Fast and effective prediction of microRNA/target duplexes (original) (raw)
Abstract
MicroRNAs (miRNAs) are short RNAs that post-transcriptionally regulate the expression of target genes by binding to the target mRNAs. Although a large number of animal miRNAs has been defined, only a few targets are known. In contrast to plant miRNAs, which usually bind nearly perfectly to their targets, animal miRNAs bind less tightly, with a few nucleotides being unbound, thus producing more complex secondary structures of miRNA/target duplexes. Here, we present a program, RNA-hybrid, that predicts multiple potential binding sites of miRNAs in large target RNAs. In general, the program finds the energetically most favorable hybridization sites of a small RNA in a large RNA. Intramolecular hybridizations, that is, base pairings between target nucleotides or between miRNA nucleotides are not allowed. For large targets, the time complexity of the algorithm is linear in the target length, allowing many long targets to be searched in a short time. Statistical significance of predicted targets is assessed with an extreme value statistics of length normalized minimum free energies, a Poisson approximation of multiple binding sites, and the calculation of effective numbers of orthologous targets in comparative studies of multiple organisms. We applied our method to the prediction of Drosophila miRNA targets in 3′UTRs and coding sequence. RNAhybrid, with its accompanying programs RNAcalibrate and RNAeffective, is available for download and as a Web tool on the Bielefeld Bioinformatics Server (http://bibiserv.techfak.uni-bielefeld.de/rnahybrid/).
Keywords: miRNA, RNA folding, minimum free energy, statistical significance, extreme value distribution, database searching
INTRODUCTION
MicroRNAs (miRNAs) are short RNAs that post-transcriptionally regulate the expression of target genes by binding to the target mRNAs. Although a large number of animal miRNAs has been defined (e.g., Lau et al. 2001), only a few targets are known. lin-4 and let-7 control developmental timing in Caenorhabditis elegans by repressing their target genes, lin-14, lin-28, and lin-41 (Lee et al. 1993; Moss et al. 1997; Reinhart et al. 2000; Slack et al. 2000; for review, see Grosshans and Slack 2002). The C. elegans hunchback homolog, hbl-1, has been identified as a probable further target of let-7 (Abrahante et al. 2003; Lin et al. 2003). The Drosophila melanogaster proapoptotic gene hid has been demonstrated to be a target of the newly identified bantam miRNA (Brennecke et al. 2003). Lewis et al. (2003) provide experimental support for at least eight human targets (SMAD-1, SDF-1, BRN-3b, ENX-1, N-MYC, PTEN, Delta1, and Notch1). In contrast to plant miRNAs, which usually bind nearly perfectly to their targets (Rhoades et al. 2002), animal miRNAs bind less tightly, with a few nucleotides being unbound, thus producing more complex secondary structures of miRNA/target duplexes. The combinatorial nature of secondary structure formation, that is, the huge number of possible bindings as a result of loops of unpaired nucleotides, makes prediction of miRNA targets by simple pattern matching or BLAST searches impossible. A number of further Drosophila miRNA targets has recently been predicted by combining information about sequence conservation between D. melanogaster and D. pseudoobscura, and secondary structure prediction by energy minimization (Stark et al. 2003). Those authors first identified potential binding sites by searching for near-perfect base comple-mentarity to the 5′-end of the miRNAs, and then computed the secondary structure by applying the standard folding program mfold (Zuker 2003) to the concatenation of potential binding site and miRNA. Similar approaches have been presented in Enright et al. (2003) and Rajewsky and Socci (2004), and for mammals and vertebrates, in Lewis et al. (2003). As noted by Stark et al. (2003), the drawbacks of this approach are, first, that the sequences have to be concatenated with a short linker sequence that can lead to artefacts in the prediction, and second, that hybridizations of the target with itself, or of the miRNA with itself, or of both with the linker, can happen. An additional drawback is that for prediction of multiple bindings in one target, the appropriate potential binding sites have to be cut out and folded separately.
Here, we present a program, RNAhybrid, that directly predicts multiple potential binding sites of miRNAs in large target RNAs. In general, the program finds the energetically most favorable hybridizations of a small RNA to a large RNA. Intramolecular hybridizations, that is, base pairings between target nucleotides or between miRNA nucleotides are not allowed. The program predicts optimal and additional suboptimal, nonoverlapping hits, either up to a user-defined number or with free energies up to a user-defined energy or _p_-value threshold. The user can force hybridizations to contain perfect helices in the 5′-part of the miRNA, for example, from nucleotides 2–7. For large targets, the time complexity of the algorithm is linear in the target length, allowing many long targets to be searched in a short time.
In the analysis of large databases of potential target sequences, a thorough statistical analysis of minimum free energies (MFEs) or other scores is of utmost importance. Current analyses do not sufficiently address this topic accurately. Stark et al. (2003) state that “folding energies of more than 3 standard deviations above the mean (Z ≥ 3) are expected to occur for only 0.3% of random matches,” which apparently assumes folding energies to be normally distributed. However, since such energies are results from an optimization procedure, this assumption is not appropriate. Rather, such energies are extreme value distributed and, for example, for a standard extreme value distribution, the probability of exceeding the mean score by more than three standard deviations is nearly 5%. Using _Z_-scores in this way thus vastly underestimates the probability of chance occurences. In addition, the authors calculate _E_values for the bantam miRNA by fitting an exponential function to the tail of a background empirical distribution function, but no such analysis is provided for the other miRNAs. In Enright et al. (2003), the authors repeated their experiment 100 times on shuffled sets of 73 miRNAs and compared the outcome for the real miRNAs with the background. Again, this assessment of false positive rates is not miRNA specific. Furthermore, the dinucleotide composition of the miRNAs is apparently not preserved, although this can be expected to be a major influence on duplex energies. In Rajewsky and Socci (2004), it has been recognized that miRNA-specific statistics are necessary due to variations in GC content or other features of sequence composition. Nevertheless, statistics of multiple target sites in a single sequence is not provided. In Lewis et al. (2003), miRNAs are carefully shuffled to preserve their dinucleotide distributions and other properties. On this basis, the authors compare numbers of predicted targets for real miRNAs (the “signal”) and shuffled miRNAs (the “noise”), but do not assign _E_-values to individual hits. None of the published approaches addresses the problem of statistical dependence between orthologous target sequences in cross-species analyses.
