Nuclear import of PKCδ is required for apoptosis: identification of a novel nuclear import sequence (original) (raw)
Abstract
We have shown previously that protein kinase Cδ (PKCδ) is required for mitochondrial-dependent apoptosis. Here we show that PKCδ is imported into the nucleus of etoposide-treated cells, that nuclear import is required for apoptosis and that it is mediated by a nuclear localization signal (NLS) in the C-terminus of PKCδ. Mutation of the caspase cleavage site of PKCδ inhibits nuclear accumulation in apoptotic cells, indicating that caspase cleavage facilitates this process. Expression of the PKCδ catalytic fragment (CFδ) in transfected cells results in nuclear localization and apoptosis. We show that the PKCδ NLS is required for nuclear import of both full-length PKCδ and CFδ, and drives nuclear localization of a multimeric green fluorescent protein. Mutations within the NLS of CFδ prevent nuclear accumulation and block apoptosis. Conversely, nuclear expression of a kinase-negative catalytic fragment (KN-CFδ) protects cells from etoposide-induced apoptosis. Mutation of the NLS blocks the ability of KN-CFδ to protect against etoposide-induced apoptosis. These results indicate that PKCδ regulates an essential nuclear event(s) that is required for initiation of the apoptotic pathway.
Keywords: apoptosis/DNA damage/nuclear localization/protein kinase Cδ
Introduction
Apoptosis is a genetically programmed form of cell death required for normal development, tissue homeostasis and the elimination of damaged cells (Utz and Anderson, 2000). Initiation of a mitochondrial-dependent pathway of apoptosis occurs in response to genotoxins, organ elle toxins, irradiation and other types of cell stress. These agents converge at the mitochondria, resulting in the release of cytochrome c and caspase activation (Chinnaiyan, 1999; Wolf and Green, 1999; Antonsson and Martinou, 2000; Kroemer and Reed, 2000). Pro- and anti-apoptotic members of the Bcl-2 family regulate this pathway, as do specific signal transduction cascades, including members of the mitogen-activated protein kinase family, the pro-survival protein kinase, AKT (Datta,S.R. et al., 1997; Widmann et al., 1997; Anderson et al., 1999) and specific isoforms of protein kinase C (PKC; Ghayur et al., 1996; Datta,R. et al., 1997; Mizuno et al., 1997; Bharti et al., 1998).
PKCδ is a member of a large superfamily of isoforms that differ based on their requirement for lipid cofactors and Ca2+ for activation. PKCδ, a Ca2+-independent isoform, has been shown to regulate the mitochondrial-dependent pathway of apoptosis, and in some cell types PKCδ overexpression can induce apoptosis in the absence of additional stimuli (Emoto et al., 1995, 1996; Ghayur et al., 1996; Denning et al., 1998; Li et al., 1999; Pongracz et al., 1999; Reyland et al., 1999, 2000). In addition, recent studies show that cells derived from PKCδ-null transgenic mice are defective in mitochondrial-dependent apoptosis (Leitges et al., 2001). Proteolytic activation of PKCδ by caspases, which results in the generation of an active kinase domain, occurs in response to a variety of stimuli including DNA-damaging agents (Emoto et al., 1995, 1996; Reyland et al., 1999), FAS ligand (Mizuno et al., 1997; Frasch et al., 2000) and mitomycin C (Emoto et al., 1996; Ghayur et al., 1996). Interestingly, when the catalytic domain of PKCδ is transiently transfected into cultured cells, it rapidly induces apoptosis (Ghayur et al., 1996; Mizuno et al., 1997; Bharti et al., 1998).
Insight into how PKCδ may regulate apoptosis has been gained from studies that have investigated the subcellular localization of PKCδ in apoptotic cells. For example, when cells transiently overexpressing PKCδ were treated with phorbol ester, PKCδ translocated to the mitochondria, resulting in loss of mitochondrial membrane potential and release of cytochrome c (Li et al., 1999; Majumder et al., 2000). However, in T cells induced to undergo apoptosis by cytokine deprivation, and in glioma cells treated with etoposide, PKCδ localized to the nucleus, consistent with a nuclear function (Scheel-Toellner et al., 1999; Blass et al., 2002). Many proteins that function in the nucleus harbor conserved nuclear localization signals (NLSs) that allow rapid import of proteins via complex formation with the importin protein family members (Nigg, 1997; Gorlich, 1998; Gama-Carvalho and Carmo-Fonseca, 2001; Hodel et al., 2001). Sequences that mediate nuclear import have been identified in PKCα and PKCζ, but not in the novel PKC isoform subfamily, which includes PKCδ (James and Olson, 1992; Perander et al., 2001).
Previous studies from our laboratory have demonstrated an essential requirement for PKCδ in apoptosis induced by agents that target the mitochondrial-dependent pathway (Reyland et al., 1999, 2000; Matassa et al., 2001). Here we show that nuclear localization of PKCδ is required for apoptosis induced by etoposide, and that caspase cleavage of PKCδ contributes to its pro-apoptotic function by facilitating nuclear import. Furthermore, we have defined a unique and functional NLS in the C-terminus of PKCδ that mediates nuclear import. These studies demonstrate that PKCδ regulates an essential nuclear event that is required for activation of the apoptotic pathway.
