A role for eIF4E and eIF4E-transporter in targeting mRNPs to mammalian processing bodies (original) (raw)
Abstract
mRNP remodeling events required for the transition of an mRNA from active translation to degradation are currently poorly understood. We identified protein factors potentially involved in this transition, which are present in mammalian P bodies, cytoplasmic foci enriched in 5′ → 3′ mRNA degrading enzymes. We demonstrate that human P bodies contain the cap-binding protein eIF4E and the related factor eIF4E-transporter (eIF4E-T), suggesting novel roles for these proteins in targeting mRNAs for 5′ → 3′ degradation. Furthermore, fluorescence resonance energy transfer (FRET) studies indicate that eIF4E interacts with eIF4E-T and the putative DEAD box helicase rck/p54 in the P bodies in vivo. RNAi-mediated knockdowns revealed that a subset of P body factors, including eIF4E-T, LSm1, rck/p54, and Ccr4 are required for the accumulation of each other and eIF4E in P bodies. In addition, treatment of HeLa cells with cycloheximide, which inhibits translation, revealed that mRNA is also required for accumulation of mRNA degradation factors in P bodies. In contrast, knockdown of the decapping enzyme Dcp2, which initiates the actual 5′ → 3′ mRNA degradation did not abolish P body formation, indicating it first functions after mRNPs have been targeted to these cytoplasmic foci. These data support a model in which mRNPs undergo several successive steps of remodeling and/or 3′ trimming until their composition or structural organization promotes their accumulation in P bodies.
Keywords: mRNA degradation, translation, P bodies, eIF4E transporter
INTRODUCTION
mRNA degradation is an important step in the regulation of gene expression in eukaryotic cells. Most mRNAs are either degraded in the 3′ → 5′ direction by the exosome, a multicomponent protein complex, or in the 5′ → 3′ direction by other degradation factors. In yeast, the major mRNA degradation pathway initiates with the deadenylation of the mRNA by Ccr4p, followed by decapping at its 5′ end by Dcp1p/Dcp2p, and concludes with 5′ → 3′ digestion by the exonuclease Xrn1p (Meyer et al. 2004). In addition to degrading enzymes, auxiliary factors such as Pat1p/Mrt1p and the Sm-like proteins Lsm1p to Lsm7p, which bind sub-sequent to deadenylation, are also required (Tharun and Parker 2001). Mammalian homologs of most of these proteins, which are structurally and functionally similar to the yeast proteins, have been identified indicating that this mRNA degradation pathway is conserved in mammals (Chen et al. 2002; Ingelfinger et al. 2002; Lykke-Andersen 2002; van Dijk et al. 2002; Wang et al. 2002). Several of these factors are not evenly distributed in the cytoplasm of mammalian cells, but rather are found in discrete foci. These include the 3′ deadenylase Ccr4, the 5′ decapping factors Dcp1 and Dcp2, the exonuclease Xrn1, and LSm proteins (Bashkirov et al. 1997; Ingelfinger et al. 2002; Lykke-Andersen 2002; van Dijk et al. 2002; Cougot et al. 2004). In addition, the mRNA-binding protein GW182 also accumulates in these foci (Eystathioy et al. 2003). Several factors required for mRNA degradation in yeast have also been reported to be enriched in similar cytoplasmic foci. The latter have been shown to contain mRNA degradation intermediates suggesting that they are mRNA processing centers (Sheth and Parker 2003). In mammals, RNAi-mediated knockdown of Xrn1 leads to the accumulation of poly(A)+ containing mRNAs in these foci indicating that, like in yeast, the mammalian structures are likely mRNA processing bodies (Cougot et al. 2004). Thus, we henceforth refer to them as mammalian P bodies.
While several factors involved in mRNA degradation are known to be present in P bodies, many unanswered questions concerning the assembly and function of these foci remain. For example, what are the factor requirements for the assembly of P bodies and how are degradation factors targeted to these sites? Are they imported independently of each other or as part of protein or mRNP complexes? Finally, what targets a translating mRNA to these structures and which factors are required to initiate the transition from translation to degradation within P bodies? The transition of an mRNA from active translation to being committed for degradation has been proposed to involve one or more mRNP remodeling events (Tharun and Parker 2001). Deadenylation is clearly a crucial determinant for initiating mRNA degradation, and thus factors involved in 3′ end trimming may play an important role. In addition, factors interacting at the 5′ end of the mRNA could also be involved. For example, a block in translation initiation could potentially trigger the degradation process and thus, translation initiation factors and proteins that repress their activity might also play a decisive role. However, whether translation factors are present in P bodies, and more importantly, whether they are required for P body formation, is currently unknown.
RESULTS
The cap-binding, translation initiation factor eIF4E is present in P bodies
To address these questions, we first checked for the presence of the translation initiation factor eIF4E in P bodies of HeLa cells by performing immunofluorescence studies with a mouse monoclonal anti-eIF4E antibody (see Materials and Methods). Cells were counterstained with antibodies against LSm1, a marker of P bodies (Ingelfinger et al. 2002). eIF4E was distributed throughout the cytoplasm, but not the nucleus, of HeLa cells and accumulated in discrete cytoplasmic foci (Fig. 1a–c ▶). A similar distribution of eIF4E in HeLa cells was observed with a rabbit anti-eIF4E serum (data not shown). The latter represent P bodies as evidenced by the colocalization of eIF4E with LSm1 in these foci (Fig. 1c ▶). eIF4E was previously detected by immunofluorescence studies in both the cytoplasm and the nucleus of mammalian cells, but not in discrete cytoplasmic foci (Dostie et al. 2000b). To rule out antibody-dependent effects and independently confirm our findings, HeLa cells were transfected with a plasmid expressing a YFP-eIF4E fusion. Significantly, the distribution of YFP-eIF4E in transfected cells (Fig. 1d ▶) was indistinguishable from that of the endogenous protein, as detected by immunofluorescence (Fig. 1 ▶ cf. f and c). The presence of eIF4E in P bodies suggested that other translation initiation factors might also be present in these structures. Thus, we subsequently tested for the presence of eIF4G, eIF4A, and eIF4B in P bodies. As shown in Figure 1g–i ▶, eIF4G did not colocalize with LSm1 in P bodies. Similar results were obtained with antibodies against eIF4A and eIF4B, as well as other components of the translation machinery, namely eEF2, PABP, and the ribosomal proteins rpS6 and rpL28; none of these factors colocalized with P body markers (data not shown). Thus, translationally active complexes are not present in P bodies.