We complement the optimization of miRNA/target duplexes by a thorough statistical analysis of MFEs. We normalize MFEs with the sequence lengths of miRNAs and targets, and model such normalized MFEs as extreme value distributed. The parameters of these distributions are estimated specifically for every miRNA with a second program, RNAcalibrate, and are subsequently used to assign _p_-values to normalized MFEs. The significance of multiple binding sites in a single target is evaluated with a Poisson statistics. For comparative studies on multiple organisms such as different Drosophila species, we combine Poisson _p_-values from the orthologous targets using the effective number of sequences. This effective number respects the fact that related sequences cannot always be treated as statistically independent. Calculation of these effective numbers is miRNA and target specific and is accomplished by a third program, RNAeffective.
We applied RNAhybrid, RNAcalibrate, and RNAeffective in the prediction of miRNA 3′UTR targets in D. melanogaster, D. pseudoobscura, and Anopheles gambiae. We were able to significantly identify previously predicted or experimentally verified targets and a number of additional new ones. Results from previous predictions are revisited and discussed from a statistical point of view. In addition to the 3′UTRs, we searched coding sequences from D. melanogaster and A. gambiae. RNAhybrid, RNAcalibrate, and RNA-effective are available for download and as a Web tool on the Bielefeld Bioinformatics Server (http://bibiserv.techfak.unibielefeld.de/rnahybrid/).
RESULTS
Algorithm and implementation
RNAhybrid is an extension of the classical RNA secondary structure prediction algorithm (Zuker and Stiegler 1981) to two sequences. The miRNA is hybridized to the target in an energetically optimal way (i.e., yielding the minimum free energy, MFE), forbidding intramolecular base pairings and branching structures (multiloops). Energy parameters are from Mathews et al. (1999). Using the Dynamic Programming technique, the program calculates the MFE hybridizations of all possible start positions in the miRNA and in the target. Bulge loops (i.e., stretches of unpaired nucleotides in either of the sequences) and internal loops (i.e., stretches of unpaired nucleotides in both sequences) are restricted to a constant maximum length in either sequence (which is set to 15 as a default value). If m and n are the lengths of the target and the miRNA, respectively, and c is the maximal length of a loop in either sequence, the space consumption of the algorithm is of the order O(mn), and the time consumption is of the order O(c_2_mn). If m is much larger than n and c, which is usually the case for miRNAs and their potential targets, the space and time consumption is linear in the target length m. Additional optimal or suboptimal binding sites are found by masking previously reported sites and running the algorithm again with the constraint that masked nucleotides must not be part of a hybridization.
The original version of RNAhybrid was implemented in an extension of the Algebraic Dynamic Programming (ADP) framework (Giegerich 2000; Giegerich et al. 2004) that can handle two input sequences directly. The ADP version was translated into the C programming language by an extension of the ADP compiler (Giegerich and Steffen 2002), and subsequently hand-tuned in a few places. Command line interface and file handling was implemented directly by hand. The graphical output uses code from the Vienna RNA package (Hofacker 2003). The Dynamic Programming recurrences are shown in Table 1 ▶. The ADP version can be tested on the ADP Web page at http://bibiserv.techfak.uni-bielefeld.de/adp/. RNAhybrid and its accompanying programs RNAcalibrate and RNAeffective (see below) are available for download and as a Web tool on the Bielefeld Bioinformatics Server (http://bibiserv.techfak.unibielefeld.de/rnahybrid/).
TABLE 1.
Dynamic Programming recurrences for miRNA/target hybridization
| Hi,j = min {0, Ti, j, Ci, j} |
|---|
| Ti,j = min {T i+1,j,Bi, j} |
| Bi,j = min {B i, j+1, eds(i + 1, j + 1, C i+1,j+1), edt(i + 1, j, C i+1, j), edb(i, j + 1, C i,j+1)} |
| Ci,j = if can_pair (x i+1, y j+1) then min {sr(i + 1, j + 1, C i+1, j+1),{bt(i + 1, j + 1, k, C k,j+1)},{bb(i + 1, j + 1, l, C i+1,l)}, min i+2≤_k_≤min(i+16,_m_−1) j+2≤_l_≤min(j+16,_n_−1) {il(i + 1, j + 1, k, l, Ck,l)}, el(i + 1, j + 1, m, n)} else ∞ |
Prevention of hybridization artefacts
Figure 1 ▶ presents two examples of artefacts that can arise from folding the concatenation of a potential target site and a miRNA with standard folding programs like mfold (Zuker 2003) or RNAfold (Hofacker 2003; data provided by A. Stark, pers. comm.). In the first example, part of the target hybridizes with the linker. In the second example, part of the target hybridizes with itself. Whereas the hybridization between a miRNA and a target may indeed form an internal hairpin in either sequence as shown, the hybridization energy calculated by the folding program would include the energy of the hairpin, and would therefore be a misleading indicator of the strength of hybridization. Although these artefacts could have been prevented by setting appropriate base-pairing constraints, it would still be difficult to assign the correct energy. Whereas the contribution from the linker is the same for each hybridization, unpaired nucleotides at the 5′-end of the miRNA or the 3′-end of the target would constitute a bulge or internal loop, thus distorting the overall free energy in a nucleotide and loop-length-dependent way. None of these considerations are necessary for RNAhybrid, which automatically gives the desired results.
FIGURE 1.

Artefacts of target/miRNA concatenation. The linker sequence is CGNNNNNNCG. Hybridized are parts of 3′UTRs from D. melanogaster and the miR-2b miRNA. (A) The structure (from RNAfold) exhibits hybridization between target and linker (arrow). (B) Corresponding prediction from RNAhybrid that shows no artefact. (C) The structure (from RNAfold) exhibits self-hybridization of the target (arrow). (D) Corresponding prediction from RNAhybrid that shows no artefact. The 3′UTRs are from CG1969-RB (A,B) and CG30120-RA (C,D). The structures are drawn counter-clockwise, with the target followed by the miRNA. For RNAhybrid, the target is shown with its 5′-end additionally marked.
