 h prior to reading the plate.
Data analysis
The apparent equilibrium dissociation constant for direct titration electrophoretic mobility shift and fluorescence-polarization assays were determined from a fit of the data using nonlinear least squares regression using IGOR (Wavemetrics). The apparent equilibrium dissociation constant of Qk1 and GLD-1 for each RNA variant was determined using the equation:
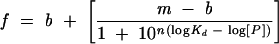
where f is the fraction bound or the mP value, [P] is the concentration of fusion protein, Kd is the equilibrium dissociation constant, n is the Hill coefficient, m is the maximum signal, and b is the base signal. The apparent Kd is equivalent to the
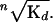
The reported Kd for each variant is the average of at least three ndependent experiments.
The IC50 for each competitor was determined by performing weighted fits to the following equation:
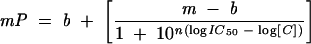
where C is the concentration of unlabelled competitor, and IC50 is the concentration of competitor RNA that displaces 50% of the labeled RNA. The equilibrium dissociation constant for each competitor (KC) was calculated by fitting the data to a quadratic solution of the Lin and Riggs equation (Lin and Riggs 1972; Weeks and Crothers 1992):
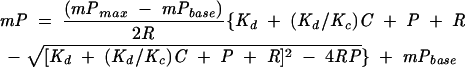
where mPmax is the polarization at saturation, mPbase is the polarization in the absence of protein, and R is the concentration of fluorescein-labeled RNA. This solution assumes that the stoichiometry of the labeled RNA and competitor are identical and hence may not apply in all situations. Therefore, comparative inhibition analyses were performed directly from the IC50 values for experiments performed on the same day. Each experiment was performed at least in duplicate. Reported errors for the Lin and Riggs fits are the 95% confidence intervals for the fitted parameters, which were typically larger than the standard deviations.
Acknowledgments
We thank Dr. Andrew Carmel and Dr. Katrina Lehmann-Blount for helpful discussions and critical comments concerning this manuscript, and Dr. Traci Tanaka-Hall for generously providing a clone of the Qk1 STAR/GSG domain. S.P.R. is supported by the Damon Runyon Cancer Research Foundation Fellowship (DRG-1723). This research was supported by a grant from the National Institutes of Health (NIH, GM53320) to J.R.W.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
REFERENCES
- Ainger, K., Avossa, D., Morgan, F., Hill, S.J., Barry, C., Barbarese, E., and Carson, J.H. 1993. Transport and localization of exogenous myelin basic protein mRNA microinjected into oligodendrocytes. J. Cell Biol. 123**:** 431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ainger, K., Avossa, D., Diana, A,S., Barry, C., Barbarese, E., and Carson, J.H. 1997. Transport and localization elements in myelin basic protein mRNA. J. Cell. Biol. 138**:** 1077–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashley Jr., C.T., Wilkinson, K.D., Reines, D., and Warren, S.T. 1993. FMR1 protein: Conserved RNP family domains and selective RNA binding. Science 262**:** 563–566. [DOI] [PubMed] [Google Scholar]
- Baehrecke, E.H. 1997. who encodes a KH RNA binding protein that functions in muscle development. Development 124**:** 1323–1332. [DOI] [PubMed] [Google Scholar]
- Barbarese, E. 1991. Spatial distribution of myelin basic protein mRNA and polypeptide in quaking oligodendrocytes in culture. J. Neurosci. Res. 29**:** 271–281. [DOI] [PubMed] [Google Scholar]
- Berglund, J.A., Chua, K., Abovich, N., Reed, R., and Rosbash, M. 1997. The splicing factor BBP interacts specifically with the pre-mRNA branchpoint sequence UACUAAC. Cell 89**:** 781–787. [DOI] [PubMed] [Google Scholar]