FIGURE 1.

eIF4E and eIF4E-T colocalize with LSm1 in distinct cytoplasmic foci. HeLa SS6 cells were grown on coverslips, fixed, and stained with antibodies specific for eIF4E (monoclonal anti-eIF4E, Santa Cruz Biotechnology) (A,N), LSm1 (B,E,H,K,Q,T), eIF4G (G,J,M), or eIF4E-T (P). Alternatively, cells were transfected with plasmids encoding YFP-eIF4E (D) or YFP-eIF4E-T (S). Panels C,F,I,L,O,R,U show the merged picture of the proceeding two panels, with overlaying signals appearing yellow. In panels J_–_O, cells were treated with 100 μM arsenite for 45 min at 37°C to induce stress granules (indicated by arrows). Cells were analyzed by confocal fluorescence microscopy. Scale bars = 10 μm.
In higher eukaryotes, under certain nonphysiological conditions such as thermal or chemical stress, inactive mRNPs accumulate in cytoplasmic structures called stress granules (SG). They store inactive translation preinitiation complexes that contain eIF4E and eIF4G, which means that both factors are part of mRNP complexes in SGs (Kimball et al. 2003). The presence of eIF4E in P bodies suggested a possible relationship to SGs. To investigate similarities and/or differences between P bodies and SGs directly, we treated HeLa cells with arsenite to induce the formation of stress granules, and compared the composition of both P bodies and SGs by immunofluorescence with antibodies against LSm1 (as a marker of P bodies) and the translation initiation factors eIF4E and eIF4G. While eIF4G solely accumulates in SGs and LSm1 solely in P bodies (Fig. 1j–l ▶), eIF4E localizes to both structures (Fig. 1m–o ▶). Consistent with these results, the SG marker TIA-1 is also excluded from P bodies (Cougot et al. 2004). Thus, P bodies and SGs are clearly distinct, but share eIF4E.
The eIF4E-binding protein eIF4E-T is a component of P bodies
The presence of eIF4E, but not eIF4G, in P bodies is a first indication that the former factor may play an early role in the transition of an mRNA from active translation to degradation. eIF4G interacts with eIF4E bound to the mRNA’s 5′ cap and with the poly A-binding protein (PABP) bound to the 3′ end of the mRNA. Thus, a closed loop, which has been shown to be necessary for optimal mRNA translation, may form between the 5′ and 3′ ends of the mRNA (for review, see Sachs and Varani 2000). Disruption of this loop either by deadenylation and/or by inhibiting the eIF4E/eIF4G interaction could potentially initiate mRNA degradation. Several eIF4E-binding proteins, including eIF4E-BP1 and eIF4E-transporter (eIF4E-T), compete with eIF4G for the same binding site on eIF4E, and their binding to eIF4E therefore inhibits translation. In addition, studies in Xenopus oocytes suggest that Xp54 (the ortholog of yeast Dhh1) may also bind eIF4E, but a direct interaction has not been conclusively demonstrated (Minshall and Standart 2004). We thus investigated whether one or more of these factors are also present in P bodies in HeLa cells.
eIF4E-BP1 immunofluorescence studies, as well as experiments carried out with YFP-eIF4E-BP1, demonstrated that eIF4E-BP1 does not accumulate in P bodies (data not shown). In contrast, studies with antibodies raised against the Xenopus counterpart of human rck/p54 (designated Xp54), which also recognize the human protein, and YFP-LSm6 as P body-marker, revealed that endogenous rck/p54 is present in P bodies in HeLa cells (data not shown.). These results are consistent with recent data localizing a GFP-rck/p54 fusion protein to discrete cytoplasmic foci in human HEK 293 cells (Cougot et al. 2004). Interestingly, immunofluorescence studies with antibodies against eIF4E-T, as well as experiments carried out with YFP-eIF4E-T, demonstrated that eIF4E-T colocalizes with LSm1 in P bodies (Fig. 1p–r and s–u ▶). In addition, eIF4E-T was found solely in P bodies, and not in SGs, after arsenite treatment (data not shown). Thus, proteins potentially interacting with eIF4E are also P body components. eIF4E-T was previously reported to play a role in the nucleocytoplasmic transport of eIF4E, and was postulated to contribute to a possible function of eIF4E in the nucleus (Dostie et al. 2000a; Strudwick and Borden 2002). Our data thus suggest that eIF4E-T potentially plays an additional role in the cytoplasm, namely in the transition of an mRNA from active translation to being committed for degradation.
eIF4E interacts with eIF4E-T and rck/p54 in P bodies
Biochemically, it has been reported that eIF4E and eIF4E-T interact in vivo and in vitro (Dostie et al. 2000a). Therefore, it was tempting to study if these proteins interact in P bodies in vivo. To determine whether the presumed protein pair eIF4E/eIF4E-T indeed forms a molecular complex in P bodies in vivo, we performed fluorescence resonance energy transfer (FRET) measurements. The fluorescent protein pairs of CFP and YFP can serve as donor and acceptor, respectively, with a calculated Förster distance, R0, of 4.9 nm for unoriented molecules (Patterson et al. 2000). Owing to the presence of endogenous eIF4E and eIF4E-T proteins in the cells and the variable expression of the FP-eIF4E/eIF4E-T constructs, it is difficult to demonstrate FRET unambiguously by using sensitized acceptor emission alone. However, the FRET efficiency can be measured by acceptor photobleaching. This method makes use of the fact that FRET quenches the donor fluorescence as the excitation energy is transferred to the acceptor. After photobleaching of the acceptor, this quenching no longer occurs, and the donor fluorescence increases. Quantification of the increase is a reliable and robust measure of FRET (Bastiaens and Jovin 1998; Miyawaki and Tsien 2000).