Searching a target database
Due to its linear time efficiency, RNAhybrid can be used to search large databases of long sequences. On an Ultra-SparcIII 900 MHz, searching C. elegans 3′UTRs from the UTR database (Pesole et al. 2002; http://bighost.area.ba.cnr.it/BIG/UTRHome/, 882 sequences with an average sequence length of 301 bases, 265,730 bases altogether) with the let-7 miRNA takes 23 sec. Figure 2 ▶ shows the four best MFE duplexes. These top-hits are the known targets of let-7 as follows: lin-14, lin-41, daf-12, and hbl-1. The next-best hybridizations are the metabotropic glutamate receptor homolog _CELF35_-1 with an MFE of −27.1 kcal/mole and the stress-responsive gene CePqM96 with an MFE of 26.6 kcal/mole (data not shown). The ability of RNAhybrid to correctly identify four bona-fide targets from a large data set of over 265 Kb indicates its potential for identifying unknown miRNA targets. The fact that two further hits were found that have free energies similar to those of the known let-7 targets, strongly suggests that these candidates may merit further experimental investigation.
FIGURE 2.

The four best minimum free energy (MFE) duplexes of the let-7 miRNA and C. elegans 3′UTRs (5′-end marked) from the UTR database (http://srs.ebi.ac.uk). The targets are 3CEL000274, lin-14, 3CEL000914 lin-41, 3CEL000790 daf-12, and 3CEL000772 hunchback-related protein hbl-1. The alignments show the complete miRNAs. The target UTRs are shown where they hybridize to the respective miRNA, plus dangling bases on either side. Each UTR was only searched for one optimal hit. Note also that in database search mode, RNAhybrid normally gives a textual representation of hybridizations to avoid the accumulation of a large number of plots.
Multiple hits per target
RNAhybrid can predict multiple potential binding sites per target. To illustrate this, Figure 3 ▶ shows the two energetically best hits between the let-7 miRNA and the 3′UTR of the C. elegans lin-41. The minimum free energies are -29.0 kcal/mole and -28.0 kcal/mole, respectively. The next-best hit has a far worse energy of −20.5 kcal/mole (data not shown). The hybridizations of the two hits are exactly the ones published (cf., for example, Grosshans and Slack 2002).
FIGURE 3.

The top two hits of the let-7 miRNA in the 3′UTR of the C. elegans lin-41 (5′-end marked). The first hit was also among the top hits in the target database search (cf. Fig. 2 ▶).
Forcing miRNA 5′-helices
It has been proposed that miRNA/target duplexes have to have perfect helices in the miRNA 5′-end to be functional. This idea is supported by the observation that known binding sites are conserved in their 3′-ends, which can thus all form helices with the miRNA 5′-ends (Lim et al. 2003), and its validity has recently been demonstrated experimentally in Doench and Sharp (2004). In Stark et al. (2003) and Lewis et al. (2003), this observation is an assumption in the prediction of further targets, in that it is used in the first step of finding sequence elements that show perfect complementarity to a 5′-part of the miRNA. This is to some extent the case in Enright et al. (2003), where 5′-complementarity is rewarded, and in Rajewsky and Socci (2004), where the presence of a nucleus of complementarity is assumed, but its position in the miRNA is determined in a training phase. Requiring such a nucleus biases the secondary structure prediction toward optimal structures that have an uninterrupted helix in the miRNA 5′-/target site 3′-region, effectively reducing the search space of secondary structures in the energy minimization procedure, and thus increasing the statistical significance of predicted binding sites. This has also been observed in Lewis et al. (2003), and the authors suggest that the importance of the “seed” in silico reflects its importance in vivo, and speculate that this segment nucleates pairing between miRNAs and their target sequences. RNAhybrid implements this structural constraint directly. The user can, if he or she wishes to do so, define which part of the miRNA has to form a perfect helix (e.g., from nt 2 to nt 7), and only structures fulfilling this constraint are considered in the Dynamic Programming optimization.
Figure 4 ▶ demonstrates the increase in statistical significance with extreme value distribution (EVD) density functions with and without a 5′-helix constraint. The distribution parameters are mean values for Drosophila miRNAs (see sections on negative normalized MFEs and extreme value statistics below). Negative normalized MFEs for 5′-helix-constrained duplexes are shifted considerably toward weaker energies, due to the search-space reduction described above. This has a strong influence on the statistical significance of moderate MFEs. For example, the _p_-value of an MFE of −25kcal/mole between a miRNA of length 22 and a target sequence of length 2000 is 0.045 with 5′-helix constraint and 0.21 without. For lower MFEs and smaller targets, however, the relative difference becomes smaller, for example, 3.7e-5 compared with 4.6e-5 for an MFE of −35kcal/mole in a target sequence of length 500.
FIGURE 4.

Extreme value distribution density functions. The location and shape parameters are mean values for Drosophila miRNAs. The left curve shows negative normalized MFEs of duplexes that are constrained to have a miRNA 5′-helix from nucleotides 2–7. The right curve shows such energies without a helix constraint, thus allowing unpaired nucleotides in all parts of the duplexes.
Length normalization of minimum free energies
Due to the shortness of miRNAs, good MFEs can occur frequently by chance. The longer a putative target sequence, the better such random energies will be. As a consequence, largely negative MFEs are meaningless if they are the result of searching large sequences. Borrowing a result from Karlin and Altschul (1990), we can normalize MFEs to eliminate the influence of sequence length as follows.
If e is the minimum free energy, m the length of the target sequence searched, and n the length of the miRNA, the negative normalized energy _e_n is defined as
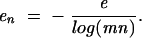 |
(1) |
|---|
Extreme value statistics of negative normalized minimum free energies
Minimum free energies (MFEs) are results from an optimization procedure, in our case, the optimization of duplexes between a miRNA and a putative target sequence. A result from probability theory states that the maximum of independent random variables follows an extreme value distribution (EVD; Gumbel 1958). Negative normalized MFEs, where high positive values correspond with low MFEs, can thus be modeled with EVDs. The distribution function of the standard EVD is
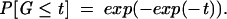 |
(2) |
|---|
With a location parameter ξ and a scale parameter θ, we get the shifted and rescaled distribution function
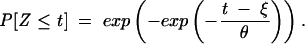 |
(3) |
|---|
The distribution function Ψ of an extreme value distribution can be transformed to a straight line by log(−log(Ψ)). If a sample distribution is approximately extreme value distributed, its cumulative density function can thus be transformed to an approximately straight line. To this line, the parameters of the EVD can be fit by least square linear regression (Waterman and Vingron 1994). If a is the slope and b the intercept of the regression line, we get the estimators
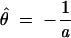 |
(4) |
|---|
and
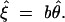 |
(5) |
|---|
To improve the estimates in the tail of the distribution, we do the fit only for normalized MFEs larger than 2.0, which corresponds to an MFE of ~20 kcal/mole from a miRNA of length 22 in a target of length 1000. We also skip the 1% best MFEs to get a more robust estimate. Figure 5 ▶ shows a fit to a distribution of negative normalized MFEs from the bantam miRNA and 5000 random target sequences.