- Berglund, J.A., Abovich, N., and Rosbash, M. 1998. A cooperative interaction between U2AF65 and mBBP/SF1 facilitates branch-point region recognition. Genes Dev. 12**:** 858–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, T. and Richard, S. 1998. Structure-function analysis of Qk1: A lethal point mutation in mouse quaking prevents homodimerization. Mol. Cell. Biol. 18**:** 4863–4871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle, J.H., Guzik, B.W., Bor, Y.C., Jin, L., Eisner-Smerage, L., Taylor, S.J., Rekosh, D., and Hammarskjold, M.L. 2003. Sam68 enhances the cytoplasmic utilization of intron-containing RNA and is functionally regulated by the nuclear kinase Sik/BRK. Mol. Cell Biol. 23**:** 92–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Ferra, F., Engh, H., Hudson, L., Kamholz, J., Puckett, C., Molineaux, S., and Lazzarini, R.A. 1985. Alternative splicing accounts for the four forms of myelin basic protein. Cell 43**:** 721–727. [DOI] [PubMed] [Google Scholar]
- Di Fruscio, M., Styhler, S., Wikholm, E., Boulanger, M.C., Lasko, P., and Richard, S. 2003. Kep1 interacts genetically with dredd/caspase-8, and kep1 mutants alter the balance of dredd isoforms. Proc. Natl. Acad. Sci. 100**:** 1814–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebersole, T.A., Chen, Q., Justice, M.J., and Artzt, K. 1996. The quaking gene product necessary in embryogenesis and myelination combines features of RNA binding and signal transduction proteins. Nat. Genet. 12**:** 260–265. [DOI] [PubMed] [Google Scholar]
- Francis, R., Barton, M.K., Kimble, J., and Schedl, T. 1995. gld-1, a tumor suppressor gene required for oocyte development in Caenorhabditis elegans. Genetics 139**:** 579–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy, R.J. 1998a. Molecular defects in the dysmyelinating mutant quaking. J. Neurosci. Res. 51**:** 417–422. [DOI] [PubMed] [Google Scholar]
- ———. 1998b. QKI expression is regulated during neuron-glial cell fate decisions. J. Neurosci. Res. 54**:** 46–57. [DOI] [PubMed] [Google Scholar]
- Hardy, R.J., Loushin, C.L., Friedrich Jr., V.L., Chen, Q., Ebersole, T.A., Lazzarini, R.A., and Artzt, K. 1996. Neural cell type-specific expression of QKI proteins is altered in quakingviable mutant mice. J. Neurosci. 16**:** 7941–7949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan, E., Motzny, C.K., Graves, L.E., and Goodwin, E.B. 1999. The STAR protein, GLD-1, is a translational regulator of sexual identity in Caenorhabditis elegans. EMBO J. 18**:** 258–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justice, M.J. and Bode, V.C. 1988. Three ENU-induced alleles of the murine quaking locus are recessive embryonic lethal mutations. Genet. Res. 51**:** 95–102. [DOI] [PubMed] [Google Scholar]
- Kwon, S., Barbarese, E., and Carson, J.H. 1999. The _cis_-acting RNA trafficking signal from myelin basic protein mRNA and its cognate _trans_-acting ligand hnRNP A2 enhance cap-dependent translation. J. Cell Biol. 147**:** 247–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larocque, D., Pilotte, J., Chen, T., Cloutier, F., Massie, B., Pedraza, L., Couture, R., Lasko, P., Almazan, G., and Richard, S. 2002. Nuclear retention of MBP mRNAs in the quaking viable mice. Neuron 36**:** 815–829. [DOI] [PubMed] [Google Scholar]
- Lee, M.H. and Schedl, T. 2001. Identification of in vivo mRNA targets of GLD-1, a maxi-KH motif containing protein required for C. elegans germ cell development. Genes Dev. 15**:** 2408–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeTilly, V. and Royer, C.A. 1993. Fluorescence anisotropy assays implicate protein–protein interactions in regulating trp repressor DNA binding. Biochemistry 32**:** 7753–7758. [DOI] [PubMed] [Google Scholar]