In cells expressing FP-tagged eIF4E and eIF4E-T we measured an average FRET efficiency of 13%. In contrast, cells expressing only YFP and CFP (negative control) did not show any FRET (Fig. 2A ▶). Figure 2B ▶ shows a color-coded image of a cell coexpressing YFP-eIF4E and CFP-eIF4E-T. Green indicates a FRET efficiency of 15%, which is the case for the two bleached P bodies in this cell. The nonbleached P bodies did not exhibit FRET, as indicated in red (~0% efficiency). In the same way, interactions between LSm proteins and Dcp1/Dcp2 were identified in P bodies, previously (Ingelfinger et al. 2002; Cougot et al. 2004). Therefore, we decided to screen for interactions between other protein pairs. We coexpressed YFP-eIF4E-T/CFP-rck/p54 and YFP-eIF4E/CFP-rck/p54 protein pairs in HeLa SS6 cells. Cells expressing FP-tagged eIF4E and rck/p54 showed an average FRET efficiency of 12.6%, whereas cells expressing FP-tagged eIF4E-T and rck/p54 did not exhibit FRET (1.6% efficiency) (Fig. 2A ▶).
FIGURE 2.

eIF4E and eIF4E-T proteins interact directly in vivo, as demonstrated by FRET. (A) Bar chart representing the mean values of the apparent FRET efficiencies of protein pairs from several P bodies in different cells. Error bars indicate the standard deviation from the mean values.The protein pair CFP-eIFE-T/YFP-eIF4E was coexpressed (B). HeLa SS6 cells were fixed 16 h after transfection with 4% PFA and confocal images of both CFP and YFP channels were taken before and after photobleaching. The difference in post- and prebleach (of the YFP acceptor) intensity of the CFP-fluorescence was divided by the postbleach CFP-fluorescence. The ratio was calculated pixel by pixel, and the result is shown color coded. Green indicates a FRET efficiency of 15%, red indicates a FRET efficiency near 0 (no FRET). The image was contrast enhanced to also display the dim P bodies, in the prebleach donor image, which is the brightness channel of this image. The total size of this image corresponds to 56 μm in the sample.
In conclusion, these studies indicate that the cap-binding translation initiation factor eIF4E is in molecular contact with eIF4E-T and rck/p54 in P bodies in vivo.
Interdependence of degradation factors for accumulation in P bodies
Currently, little is known about the factor requirements for the accumulation of P body components in P bodies. mRNA degradation factors could be targeted independently to P bodies or, alternatively, together as part of a complex. To identify proteins required for the accumulation of degradation factors in P bodies, we depleted cells of a given P body factor using RNAi and then assayed for LSm1, rck/p54, eIF4E, eIF4E-T, and Ccr4 in P bodies by immunofluorescence. The efficiency of the knockdowns could be demonstrated by real-time RT-PCR showing an 82%, 90%, 79%, 85%, and 84% reduction in mRNA levels in the Ccr4, LSm1, eIF4E-T, rck/p54, and Dcp2 cells, respectively (Fig. 3 ▶). Efficient knockdown of eIF4E-T, LSm1, rck/p54, and Ccr4 is also shown by the reduction in their own immunofluorescence signal (Fig. 4 ▶, panels 9,11,17,25). The distribution of the tested proteins was indistinguishable from that observed with untreated cells when a control siRNA (GL2) directed against the firefly luciferase mRNA was used (Fig. 4 ▶, panels 1–5; cf. Fig. 1 ▶). In contrast, RNAi-mediated knockdown of eIF4E-T resulted in a dramatic reduction of the localization of rck/p54, eIF4E, and Ccr4 in P bodies (Fig. 4 ▶, panels 6–10). Similarly, LSm1 was now more evenly distributed in the cytoplasm and no longer accumulated in cytoplasmic foci. These results demonstrate for the first time that eIF4E-T is indeed functionally important for the accumulation of mRNA degradation factors in P bodies.
FIGURE 3.

Efficient knockdown of Ccr4, LSm1, eIF4E-T, Dcp2, and rck/p54 mRNA levels revealed by real-time RT-PCR. HeLa SS6 cells growing in six-well cell cultures dishes were transfected with either the GL2 control siRNA or with siRNAs against Ccr4, LSm1, eIF4E-T, Dcp2, and rck/p54 at a concentration of 120 nM. After 48 h cells were harvested and total RNA was extracted and incubated with RQ1 Dnase; 50 ng of total RNA was used in one-step RT-PCR (QuantiTec SYBR Green RT-PCR Kit). The graph shows the reduction in target mRNA levels calculated from the real-time RT-PCR data compared to mRNA levels from cells transfected with GL2 control siRNA. Error bars indicate the standard deviation from several independent experiments.
FIGURE 4.

eIF4E-T, LSm1, rck/p54, and Ccr4, but not Dcp2, are required for the accumulation of each other in P bodies. HeLa cells were transfected with GL2 luciferase control (1–5), eIF4E-T (6_–_10), LSm1 (11–15), rck/p54 (16–20), Ccr4 (21–25), and Dcp2 (26–30) siRNA duplexes. Cells were immunostained with antibodies against LSm1 (panels 1,6,11,16,21,26), rck/p54 (panels 2,7,12,17,22,27), eIF4E (panels 3,8,13,18,23,28), eIF4E-T (panels 4,9,14,19,24,29), and Ccr4 (panels 5,10,15,20,25,30). Cells immunostained for LSm1 were counterstained with CybrGold showing cell nuclei (green). The panels show two-dimensional projections (Zeiss Software) of a series of confocal fluorescence images in order to obtain sharp images displaying all P bodies in every cell. To highlight the important structures, different magnifications were used for the various panels.
Similar results were obtained when cells were depleted of LSm1 (Fig. 4 ▶, panels 11–15) or Ccr4 (Fig. 4 ▶, panels 21–25). Knockdown of rck/p54 had a slightly less pronounced effect, presumably due to a less efficient reduction in rck/p54 protein levels (Fig. 4 ▶, panels 16–20). That is, small LSm-containing foci were still observed in the cytoplasm (Fig. 4 ▶, panel 16). However, a significant amount of LSm1 was already redistributed throughout the cytoplasm (Fig. 4 ▶, panel 16) as observed with the eIF4E-T knockdown (Fig. 4 ▶, panel 6). Additionally, a readily visible increase in nuclear eIF4E-T protein levels could be observed in the rck/p54 and Ccr4 knockdown cells. Due to insufficient knockdown efficiency, the effect of depletion of eIF4E could not be determined. These results demonstrate an interdependence of each of these factors for their accumulation in P bodies and are consistent with the idea that they are targeted together to these structures as a complex.