FIGURE 5.

Fitting extreme value distribution parameters. The crosses show the log(−log) transformed empirical cumulative density function of negative normalized MFEs from the bantam miRNA and 5000 random target sequences. The straight line is fitted to negative normalized MFEs larger than 2.0 without the top 1% of data points.
For each predicted duplex between a miRNA and a target sequence with a certain MFE, we can now calculate the probability that such an MFE or a better one occurs by chance, the _p_-value:
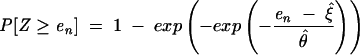 |
(6) |
|---|
where _e_n is the negative normalized MFE according to Equation 1, and ξ̂ and θ̂ are the estimated EVD parameters.
The expected number of such chance MFEs, the _E_-value, is the product of _p_-value and number of sequences in the target database, M:
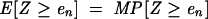 |
(7) |
|---|
_p_-values and _E_-values assess the statistical significance of observed (normalized) MFEs. If a _p_-value and its corresponding _E_-value are small, it is considered unlikely that the observed MFE is the result of a random complementarity between miRNA and target, and a biological meaning can be assumed. _p_- and _E_-values thus guide the user as to which results are likely to be correct predictions. Estimation of EVD parameters based on MFEs from random target sequences is implemented in the accompanying program RNAcalibrate.
Linear correlation between minimal duplex energies and extreme value distribution parameters
Figure 6 ▶ shows a scatter plot of minimal duplex energies (MDEs) of Drosophila miRNAs and location and scale parameters of fitted EVDs. The MDE of a miRNA is the best energy that can be achieved, and is easily calculated by hybridizing the miRNA with its reverse complement. Note that we are not saying here that such a complete hybridization would be functional in the miRNA pathway. The plots show a strong linear correlation between MDEs and location and scale parameters. For both data sets, the correlation coefficient is −0.86. This is in itself an interesting observation, but can also be used to estimate extreme value parameters quickly. Instead of searching a large set of random sequences, it suffices to calculate the MDE of a miRNA and then calculate location and scale parameters on the basis of a linear regression line of the scatter plots. The linear regression only has to be done once and can subsequently be used for all miRNAs. In fact, slopes and intercepts of these lines are part of RNAhybrid, thus allowing the immediate calculation of _p_-values without calibration. To be most accurate, however, one is advised to perform a calibration, as individual data points might deviate strongly from the regression lines. This is more important for human, with correlation coefficients of −0.46 and −0.53, and worm, with correlation coefficients of −0.47 and −0.45.
FIGURE 6.

Linear correlation between minimal duplex energies (MDEs) and extreme value distribution parameters. The top plot shows MDEs of Drosophila miRNAs (x-axis) and corresponding fitted location parameters (y-axis). The bottom plot shows the same MDEs and corresponding fitted scale parameters. The data points are highly linearly correlated with a correlation coefficient of −0.86 for both data sets.
Poisson statistics of multiple binding sites
Recent publications suggest that multiple potential binding sites of a miRNA in a single target are good evidence for the target being regulated by the miRNA (Enright et al. 2003; Lewis et al. 2003; Stark et al. 2003). If we consider a potential binding site being a rare event in our random model, the number of binding sites can be approximated by a Poisson distribution. Then, the probability that the number N of binding sites equals k, is
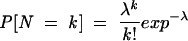 |
(8) |
|---|
with λ being the expectation _E_[_N_]. For small _p_-values, we have _E_[_N_]≈ p, thus, we can set λ= p, where p is the largest _p_-value of the k binding sites. We define k as the number of binding sites with a _p_-value not larger than 0.1. The probability of at least k binding sites is then
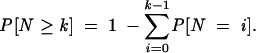 |
(9) |
|---|
Comparative analysis of orthologous targets
It has been noted that it is difficult to make significant target predictions when searching sequences from a single organism, and that targets should be predicted in a comparative analysis of multiple organisms (Enright et al. 2003; Lewis et al. 2003; Stark et al. 2003; Rajewsky and Socci 2004). If two orthologous sequences, for example, one from D. melanogaster and the other from D. pseudoobscura, are searched with the same miRNA, resulting in two optimal duplexes with negative normalized MFEs of _e_1 and _e_2, respectively, the joint probability of such energies occuring by chance, the joint _p_-value, is defined as
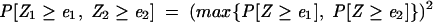 |
(10) |
|---|
assuming that in both organisms, random energies are identically distributed, and that the targets are independent. In the general case of an arbitrary number of orthologous sequences, the maximum is taken over all individual _p_-values and raised to the appropriate power:
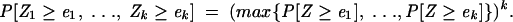 |
(11) |
|---|
The treatment of orthologous targets as statistically independent sequences is not always justified. For example, two 3′UTR sequences can share large blocks of similar sequence that might have been conserved during evolution, because they are functionally important in regulatory processes that are independent of the miRNA pathway. If no such regions in a given set of orthologous sequences exist, there can still be a strong dependence due to a very similar nucleotide or dinucleotide composition that gives the miRNA at hand plenty of opportunity for energetically good bindings. In general, the effective number of sequences, _k_eff, lies between 1 and the actual number k:
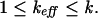 |
(12) |
|---|
To assess the degree of dependence in the context of miRNA/target duplex optimization, we do the following. We generate random miRNAs following the same dinucleotide distribution as the given miRNA and search the given orthologous target sequences (usually two or three). For each of the targets, this gives rise to an empirical distribution of normalized MFEs, on the basis of which we estimate target-specific EVD parameters as described above. Using these parameters, the normalized MFEs can be transformed into _p_-values. The _p_-values are then combined into joint _p_-values as in Equation 11, but with _k_′ instead of k, ranging between 1 and k. For each of these _k_′, we evaluate its goodness by the following rationale: If the joint _p_-values are good estimates, the empirical cumulative density function (CDF) is more or less a straight line. Thus, the best _k_′ (which is then our _k_eff), is the one that makes the empirical CDF as straight as possible under a squared error measure. Because we are especially interested in small _p_-values, the errors are weighted with a reverse function. Together:
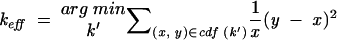 |
(13) |
|---|
where CDF(_k_′) is the empirical CDF that results from using _k_′ in the calculation of joint _p_-values.