- Li, Z., Zhang, Y., Li, D., and Feng, Y. 2000. Destabilization and mislocalization of myelin basic protein mRNAs in quaking dysmyelination lacking the QKI RNA-binding proteins. J. Neurosci. 20**:** 4944–4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, S.Y. and Riggs, A.D. 1972. Lac repressor binding to non-operator DNA: Detailed studies and a comparison of equilibrium and rate competition methods. J. Mol. Biol. 72**:** 671–690. [DOI] [PubMed] [Google Scholar]
- Liu, Z., Luyten, I., Bottomley, M.J., Messias, A.C., Houngninou-Molango, S., Sprangers, R., Zanier, K,, Kramer, A., and Sattler, M. 2001. Structural basis for recognition of the intron branch site RNA by splicing factor 1. Science 294**:** 1098–1102. [DOI] [PubMed] [Google Scholar]
- Lu, Z., Zhang, Y., Ku, L., Wang, H., Ahmadian, A., and Feng, Y. 2003. The quakingviable mutation affects qkI mRNA expression specifically in myelin-producing cells of the nervous system. Nucleic Acids Res. 31**:** 4616–4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin, V.A. and Evans, T.C. 2003. Translational repression of a C. elegans Notch mRNA by the STAR/KH domain protein GLD-1. Development 130**:** 2623–2632. [DOI] [PubMed] [Google Scholar]
- Mikoshiba, K., Okano, H., Tamura, T., and Ikenaka, K. 1991. Structure and function of myelin protein genes. Annu. Rev. Neurosci. 14**:** 201–217. [DOI] [PubMed] [Google Scholar]
- Peled-Zehavi, H., Berglund, J.A., Rosbash, M., and Frankel, A.D. 2001. Recognition of RNA branch point sequences by the KH domain of splicing factor 1 (mammalian branch point binding protein) in a splicing factor complex. Mol. Cell. Biol. 21**:** 5232–5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrone-Bizzozero, N. and Bolognani, F. 2002. Role of HuD and other RNA-binding proteins in neural development and plasticity. J. Neurosci. Res. 68**:** 121–126. [DOI] [PubMed] [Google Scholar]
- Richter, J.D. and Lorenz, L.J. 2002. Selective translation of mRNAs at synapses. Curr. Opin. Neurobiol. 12**:** 300–304. [DOI] [PubMed] [Google Scholar]
- Ryder, S.P., Frater, L., Abramovitz, D.L., Goodwin, E.B., and Williamson, J.R. 2004. RNA target specificity of the STAR/GSG domain post-transcriptional regulatory protein GLD-1. Nat. Struct. Mol. Biol. 11**:** 20–28. [DOI] [PubMed] [Google Scholar]
- Saccomanno, L., Loushin, C., Jan, E., Punkay, E., Artzt, K., and Goodwin, E.B. 1999. The STAR protein QKI-6 is a translational repressor. Proc. Natl. Acad. Sci. 96**:** 12605–12610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidman, R.L., Dickie, M.M., and Appel, S.H. 1964. Mutant mice (Quaking and Jimpy) with deficient myelination in the central nervous system. Science 144**:** 309–311. [DOI] [PubMed] [Google Scholar]
- Szabo, A., Dalmau, J., Manley, G., Rosenfeld, M., Wong, E., Henson, J., Posner, J.B., and Furneaux, H.M. 1991. HuD, a paraneoplastic encephalomyelitis antigen, contains RNA-binding domains and is homologous to Elav and Sex-lethal. Cell 67**:** 325–333. [DOI] [PubMed] [Google Scholar]
- Vernet, C. and Artzt, K. 1997. STAR, a gene family involved in signal transduction and activation of RNA. Trends Genet 13**:** 479–484. [DOI] [PubMed] [Google Scholar]
- Weeks, K.M. and Crothers, D.M. 1992. RNA binding assays for Tat-derived peptides: Implications for specificity. Biochemistry 31**:** 10281–10287. [DOI] [PubMed] [Google Scholar]
- Wu, J., Zhou, L., Tonissen, K., Tee, R., and Artzt, K. 1999. The quaking I-5 protein (QKI-5) has a novel nuclear localization signal and shuttles between the nucleus and the cytoplasm. J. Biol. Chem. 274**:** 29202–29210. [DOI] [PubMed] [Google Scholar]
- Wu, J.I., Reed, R.B., Grabowski, P.J., and Artzt, K. 2002. Function of quaking in myelination: regulation of alternative splicing. Proc. Natl. Acad. Sci. 99**:** 4233–4238. [DOI] [PMC free article] [PubMed] [Google Scholar]