We and others have previously shown that Xrn1 and Dcp1/Dcp2 are also present in P bodies (Ingelfinger et al. 2002; Cougot et al. 2004). As these enzymes are carrying out the actual 5′ → 3′ degradation of the mRNA (in contrast to the factors analyzed above), we were interested in determining whether their depletion also affected the accumulation of other factors in P bodies. In striking contrast, RNAi-mediated knockdown of the decapping enzyme Dcp2 (for details, see Materials and Methods), which initiates the actual 5′ → 3′ degradation, did not have an inhibitory effect, but rather stimulated the accumulation of LSm1, rck/p54, eIF4E, and Ccr4 in P bodies (i.e., an increase in the number of P bodies was observed) (Fig. 2 ▶, panels 26–30). A similar effect has been observed for the knockdown of the 5′ → 3′ exoribonuclease Xrn1 (data not shown; see also Cougot et al. 2004). Thus, these enzymes are dispensable for the targeting of other degradation factors to P bodies.
mRNA is required for the accumulation of mRNA degradation factors in P bodies
The loss of accumulation of multiple factors in P bodies upon RNAi-mediated depletion of eIF4E-T, LSm1, rck/p54, or Ccr4, suggested that these proteins may be targeted to these foci together, either as part of one or more heteromeric protein complexes or a larger mRNP complex. To inspect whether mRNA is required for the accumulation of all tested P body factors, we treated HeLa cells with cycloheximide, which inhibits translation and stabilizes mRNA by preventing its release from polysomes (for reviews, see Ross, 1995; Jacobson and Peltz 1996), and checked by immunofluorescence whether the accumulation of various factors in P bodies was affected. Indeed, as shown in Figure 5 ▶, treatment of cells with cycloheximide led to a rapid loss of LSm1 (Fig. 5a–c ▶), eIF4E (Fig. 5d–f ▶), rck/p54 (Fig. 5g–i ▶), and eIF4E-T (Fig. 5j–l ▶) from P bodies. Time-lapse imaging with cells expressing an YFP-eIF4E-T fusion revealed that 50% of the P bodies disappeared within 17 min of cycloheximide incubation (data not shown). Incubation with the solvent (DMSO) had no effect on P body formation. Thus translation (which is inhibited by cycloheximide) is required for the accumulation of these factors in P bodies, supporting a model in which they are targeted to these foci together with mRNA as an mRNP complex.
FIGURE 5.

Effect of cycloheximide on the accumulation of LSm1, eIF4E, rck/p54, and eIF4E-T in P bodies. Cells were incubated with cycloheximide (20 μg/mL) and fixed at 0 min, 10 min, and 40 min after addition of inhibitor. Cells were immunostained with antibodies against LSm1 (a–c), eIF4E (d–f), rck/p54 (g–i), and eIF4E-T (j–l). The panels represent two-dimensional projections (Zeiss Software) of a series of confocal fluorescence images (see legend to Fig. 2 ▶).
FRAP reveals a rapid exchange of P body components
The rapid loss of LSm1, eIF4E, and eIF4E-T from P bodies upon cycloheximide treatment suggested that factors exchange rapidly with these structures and that they are very dynamic in nature. To learn more about the exchange of factors involved in P body formation, we performed fluorescence recovery after photobleaching (FRAP) experiments using YFP-LSm6, YFP-eIF4E, and YFP-eIF4E-T fusion proteins. The data were acquired 20 h after transfection, when the P bodies were readily visible (see Fig. 6 ▶, “before” panel). Individual P bodies were bleached and fluorescence recovery was measured in a time series up to 400 sec. The fluorescence of YFP-LSm6 and YFP-eIF4E-T recovered to a maximum level of 73 ± 5% (_t_1/2 = 25 ± 5 sec) and 89 ± 3% (_t_1/2 = 60 ± 7 sec), respectively, of the prebleaching intensity within the acquisition time (Fig. 6 ▶, graph). YFP-eIF4E recovered to 45 ± 2% with a calculated _t_1/2 of 46 ± 8 sec (Fig. 6 ▶, graph). These results demonstrate that YFP-eIF4E-T, YFP-LSm6, and YFP-eIF4E exchange quickly, supporting the notion that P bodies are dynamic structures rather than static aggregates.
FIGURE 6.

Fluorescence recovery after photobleaching indicates that P bodies are highly dynamic in nature. HeLa cells were transfected with plasmids encoding YFP-LSm6, YFP-eIF4E, or YFP-eIF4E-T. (A) In an YFP-LSm6-transfected cell a P body (see rectangle) was recorded (“before”), bleached (“0 s”), and images were collected every 20 sec during the course of recovery. (B) For each time point fluorescence intensity within the region of interest was measured and plotted in the graph shown relative to the intensity before bleaching. From each data set, fluorescence recovery curves were calculated using nonlinear regression. Fitting was carried out by the Marquardt method (GraphPad Software). Scale bar = 10 μm.
DISCUSSION
In this manuscript we demonstrate for the first time that the m7G cap-binding protein eIF4E and one of its interaction partners, eIF4E-T, are also components of mammalian P bodies (Fig. 1 ▶). We further show, by FRET, that they form a molecular complex with each other in P bodies in vivo (Fig. 2 ▶). In contrast, other translation initiation factors such as eIF4G or components of the translation machinery were not detected in these cytoplasmic foci, ruling out the possibility that active translation occurs in P bodies. Furthermore, RNAi-mediated knockdowns revealed that a subset of P body factors, including eIF4E-T, LSm1, rck/p54, and Ccr4 are required for the accumulation of each other and eIF4E in P bodies. In addition, cycloheximide treatment of cells revealed that the accumulation of LSm1, eIF4E, eIF4E-T, and rck/p54 in P bodies also requires that mRNA be available for degradation. These results suggest that these factors are targeted to P bodies as part of a larger mRNP complex and that they are essential for P body formation. In this respect it is interesting to note that siRNA-mediated knockdown of the RNA binding protein GW182 inhibits the accumulation of LSm4 in P bodies, indicating that GW182 is also required for P body formation (Yang et al. 2004).