The statistics of orthologous targets can be combined with the statistics of multiple binding sites in a straightforward way. For each of the orthologous sequences, Poisson _p_-values are calculated with the Poisson approximation as in Equation 9, and the results are combined into joint _p_-values following Equation 11, where the _P_[_Z≥e_i] are replaced by the corresponding Poisson _p_-values, and the exponent is replaced by _k_eff from Equation 13.
Prediction of Drosophila miRNA targets
As an application more challenging than the small C. elegans data set, we used RNAhybrid, RNAcalibrate, and RNAeffective for the prediction of miRNA targets in Drosophila and Anopheles. To this end, we searched 3′UTRs from D. melanogaster, D. pseudoobscura, and A. gambiae. The Drosophila sequences were the same as in Stark et al. (2003; kindly provided by A. Stark, pers. comm.). The Anopheles sequences were downloaded from the Ensembl database (Hubbard et al. 2002). In analogy to the construction of the D. pseudoobscura data set, the Anopheles set consists of sequences that have an ortholog in D. melanogaster. Where no 3′UTR was known, we selected 2 kb of the downstream sequence instead.
The three data sets were searched with 78 Drosophila miRNAs, forcing the duplexes to form perfect helices from nt 2 to 7 in the miRNAs. The MFEs were normalized by their lengths, and individual hit _p_-values, Poisson _p_-values for whole sequences, and joint _p_-values for orthologous sequences were calculated. The individual _p_-values were based on miRNA-specific simulations on 5000 random target sequences per miRNA using RNAcalibrate. The random target sequences were generated according to the dinucleotide distribution of the D. melanogaster 3′UTR data set, with lengths normally distributed with mean 500 and standard deviation 100. To enhance sensitivity and selectivity in the twilight zone, we first collected hits with joint _E_-values of up to 10, and then repeated the calibration for each of the hits, generating random sequences following target-specific dinucleotide frequencies. This procedure avoids artefactually good or bad _E_-values that are due to deviating dinucleotide distributions in individual target sequences. Further, for all hits, we calculated the effective number of orthologous sequences with RNAeffective. This avoids artefactually good _E_-values that are due to statistical dependences between orthologous targets. We also restricted the analysis to those cases in which a miRNA has at least one hit each in D. melanogaster and D. pseudoobscura. Due to the multiple-testing scenario, we adjusted _E_-values conservatively by a factor of 6, corresponding to three Poisson tests (one, two, or at least three hits per target of various quality) times two tests from the comparative study (with or without a hit in A. gambiae).
Previously and newly predicted miRNA targets in 3′UTRs
We were able to predict known and new miRNA targets in Drosophila. For the bantam miRNA, the previously identified target hid (Brennecke et al. 2003) has an _E_-value of 0.29 (with two significant hits in D. melanogaster, two in D. pseudoobscura, and none in A. gambiae, abbreviated as 2/2/0). The best prediction (E = 8.5e-4, 2/1/1) is nervous fingers 1, which is required for proper CNS axon guidance (Kuzin et al. 2003). We also identified Distal-less (E = 0.18, 2/2/0), which is specifically expressed early in developing insect limbs, encoding a homeodomain transcription factor (Panganiban and Rubenstein 2002).
The proapoptotic genes grim, reaper, and sickle have been shown experimentally to be targets of miR-2 in Stark et al. (2003). There, however, only grim and reaper are among the top predictions, whereas we predicted all three targets with significant _E_-values. grim was predicted as miR-2a target (E = 0.082, 1/1/0), miR-2b target (E = 0.43, 1/2/0), and miR-2c target (E = 0.27, 1/1/0), reaper as miR-2a target (E = 0.0037, 1/1/0), miR-2b target (E = 0.11, 1/1/0), and miR-2c target (E = 0.057, 1/1/0), and sickle as miR-2b target (E = 0.33, 2/2/0). A very interesting candidate is spastin (miR-2b, E = 4.8, 1/1/0; miR-2c, E = 0.35, 1/1/0) whose human homolog plays a major role in dominant hereditary spastic paraplegia, where spastin overexpression causes massive death of cells (Orso et al. 2003). This apoptotic phenotype fits well into the picture of miR-2 as a regulator of proapoptotic genes. Weak hits are Hairy/E(spl)-related with YRPW motif (miR-2c, E = 2.2, 1/1/0; Lai 2002) and the antiapoptotic gene tartan, which supports cell survival in the Drosophila wing imaginal disc (Milan et al. 2002; miR-2a, E = 3.14, 1/1/0). The latter hit suggests the possibility that miR-2 regulates apoptotic genes in a more general sense.
Among the top scoring hits for miR-7, we find members of the Notch signaling pathway that were described in Stark et al. (2003) as follows: E(spl) region transcript m3 (E = 0.05, 1/1/0) and Twin of m4 (E = 8.6e-4, 2/2/0). Not predicted by Stark et al. (2003) was the E(spl) region transcript mγ (E = 0.56, 1/2/0). We also weakly identified Him (E = 1.8, 1/2/0), which shows a highly restricted expression pattern in the Drosophila wing disc (Butler et al. 2003) and hairy (E = 6.4, 1/1/0), which in Stark et al. (2003) was among the top 10 hits and experimentally verified. Our best prediction was CG8394 (E = 5.7e-4, 2/3/3), a homolog of unc-47, which is localized to synaptic vesicles (Eastman et al. 1999), suggesting that in addition to transcriptional regulation, unc-47 might also undergo post-transcriptional regulation.
Stark et al. (2003) suggest the possibility that miR-277 might function as a metabolic switch in the valine, leucine, and isoleucine catabolic pathway. This was supported by their prediction of seven members from that pathway as putative targets. In our analysis, however, we only identified one target with a small _E_-value, CG1673 (E = 0.09, 2/2/0), and two other previously reported hits with weak _E_-values, CG15093 (E = 8.3, 2/1/1) and CG1140 (E = 17, 1/1/0). The D. melanogaster and D. pseudoobscura 3′UTRs show high sequence similarity. For example, the effective number of orthologous sequences for CG15093 is 2.3. Had this not been taken into account, the _E_-value would have been 0.96. For CG1140, with an effective number of 1.6 (for two sequences), the _E_-value would have been 3.5. In such cases, a conserved miRNA binding site cannot be interpreted as evidence of miRNA regulation, as it might be an artefact of the overall sequence conservation. However, the question would be why several of these functionally related targets appear among the top, if not significant predictions. Whereas a miRNA-independent regulation that is directed at conserved elements in the 3′UTRs of the genes in question might be the answer, the mechanism of regulation by miR-277 proposed by Stark et al. (2003) cannot be excluded. As the authors point out, it remains to be determined whether this is the case.