The presence of eIF4E in mammalian P bodies is a first indication that, in addition to its involvement in translation initiation, eIF4E may also play a key role in mRNP remodeling events that target an mRNA for degradation in P bodies. Our data also indicate that eIF4E-T likewise plays a key role in this process. Significantly, RNAi-mediated knockdown of eIF4E-T inhibited the accumulation of eIF4E, LSm1, rck/p54, and Ccr4 in P bodies, suggesting that the binding of this factor to eIF4E is an important remodeling event for the accumulation of mRNA and degradation factors in these structures. It is appealing to speculate that eIF4E-T could be involved in the earliest stages of the transition of an actively translating mRNA to one earmarked for degradation in P bodies. For example, it could potentially inhibit the interaction of eIF4E with eIF4G, which would disrupt the communication between the 5′ end of the mRNA and the poly(A) tail, and thereby facilitate the inactivation of translation complexes. Consistent with this idea, eIF4E-T binds a region of eIF4E that is a hot spot for communication with several other factors, including eIF4G. Interestingly, there is no obvious counterpart for eIF4E-T in yeast cells, suggesting that not all aspects of this mRNA degradation pathway are conserved between higher and lower eukaryotes.
Data presented here and elsewhere demonstrate that the DEAD-box RNA helicase rck/p54 is also a component of P bodies (Fig. 4 ▶) (see also Cougot et al. 2004). Significantly, RNAi-mediated knockdown of rck/p54 also led to the loss of accumulation of P body factors in cytoplasmic foci (Fig. 4 ▶). It has recently been shown that rck/p54 is present together with eIF4E in RNP complexes in Xenopus oocytes, and that it can repress translation (Minshall and Standart 2004). Likewise, our FRET studies indicate an interaction of rck/p54 and eIF4E in P bodies in vivo (Fig. 2 ▶). Thus, rck/p54 could also play an early role in the transition from translation to mRNA degradation in mammalian cells and modulate the interaction of eIF4E with components of the translation machinery. Previous studies indicated that in yeast, rck/p54 (Dhh1p) interacts with Dcp1 and may facilitate decapping (Coller et al. 2001). However, whereas knockdown of Dcp2 in HeLa cells revealed that it is not required for the accumulation of other RNA degradation factors in P bodies, rck/p54 was required (Fig. 4 ▶). This suggests that rck/p54’s function in higher eukaryotes is not limited to the later stages of the degradation process, but rather that it plays an earlier role.
Knockdown of two factors that bind the mRNA’s 3′ end (e.g., Ccr4 and likely, also LSm1) prevented the accumulation of other degradation factors in P bodies (Fig. 4 ▶). This suggests that mRNAs must first be trimmed and then remodeled at their 3′ end to convert the mRNP into a state that targets it to P bodies. Whether these events occur prior to remodeling events at the 5′ end of the mRNA is presently not clear. In yeast, Lsm1–7p plays a role in deadenylation which leads to displacement of translation factors from mRNA (Tharun and Parker 2001), suggesting they (together with Ccr4) might also play an early role in the transition from active translation to degradation. In contrast, knockdown of Dcp2 did not inhibit the accumulation of eIF4E, LSm1, rck/p54, or Ccr4 in P bodies (Fig. 4 ▶). Thus, these factors, which catalyze decapping and subsequent 5′ → 3′ degradation, do not appear to play a role in the targeting of mRNPs to P bodies, but appear to act at a later stage (i.e., after mRNPs accumulate in P bodies).
Taken together, these data support a model in which mRNPs undergo several successive steps of 3′ trimming and/or remodeling until their composition or structural organization promotes their accumulation in P bodies and subsequent degradation. One model would be that several factors sequentially interact with the mRNA and that only after the correct mRNP structure is formed would these factors be transported together to P bodies as part of the mRNP. RNAi knockdowns demonstrating the interdependence of eIF4E-T, LSm1, rck/p54, and Ccr4 for the accumulation of each other and eIF4E in P bodies are consistent with this idea. Conceivably, mRNP reorganization could involve an allosteric cascade of interactions where the interaction of one factor is required for the subsequent recruitment of the next. However, although we favor an initial role for eIF4E-T and/or rck/p54, the order of mRNA remodeling events is presently not clear.
The structure of the rearranged mRNP that is ultimately targeted to P bodies is also presently not known. However, based on the fact that P bodies contain factors binding the 5′ and 3′ end of the mRNA, one possible scenario is that there is a molecular bridge formed between both ends of the mRNA. In this model, rck/p54 or eIF4E-T, which interact with eIF4E, might play a key bridging role between components at the 5′ and 3′ end of the remodeled mRNA. This type of mRNA circularization could potentially replace the molecular bridge observed in active translation complexes, involving eIF4E, eIF4G, and PABP.
While several remodeling events involving eIF4E-T, LSm1, rck/p54, and Ccr4 appear to target mRNPs to P bodies, additional restructuring may be required to initiate actual degradation by Xrn1. In yeast, the 5′ → 3′ mRNA degradation pathway requires the dissociation of eIF4E from the 5′ cap to make it accessible to Dcp1p (Schwartz and Parker 2000). In vitro experiments suggested that Dcp1p interacts with eIF4G and removes the eIF4F complex from the mRNA (Vilela et al. 2000) to promote degradation (Ramirez et al. 2002). However, this does not necessarily imply the displacement of eIF4E (Vilela et al. 2000). The presence of eIF4E in P bodies suggests that some mRNPs, while translationally repressed, may not immediately undergo 5′ → 3′ degradation in these structures due to protection of the 5′ cap by eIF4E. Thus, mRNAs could be stored in P bodies in the form of an inactive mRNP before their fate is determined in a subsequent remodeling step. The presence of oligoadenylated mRNA in P bodies and the lack of PABP also suggest that an mRNA could be reversibly stored, as all of the elements needed to reinitiate translation are still present. To establish whether P bodies, in addition to functioning as degradation sites, also represent mRNP storage sites or the place where the ultimate decision regarding the fate of an mRNA is made, requires additional studies. As with many other mRNP processing events it is likely that many additional factors play a role in P body function. The precise biochemical analysis of the complete set of proteins present in P bodies should help shed light on the complex journey of an mRNA from translationally active complexes to degradation.