In the overall analysis, 227 predicted targets had good adjusted _E_-values of up to 1.0 per miRNA, thus, with an overall expected number of 78. This is a signal-to-noise ratio of 2.9:1, which is very close to the signal-to-noise ratio of 3.2:1 reported in Lewis et al. (2003) on mammalian sequences, although these might not be comparable to the insect sequences analyzed here.
A total of 22 predicted targets had very good _E_-values of <0.01 per miRNA, thus, with an overall expected number of 0.78 and a signal-to-noise ratio of 28:1. In addition to nervous fingers 1, reaper, and Twin of m4 (see above), this list contains Cytochrome P450-18a1 as target of miR-276b (E = 3.7e-4, 1/1/0), I channel as target of miR-2b (E = 3.4e-3, 1/1/2), Kinesin heavy chain as target of miR-280 (E = 5.1e-3, 1/1/0), Smg5 as target of miR-317 (E = 4.3e-4, 3/3/0), Tim13 as target of miR-314 (E = 6.7e-5, 1/1/0), comm2 as target of miR-2a (E = 7.3e-3, 2/2/0), miR-2b (E = 4.4e-3, 2/2/0), and miR-2c (E = 8.1e-3, 2/2/0), and CG10005, CG15125, CG2118, CG7713, CG8394, CG9298, and CG9746.
Predicted targets for all miRNAs are available as Online Supplemental Material at http://www.techfak.uni-bielefeld.de/persons/marc/mirna/targets/drosophila/.
Restriction to genes important for fly body patterning
One major insight from the above statistical analysis is that only a small number of miRNA targets might be significantly predictable. This is not only due to the shortness of the miRNAs, but also to the large data set of 3′UTRs. The significance can be increased by restricting the potential target set to a smaller number of genes. This is done in Rajewsky and Socci (2004), where the analysis focuses on 31 genes important for fly body patterning. Because the number of trials drops from roughly 10,000 3′UTRs to 31, _E_values of predicted binding sites in these 31 genes are decreased about 320-fold. In Rajewsky and Socci (2004), 39 high-scoring putative target sites are reported. We repeated our experiment described above with _E_-value cutoffs corrected to reflect the data size of 31 3′UTRs, demanding at least one D. melanogaster and one D. pseudoobscura hit, without recalibration and calculation of effective numbers of orthologous sequences, thus being close to the specificity of the statistical approach in Rajewsky and Socci (2004), which is miRNA, but not target-sequence specific. Our search resulted in 39 hits with individual _E_-values (adjusted by a factor of 6) up to 1.0, which is relatively close to the expected number of 78, and incidentally the same as in the above study. A total of 11 hits in four genes have _E_-values of <0.1, which is roughly the same as the expected number of 7.8. We repeated the analysis with a target-specific recalibration as described above, which resulted in 51 hits up to E = 1.0, 18 hits up to E = 0.1, and five hits up to E = 0.01. These five top hits are tailless (hit by miR-2b, 1/1/0; by miR-2c, 1/1/0; and by miR-92a, 1/2/0), empty spiracles (hit by miR-276a, 1/2/0), and hairy (hit by miR-210, 1/1/0).
A reinvestigation of Rajewsky and Socci (2004) leads to the following results. The authors define score thresholds, such that they discover 84% of the known targets in their training data set, at which random matches are expected to occur every 4000 bases of scanned sequence. For 31 sequences of average length 780 [estimated from 30 3′UTRs of the genes in question that we downloaded from the Ensembl database (Hubbard et al. 2002)], one can thus expect 6.1 random matches per miRNA. The expected number of matches in the orthologous sequences would then be 1.2 (6.1 × 780 over 4000), which leads to an overall expected number of orthologous hit pairs of 89 for 74 miRNAs. These are again more than the 39 predicted binding sites.
Our two analyses confirm each other in their conclusion that the majority of binding-site predictions in the set of 31 fly body-patterning genes are not significant. However, this does not unequivocally mean that these predicted binding sites are not functional, and hairy is evidence to the contrary, as it has been shown in Stark et al. (2003) to be a target of miR-7, and this combination is predicted by Rajewsky and Socci (2004) as well as by our method (E = 0.01, 1/1/0). Also, using RNAhybrid with recalibration, the output is slightly enriched in hits with _E_-values smaller than 0.01, and these hits are different from the ones reported by Rajewsky and Socci (2004) for the genes in question. Because we have shown our method to predict known target sites significantly, we propose that our top predictions are worth testing in the appropriate wet-lab experiments.
Prediction of miRNA targets in coding sequence
In addition to 3′UTRs, we analyzed coding sequences from D. melanogaster and A. gambiae, downloaded from the Ensembl database (Hubbard et al. 2002). Duplexes were not forced to have perfect 5′-helices. A search with 78 miRNAs resulted in coding sequence hits in two genes or orthologous gene pairs with individual _E_-values of up to 1.0 (adjusted by a factor of 9, corresponding to three Poisson tests times three tests from the comparative study), which is 39-fold lower than the expected number of 78 hits. This strong under-representation suggests that not only animal miRNAs do not regulate their targets by binding the coding part of mRNAs, but that the evolution of coding sequence might have actively selected against random binding sites.
Because coding sequence can be expected to be conserved to a higher degree than sequence from untranslated regions, orthologous sequences cannot be assumed to be statistically independent in general. Calculating the effective number of orthologous sequences as in Equation 13 is thus a necessity in a comparative study of multiple organisms. In the above analysis, 10 miRNAs had hits in both a D. melanogaster gene and its A. gambiae ortholog with an _E_-value of up to 10 (adjusted by a factor of 3). The same analysis without calculating the effective number of orthologous sequences resulted in 54 such hits.
DISCUSSION
We have presented a method for the prediction of miRNA/target duplexes. A study of 3′UTRs from D. melanogaster, D. pseudoobscura, and A. gambiae resulted in the significant prediction of known and new miRNA targets, thus demonstrating its usefulness. An analysis of coding sequences from D. melanogaster and A. gambiae produced far less hits than expected, suggesting that such binding sites might be evolutionarily under-represented to protect the coding parts of mRNA.
The core program of our method, RNAhybrid, is fast enough to allow large databases of long potential target sequences to be searched, and is effective, in that it solves the addressed problem directly without having to use makeshift adaptations of existing RNA secondary structure prediction programs. Similar programs exist, but exhibit several disadvantages in this context. RNAfold from the Vienna RNA package can consider user-defined constraints that specify the kind of base pairings at certain positions. For example, one can concatenate the target and the miRNA with a small linker sequence, as in Stark et al. (2003), and set constraints such that all target nucleotides can pair only downstream and all miRNA nucleotides can pair only upstream, thus forbidding intramolecular base pairings. This still leaves the necessity and potential artefacts of a linker sequence. RNAcofold, which is part of the latest 3-version of the same package, can hybridize two sequences, which are concatenated internally without an explicit linker; however, constraints have no effect here, thus leaving the problem of intramolecular hybridizations. One could argue that such hybridizations should be allowed to model competition between self-hybridization and miRNA/target hybridization. However, if a miRNA binds to a loop region of the target, the corresponding structure of the concatenated sequences would constitute a pseudoknot that, for complexity reasons, cannot be handled by standard folding programs (for recent developments on special classes of pseudoknots, compare Rivas and Eddy 1999 and J. Reeder and R. Giegerich, “Design, Implementation and Evaluation of a Practical Pseudoknot Folding Algorithm based on Thermodynamics,” in prep.). A new version of the mfold program allows hybridization of two sequences, which are input concatenated with three consecutive letters L. Whenever these L’s occur in a hairpin loop, this loop is treated not as a hairpin loop, but as an external loop. However, although base-pair constraints can be given, they have no effect, thus, self-hybridizations cannot be avoided. Another program, PairFold from the RNAsoft suite (http://www.rnasoft.ca) also allows self-hybridizations.
All of the above programs have the drawback that they are adaptations of the original folding algorithm, thus preserving the O(_n_3) time complexity, even if calculations are sped up by base-pair constraints. For example, running RNAfold with appropriate base-pair constraints on the C. elegans UTR database with the let-7 miRNA takes 5 min, which is 13 times slower than RNAhybrid. The difference between the two methods grows larger with larger target sequences. Running RNAhybrid on the longest sequence from this database, 3CEL000087 with 2837 bp, takes 0.21 sec, whereas RNAfold needs 38 sec, which is 181 times slower. Although the absolute values in this comparison are relatively small, the speed of RNAhybrid opens new vistas for the analysis of large data sets. In addition, none of the available programs offers the calculation of suboptimal, nonoverlapping binding sites.
Searching large databases inevitably means generating random hits. This has long been recognized in other areas of sequence analysis, such as protein database searching, and also in the recent literature about miRNA target prediction. The latter, however, lacks in places the thorough approach presented here. We have addressed a large number of statistical issues in a rigorous way, providing length normalizations of binding energies, modeling normalized energies with extreme value distributions in a miRNA and target-specific way, modeling multiple binding sites with a Poisson approximation, estimating statistical dependences between orthologous genes, and assigning significance estimates (_E_values) to individual target predictions. Some aspects of our approach, however, are still computationally expensive. In our study of Drosophila 3′UTRs, we recalibrated the EVD parameters specific for targets that looked promising with more general parameters. Each recalibration, corresponding to one miRNA/target pair, meant searching 5000 random sequences. The calculation of the effective number of or-thologous sequences meant searching two or three putative orthologous targets with 5000 random miRNAs. Even with our fast algorithm and implementation, the whole analysis took a number of days on a cluster of 100 netra CPUs. Nevertheless, even without recalibration and effectiveness calculations, the predictions should be useful, and in the above analysis, the high-scoring hits were still present (data not shown). RNAhybrid can also be used in a very simple version to find binding sites in an interesting gene that is supposed to be regulated in the miRNA pathway. For larger experiments, however, the statistical issues discussed in this study provide a rationale for a coherent evaluation of significance.
Acknowledgments
M.R. thanks Julius Brennecke, Stephen M. Cohen, and Alexander Stark for their extensive testing of RNAhybrid and for contributing examples and the Drosophila 3′UTR sequence set, Sven Rahman for discussions on _p_-values, Jan Krüger for implementing the Web server, and Leonie Ringrose for advice concerning the language of this article. R.G. thanks John Mattick for discussing the importance of sequence matching under RNA rules.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
REFERENCES
- Abrahante, J.E., Daul, A.L., Li, M., Volk, M.L., Tennessen, J.M., Miller, E.A., and Rougvie, A.E. 2003. The Caenorhabditis elegans hunchback-like gene lin-57/hbl-1 controls developmental time and is regulated by microRNAs. Dev. Cell 4**:** 625–637. [DOI] [PubMed] [Google Scholar]
- Brennecke, J., Hipfner, D.R., Stark, A., Russell, R.B., and Cohen, S.M. 2003. bantam encodes a developmentally regulated microRNA that controls cell proliferation and regulates the proapoptotic gene hid in Drosophila. Cell 113**:** 25–36. [DOI] [PubMed] [Google Scholar]
- Butler, M.J., Jacobsen, T.L., Cain, D.M., Jarman, M.G., Hubank, M., Whittle, J.R.S., Phillips, R., and Simcox, A. 2003. Discovery of genes with highly restricted expression patterns in the Drosophila wing disc using DNA oligonucleotide microarrays. Development 130**:** 659–670. [DOI] [PubMed] [Google Scholar]
- Doench, J.G. and Sharp, P.A. 2004. Specificity of microRNA target selection in translational repression. Genes & Dev. 18**:** 504–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastman, C., Horvitz, H., and Jin, Y. 1999. Coordinated transcriptional regulation of the unc-25 glutamic acid decarboxylase and the unc-47 GABA vesicular transporter by the Caenorhabditis elegans UNC-30 homeodomain protein. J. Neurosci. 19**:** 6225–6234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enright, A.J., John, B., Gaul, U., Tuschl, T., Sander, C., and Marks, D.S. 2003. MicroRNA targets in Drosophila. Genome Biol. 5**:** R1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giegerich, R. 2000. A systematic approach to dynamic programming in bioinformatics. Bioinformatics 16**:** 665–677. [DOI] [PubMed] [Google Scholar]
- Giegerich, R. and Steffen, P. 2002. Implementing algebraic dynamic programming in the functional and the imperative programming paradigm. In Mathematics of program construction (eds. E. Boiten and B. Möller), pp. 1–20. Springer LNCS 2386, Heidelberg, Germany.