MATERIALS AND METHODS
Plasmids
Plasmids encoding the YFP-LSm fusion proteins were described previously (Ingelfinger et al. 2002). The human eIF4E ORF (Gen-Bank Acc. number NM_001968) was amplified by RT-PCR from HeLa total RNA and subcloned into EcoRI and BamHI sites of the pEYFP-C1 vector (Clontech). eIF4E-T ORF was PCR amplified from cDNA (ATCC, MGC-32981) and was subcloned into the HindIII and KpnI sites of the pEYFP-C1 vector (Clontech).
Cell culture, transient transfection, and fluorescence microscopy
HeLa SS6 cells were grown on glass coverslips (Fisher Scientific) in Dulbecco’s modified Eagle’s medium (GibcoBRL) supplemented with 10% fetal calf serum (GibcoBRL) and 100 U mL−1 penicillin/streptomycin (Biochrom KG) at 37°C, 5% CO2. Plasmid transfections were performed after cells had reached ~90% confluency using FuGENE 6 (Roche) as recommended by the manufacturer. Sixteen hours after transfection cells were washed with PBS (130 mM NaCl, 20 mM potassium phosphate, pH 7.4), fixed for 20 min with PBS pH 7.4/4% w/v paraformaldehyde and mounted in antifade (Mowiol, Calbiochem).
Immunofluorescence
Cells were fixed as described above, washed with PBS pH 7.4, and permeabilized in PBS pH 7.4/0.2% Triton X-100 (Sigma) for 20 min. Cells were then rinsed with PBS, blocked in PBS pH 7.4/10% fetal calf serum (FCS) for 30 min, and incubated with the primary antibody diluted in PBS pH 7.4/10% FCS for 60 min. Subsequently, cells were washed with PBS pH 7.4 (4 × 15 min) and incubated with the secondary antibody diluted in PBS pH 7.4/10% FCS for 45 min. The cells were again washed with PBS pH 7.4 (4 × 15 min) and mounted in antifade (Mowiol, Calbiochem). All steps were performed at room temperature.
Confocal laser scanning microscopy
Samples were visualized using a Zeiss LSM 510 Meta inverted confocal laser scanning microscope. Fluorophores were excited using standard laser lines. Images were successively acquired in 0.5–0.7-μm optical sections. Images were processed and merged using Adobe Photoshop (version 6.0). The following primary antibodies were used in this study: affinity-purified anti-LSm1 polyclonal peptide antibodies (1:500 dilution) (see Ingelfinger et al. 2002); rabbit anti-Xp54 (gift from J. Sommerville, 1:200); rabbit anti-Ccr4 (gift from E. Wahle, 1:100); rabbit anti-eIF4E serum (Naegele and Morley 2004), goat anti-eIF4G (1:400); mouse anti-eIF4E (P-2, 1:800); and goat anti-eIF4E-T (E-18, 1:400) (purchased from Santa Cruz Biotechnology). The following secondary antibodies were used: Texas Red goat anti-mouse and goat anti-rabbit IgG (H + L) (1:500 dilution) conjugates (purchased from Molecular Probes); Cy2 donkey anti-rabbit (1:200), Cy3 goat anti-mouse (1:400), Cy3 donkey anti-goat (1:400), and Cy5 donkey anti-mouse (1:500) conjugates (purchased from Jackson ImmunoResearch Laboratories).
Acceptor photobleaching FRET
All data were obtained on a LSM 510 Meta (Zeiss). Samples were fixed with 4% PFA for 20 min and images acquired with a C-APOCHROMAT 63×/1.4 W Korr objective (Zeiss). Specific excitation and emission of the CFP-fusion proteins were effected by excitation at 458 nm with a 30 mW Argon/2 laser (AOTF transmission 15%) and collection of emitted light with a 480/20 nm bandpass filter. No emission from YFP fusion proteins was detected in this channel. CFP images were taken before and after photobleaching of the YFP signal by using exactly the same sensitivity settings. YFP signals were photobleached by full-power excitation at 532 nm with a 50-mW solid-state laser. Images of the YFP-fusion protein expressing cells were obtained before and after photobleaching by excitation with a 30-mW Argon/2 laser (transmission 5%) at 514 nm excitation and emission collected from 531 to 606 nm (LSM 510 Meta Detector, Zeiss). No photobleaching of the CFP signal was observed under conditions of >90% photode-struction of the YFP signal. To enhance the reliability of the acquired data, two P bodies and one square-shaped region in the cytoplasm were bleached per acquired sample region. The intensity of the remaining, unperturbed P bodies served as internal control to correct for image-induced donor bleaching and fluctuations in laser intensity.
FRET efficiencies were calculated by using a MATLAB and DIP-image-based (Quantitative Imaging Group, Delft University of Technology) script. After cross-correlation-based image alignment and brightness normalization estimated from intensity in the unperturbed P bodies, the FRET efficiency was determined for each P body separately:
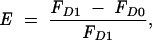
with _F_D0 and _F_D1 denoting the sum of the respective pre- and postbleach donor intensity in a P body. P bodies were segmented using a fully automatic global thresholding based on the chord method (Zack et al. 1977).
RNA Interference and validation of siRNA knockdown efficiencies
LSm1, eIF4E-T, rck/p54, and Ccr4 siRNA duplexes were designed and synthesized in-house as 21-mers with 3′dTdT overhangs as described previously (Elbashir et al. 2002). The Dcp2 siRNA duplex was purchased from Ambion. A BLAST search against the human genome sequence (NCBI UniGene database) was used to confirm that only the gene of interest would be targeted. The sense-strand sequences used to target each gene were as follows (the indicated accession numbers are from GenBank):
LSm1 (NM_014462): BC4 5′-AAGTGACATCCTGGCCACCTCAC-3′;
eIF4E-T (NM_019843): FK2 5′-CAGTCGAGTGGAGTGTACATTGT-3′;
rck/p54 (NM_004397): FA2 5′-AAAAGGCTCGTTTGGATCTGTGA-3′;
Ccr4 (NM_015455): FF2 5′-AAATGTGTGAACAGCGTATTCTC-3′; and
Dcp2 (NM_152624): DR3 5′-GTGGCATGTAATGGACATTGC-3′.