- Giegerich, R., Meyer, C., and Steffen, P. 2004. A discipline of dynamic programming over sequence data. Sci. Comput. Prog. 51**:** 215–263. [Google Scholar]
- Grosshans, H. and Slack, F.J. 2002. Micro-RNAs: Small is plentiful. J. Cell Biol. 156**:** 17–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumbel, E.J. 1958. Statistics of extremes. Columbia University Press, New York.
- Hofacker, I.L. 2003. Vienna RNA secondary structure server. Nucleic Acids Res. 31**:** 3429–3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard, T., Barker, D., Birney, E., Cameron, G., Chen, Y., Clark, L., Cox, T., Cuff, J., Curwen, V., Down, T., et al. 2002. The Ensembl genome database project. Nucleic Acids. Res. 30**:** 38–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin, S. and Altschul, S.F. 1990. Methods for assessing the statistical significance of molecular sequence features by using general scoring schemes. Proc. Natl. Acad. Sci. 87**:** 2264–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzin, A., Stivers, C., Brody, T., and Odenwald, W.F. 2003. nerfin-1, a member of a conserved Zn-finger gene subfamily, is required for proper CNS axon guidance. In A. Dros. Res. Conf. 44, page 710B. The Genetics Society of America, Bethesda, MD.
- Lai, E. 2002. Micro RNAs are complementary to 3′ UTR sequence motifs that mediate negative post-transcriptional regulation. Nat. Genet. 30**:** 363–364. [DOI] [PubMed] [Google Scholar]
- Lau, N.C., Lim, L.P., Weinstein, E.G., and Bartel, D.P. 2001. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 294**:** 858–862. [DOI] [PubMed] [Google Scholar]
- Lee, R.C., Feinbaum, R.L., and Ambros, V. 1993. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75**:** 843–854. [DOI] [PubMed] [Google Scholar]
- Lewis, B.P., Shih, I., Jones-Rhoades, M.W., Bartel, D.P., and Burge, C.B. 2003. Prediction of mammalian microRNA targets. Cell 115**:** 787–798. [DOI] [PubMed] [Google Scholar]
- Lim, L., Lau, N., Weinstein, E., Abdelhakim, A., Yekta, S., Rhoades, M., Burge, C., and Bartel, D. 2003. The microRNAs of Caenorhabditis elegans. Genes & Dev. 17**:** 991–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, S.Y., Johnson, S.M., Abraham, M., Vella, M.C., Pasquinelli, A., Gamberi, C., Gottlieb, E., and Slack, F.J. 2003. The C. elegans hunchback homolog, hbl-1, controls temporal patterning and is a probable microRNA target. Dev. Cell 4**:** 639–650. [DOI] [PubMed] [Google Scholar]
- Mathews, D.H., Sabina, J., Zuker, M., and Turner, D.H. 1999. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol. 288**:** 911–940. [DOI] [PubMed] [Google Scholar]
- Milan, M., Perez, L., and Cohen, S. 2002. Short-range cell interactions and cell survival in the Drosophila wing. Dev. Cell 2**:** 797–805. [DOI] [PubMed] [Google Scholar]
- Moss, E.G., Lee, R.C., and Ambros, V. 1997. The cold shock domain protein LIN-28 controls developmental timing in C. elegans and is regulated by the lin-4 RNA. Cell 88**:** 637–646. [DOI] [PubMed] [Google Scholar]
- Orso, G., Rossetto, M., Sartori, E., and Daga, A. 2003. Functional analysis of the Drosophila spastin gene. In A. Dros. Res. Conf. 44, page 786C. The Genetics Society of America, Bethesda, MD.
- Panganiban, G. and Rubenstein, J. 2002. Developmental functions of the Distal-less/Dlx homeobox genes. Development 129**:** 4371–4386. [DOI] [PubMed] [Google Scholar]
- Pesole, G., Liuni, S., Grillo, G., Licciulli, F., Mignone, F., Gissi, C., and Saccone, C. 2002. UTRdb and UTRsite: Specialized databases of sequences and functional elements of 5′ and 3′ untranslated regions of eukaryotic mRNAs. Update 2002. Nucleic Acids Res. 30**:** 335–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajewsky, N. and Socci, N.D. 2004. Computational identification of microRNA targets. Dev. Biol. 267**:** 529–535. [DOI] [PubMed] [Google Scholar]
- Reinhart, B.J., Slack, F.J., Basson, M., Pasquinelli, A.E., Bettinger, J.C., Rougvie, A.E., Horvitz, H.R., and Ruvkun, G. 2000. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 403**:** 901–906. [DOI] [PubMed] [Google Scholar]
- Rhoades, M.W., Reinhart, B.J., Lim, L.P., Burge, C.B., Bartel, B., and Bartel, D.P. 2002. Prediction of plant microRNA targets. Cell 110**:** 513–520. [DOI] [PubMed] [Google Scholar]
- Rivas, E. and Eddy, S.R. 1999. A dynamic programming algorithm for RNA structure prediction including pseudoknots. J. Mol. Biol. 285**:** 2053–2068. [DOI] [PubMed] [Google Scholar]
- Slack, F.J., Basson, M., Liu, Z., Ambros, V., Horvitz, H.R., and Ruvkun, G. 2000. The lin-41 RBCC gene acts in the C. elegans heterochronic pathway between the let-7 regulatory RNA and the LIN-29 transcription factor. Mol. Cell 5**:** 659–669. [DOI] [PubMed] [Google Scholar]
- Stark, A., Brennecke, J., Russell, R.B., and Cohen, S.M. 2003. Identification of Drosophila microRNA targets. PLoS Biol. 1**:** 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman, M.S. and Vingron, M. 1994. Rapid and accurate estimates of statistical significance for sequence database searches. Proc. Natl.Acad. Sci. 91**:** 4625–4628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker, M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31**:** 3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker, M. and Stiegler, P. 1981. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Res. 9**:** 133–148. [DOI] [PMC free article] [PubMed] [Google Scholar]