The GL2 siRNA, which targets the firefly (Photinus pyralis) luciferase gene, was used as a control (Elbashir et al. 2002). Transient transfections of siRNA into cultured HeLa SS6 cells were performed at a cell confluence of ~20% by using Oligofectamine (Invitrogen) as described by Elbashir et al. (2002). Different effects of the knockdowns on cell proliferation and cell survival were observed over time. Therefore, cells were assayed 32 h (Ccr4 knockdown), 50 h (LSm1 knockdown), and 60 h (eIF4E-T, rck/p54, and Dcp2 knockdowns) after transfection. At these points in time P body formation was clearly affected but no apoptosis was observed by the TUNEL assay performed according to the manufacturer’s instruction (Roche).
Efficient knockdown of eIF4E-T, LSm1, rck/p54, and Ccr4 was confirmed by the reduction in their own immunofluorescence signal (Fig. 4 ▶. panels 9,11,17,25, respectively). In addition, Ccr4, LSm1, eIF4E-T, rck/p54, and Dcp2 knockdowns were determined by the relative quantification based on the relative amount of target mRNA in knockdown cells versus target mRNA in control cells with real-time RT-PCR. For normalization of the target genes glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an endogenous standard. Total RNA was isolated from HeLa SS6 cells treated with either GL2 control siRNA or Ccr4, hLSm1, eIF4E-T, rck/p54, and Dcp2 siRNAs (RNeasy Mini Kit, Qiagen) and treated with RQ1 DNase. RNA integrity was electrophoretically verified by ethidium bromide staining and by OD260/OD280 nm absorption ratio >1.95. The following exon/exon-spanning PCR primers were used:
Ccr4–5′: 5′-TAAGTGGAAAAGTAAGAAGCTTAAG-3′; Ccr4–3′: 5′-AAGCTACGAATTTTATTAGATGACA-3′; LSm1–5: 5′-CTATATGCCTGGCACCGCCAGCCTC-3′; LSm1–3: 5′-CCTCGAGGAATATCACCGTATTTTTTGC-3′; eIF4E-T_for: 5′-ACAAGTCAGTCTGAAACCATCGAAC-3′; eIF4E-T_rev: 5′-CTTCATCCTCTTCGGCCACTCCTCC-3′; Dcp2–5_1: 5′-GAACTTGCCCATTGGTTTTACTTGG-3′; Dcp2–3_1: 5′-TATATTCCTTCCATTCATCCAAAAC-3′; rck/p54–5: 5′-GACACAGCAACAGATGAACCAGCTG-3′; and rck/p54–3: 5′-GAGGTCACATCCGAAGTTTTGATTC-3′.
Specificity of RT-PCR products was documented with high-resolution gel electrophoresis, and resulted in a single product with the desired length. In addition, an OPTICON melting curve analysis was performed, which resulted in single product specific melting curves. Investigated transcripts showed real-time PCR efficiency rates (E) ranging from 1.8 to 2.1 (Pfaffl 2001) in the investigated range from 6.25 to 100-ng total RNA input with high linearity (Pearson correlation coefficient r > 0.98). Relative quantification of knockdown versus control mRNA levels was accomplished according to the Pfaffl-method (Pfaffl 2001).
Fluorescence recovery after photobleaching (FRAP)
All data were obtained on a LSM 510 Meta scanning laser confocal microscope (Zeiss) using the 488-nm line of a 30-mW argon ion laser with a pinhole adjustment resulting in a 2-μm optical slice. A circular region of interest (ROI) with a diameter of 1.5 μm containing one P body was recorded five times for 5 sec with a laser power 0.6 mW and maximum scan speed (pixel time 1.28 μsec). The ROI was then bleached with 40 scan iterations at 12 mW. Fluorescence recovery was measured every 20 sec for 400 sec. An additional ROI in the cytoplasm of another cell was monitored in parallel to detect fluorescence fluctuations independent of bleaching. A third ROI was placed outside the cell to measure background fluorescence. Average fluorescence intensities within ROIs were measured under the same conditions for each data set and exported into Microsoft Excel. For each fusion protein this procedure was repeated five times in different cells and the mean intensity value for each time point was calculated. The half-time of fluorescence recovery (_t_1/2) was determined by curve fitting of experimental data using nonlinear regression to a single-exponential curve (Marquart method, GraphPad Software).
Acknowledgments
We thank S. Morley, J. Sommerville, and E. Wahle for kind gifts of anti-eIF4E, anti-Xp54 (DDX6), and anti-Ccr4 antibodies, respectively. We are grateful to M. Hossbach for help in the synthesis of the siRNAs and the real-time RT-PCR, C.L. Will for helpful discussions and critical comments on the manuscript, B. Rieger for contribution to data evaluation, and Elena Kardash for help with the FRET experiments. This work was supported by grants from the Deutsche Forschungsgemeinschaft (SFB526), the BMBF (031U215B), and the Fonds der Chemischen Industrie to R.L., and a grant from the BMBF (031U215B) to R.R-P. M.A.A. was supported by a fellowship of the GRK 521 of the Deutsche For-schungsgemeinschaft.
REFERENCES
- Bashkirov, V.I., Scherthan, H., Solinger, J.A., Buerstedde, J.M., and Heyer, W.D. 1997. A mouse cytoplasmic exoribonuclease (mXRN1p) with preference for G4 tetraplex substrates. J. Cell. Biol. 136**:** 761–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastiaens, P.I. and Jovin, T.M. 1998. Fluorescence energy transfer microscopy (FRET). In Cell biology: A laboratory handbook (ed. J.E. Celis), Vol. III, pp. 136–146. Academic Press, New York.
- Chen, J., Chiang, Y.C., and Denis, C.L. 2002. CCR4, a 3′-5′ poly(A) RNA and ssDNA exonuclease, is the catalytic component of the cytoplasmic deadenylase. EMBO J. 21**:** 1414–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coller, J.M., Tucker, M., Sheth, U., Valencia-Sanchez, M.A., and Parker, R. 2001. The DEAD box helicase, Dhh1p, functions in mRNA decapping and interacts with both the decapping and deadenylase complexes. RNA 7**:** 1717–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cougot, N., Babajko, S., and Seraphin, B. 2004. Cytoplasmic foci are sites of mRNA decay in human cells. J. Cell Biol. 165**:** 31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dostie, J., Ferraiuolo, M., Pause, A., Adam, S.A., and Sonenberg N. 2000a. A novel shuttling protein, 4E-T, mediates the nuclear import of the mRNA 5′ cap-binding protein, eIF4E. EMBO J. 19**:** 3142–3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dostie, J., Lejbkowicz, F., and Sonenberg, N. 2000b. Nuclear eukaryotic initiation factor 4E (eIF4E) colocalizes with splicing factors in speckles. J. Cell Biol. 148**:** 239–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbashir, S.M., Harborth, J., Weber, K., and Tuschl, T. 2002. Analysis of gene function in somatic mammalian cells using small interfering RNAs. Methods 26**:** 199–213. [DOI] [PubMed] [Google Scholar]
- Eystathioy, T., Jakymiw, A., Chan, E.K., Seraphin, B., Cougot, N., and Fritzler, M.J. 2003. The GW182 protein colocalizes with mRNA degradation associated proteins hDcp1 and hLSm4 in cytoplasmic GW bodies. RNA 9**:** 1171–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingelfinger, D., Arndt-Jovin, D.J., Lührmann, R., and Achsel, T. 2002. The human LSm1–7 proteins colocalize with the mRNA-degrading enzymes Dcp1/2 and Xrnl in distinct cytoplasmic foci. RNA 8**:** 1489–1501. [PMC free article] [PubMed] [Google Scholar]
- Jacobson, A. and Peltz, S.W. 1996. Interrelationships of the pathways of mRNA decay and translation in eukaryotic cells. Annu. Rev. Biochem. 65**:** 693–739. [DOI] [PubMed] [Google Scholar]
- Kimball, S.R., Horetsky, R.L., Ron, D., Jefferson, L.S., and Harding, H.P. 2003. Mammalian stress granules represent sites of accumulation of stalled translation initiation complexes. Am. J. Physiol. 284**:** C273–C284 [DOI] [PubMed] [Google Scholar]
- Lykke-Andersen, J. 2002. Identification of a human decapping complex associated with hUpf proteins in nonsense-mediated decay. Mol. Cell. Biol. 22**:** 8114–8121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer, S., Temme, C., and Wahle, E. 2004. Messenger RNA turnover in eukaryotes: Pathways and enzymes. Crit. Rev. Biochem. Mol. Biol. 39**:** 197–216. [DOI] [PubMed] [Google Scholar]
- Minshall, N. and Standart, N. 2004. The active form of Xp54 RNA helicase in translational repression is an RNA-mediated oligomer. Nucleic Acids Res. 32**:** 1325–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyawaki, A. and Tsien, R.Y. 2000. Monitoring protein conformations and interactions by fluorescence resonance energy transfer between mutants of green fluorescent protein. Methods Enzymol. 327**:** 472–500. [DOI] [PubMed] [Google Scholar]
- Naegele, S. and Morley, S.J. 2004. Molecular cross-talk between MEK1/2 and mTOR signaling during recovery of 293 cells from hypertonic stress. J. Biol. Chem. 44**:** 46023–46034 [DOI] [PubMed] [Google Scholar]
- Patterson, G.H., Piston, D.W., and Barisas, B.G. 2000. Förster distances between green fluorescent protein pairs. Anal. Biochem. 284**:** 438–440. [DOI] [PubMed] [Google Scholar]
- Pfaffl, M.W. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29**:** e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez, C.V., Vilela, C., Berthelot, K., and McCarthy J.E. 2002. Modulation of eukaryotic mRNA stability via the cap-binding translation complex eIF4F. J. Mol. Biol. 318**:** 951–962. [DOI] [PubMed] [Google Scholar]
- Ross, J. 1995. mRNA stability in mammalian cells. Microbiol. Rev. 59**:** 423–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs, A.B. and Varani, G. 2000. Eukaryotic translation initiation: There are (at least) two sides to every story. Nat. Struct. Biol. 7**:** 356–361. [DOI] [PubMed] [Google Scholar]
- Schwartz, D.C. and Parker, R. 2000. mRNA decapping in yeast requires dissociation of the cap binding protein, eukaryotic translation initiation factor 4E. Mol. Cell. Biol. 20**:** 7933–7942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheth, U. and Parker, R. 2003. Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science 300**:** 805–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strudwick, S. and Borden, K.L. 2002. The emerging roles of translation factor eIF4E in the nucleus. Differentiation 70**:** 10–22. [DOI] [PubMed] [Google Scholar]
- Tharun, S. and Parker, R. 2001. Targeting an mRNA for decapping: Displacement of translation factors and association of the Lsm1p–7p complex on deadenylated yeast mRNAs. Mol. Cell. 8**:** 1075–1083. [DOI] [PubMed] [Google Scholar]
- Van Dijk, E., Cougot, N., Meyer, S., Babajko, S., Wahle, E., and Seraphin, B. 2002. Human Dcp2: A catalytically active mRNA de-capping enzyme located in specific cytoplasmic structures. EMBO J. 21**:** 6915–6924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilela, C., Velasco, C., Ptushkina, M., and McCarthy, J.E. 2000. The eukaryotic mRNA decapping protein Dcp1 interacts physically and functionally with the eIF4F translation initiation complex. EMBO J. 19**:** 4372–4382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Z., Jiao, X., Carr-Schmid, A., and Kiledjian, M. 2002. The hDcp2 protein is a mammalian mRNA decapping enzyme. Proc. Natl. Acad. Sci. 99**:** 12663–12668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z., Jakymiw, A., Wood, M.R., Eystathioy, T., Rubin R.L., Fritzler, M.J., and Chan, E.K. 2004. GW182 is critical for the stability of GW bodies expressed during the cell cycle and cell proliferation. J. Cell Sci. 117**:** 5567–5578. [DOI] [PubMed] [Google Scholar]
- Zack, G.W., Rogers, W.E., and Latt, S.A. 1977. Automatic measurement of sister chromatid exchange frequency. J. Histochem. Cytochem. 25**:** 741–753. [DOI] [PubMed] [Google Scholar]