Results
Endogenous PKCδ translocates to the nucleus after etoposide treatment
To determine where PKCδ localizes in apoptotic cells, C5 cells were treated with etoposide, and endogenous PKCδ was visualized using an antibody that recognizes the C-terminal portion of the protein. As shown in Figure 1A, panels a and b, in untreated cells PKCδ localized primarily to the cytoplasm, showing a punctate, peri-nuclear distribution. Upon treatment with etoposide, peri-nuclear PKCδ decreased by 4 h, while the abundance of PKCδ in the nucleus increased, indicating translocation of PKCδ into the nucleus (Figure 1A, panels c and d). By 8 h of etoposide treatment, the majority of PKCδ appeared to be nuclear (Figure 1A, panels e and f). Staining with Mitotracker red indicated that PKCδ did not co-localize strongly with the mitochondria in either untreated or etoposide-treated cells.



Fig. 1. PKCδ localizes to the nucleus during etoposide treatment. (A) C5 cells were treated with 50 µM etoposide for 0, 4 and 8 h. The cells were stained with an FITC-conjugated antibody specific for PKCδ (green), with DAPI (blue) to identify the nuclei, and with Mitotracker red to identify the mitochondria. The cells were visualized by confocal microscopy (magnification, ×100). The white bar represents 10 µm. Panels a, c and e are an overlay of FITC and Mitotracker red. Panels b, d and f are an overlay of FITC, Mitotracker red and DAPI. (B) C5 cells were transiently transfected with pGFP–PKCδ. After 18 h, cells were left untreated or treated with etoposide for an additional 8 h, stained with Mitotracker red and then viewed by confocal microscopy (magnification, ×100). Panels are an overlay of GFP and Mitotracker red. The white bar represents 10 µm. (C) C5 cells were left untreated or treated with LMB (5 ng/ml) for 6 h, fixed, permeabilized and co-stained with an FITC-conjugated antibody specific for PKCδ (green, panels a and c), and a Cy3-conjugated antibody specific for cyclin B1 (red, panels b and d). Cells were viewed by confocal microscopy (magnification, ×100). Images were obtained from the same field in untreated and LMB-treated cells in red or green channels.
To understand how PKCδ is localized to the nucleus, and the contribution of nuclear localization of PKCδ to apoptosis, we have used a series of C-terminal green fluorescent protein (GFP)-tagged wild-type and mutant PKCδ proteins, which are illustrated in Figure 4A. As observed in Figure 1B, like endogenous PKCδ, GFP– PKCδ localized to the peri-nuclear region in untreated cells and accumulated in the nucleus after treatment of cells with etoposide. Staining with Mitotracker red indicated that GFP–PKCδ did not co-localize with mitochondria. Transfection with a plasmid that expresses GFP alone resulted in a diffuse distribution throughout the cytoplasm and nucleus in both untreated and etoposide-treated cells (data not shown).



Fig. 4. Conservation of basic amino acids in the putative PKCδ nuclear localization motif is found in other PKC isoforms. (A) A schematic representation of mutations generated in the PKCδ full-length and catalytic fragment GFP fusion constructs used in subsequent experiments. (B) A schematic representation of mutations generated in the putative NLS of the GFP–PKCδ catalytic fragment fusion constructs used in subsequent experiments. (C) Alignment of amino acid sequences in the NLS domain of Myo-D (Vandromme et al., 1995), and the putative NLS domains of PKC (DDBJ/EMBL/GenBank accession Nos: 125554, PKCδ rat; 423039, PKCθ human; 6755084, PKCε mouse; 1346393, PKCη human; 6755078, PKCα mouse; 125540, PKCβI rat; 125546, PKCβII mouse; 6981400, PKCγ rat; 400137, PKCζ mouse; and 4506071, PKCι human). The far right column indicates the number of basic amino acids in this region that are conserved between the PKC sequences and PKCδ.
Cytoplasmic distribution of PKCδ in untreated cells is not sensitive to leptomycin B
Since even in untreated cells a small amount of PKCδ is seen in the nucleus (Figures 1A, panels a and b), the nuclear accumulation of PKCδ in response to etoposide could result from either import into the nucleus or the inhibition of nuclear export. To address this latter possibility, cells were treated with an inhibitor of the CRM1–exportin pathway, leptomycin B (LMB), and the subcellular localization of endogenous PKCδ was assessed. Cyclin B1, which is exported from the nucleus by the CRM1–exportin pathway, was used as a positive control (Yang et al., 1998). In untreated cells, PKCδ was predominantly cytoplasmic (Figure 1C, panel a), while cyclin B1 was mainly cytoplasmic, with some nuclear localization (Figure 1C, panel b). Treatment with LMB resulted in the nuclear accumulation of cyclin B1 (Figure 1C, panel d), while PKCδ retained a predominantly cytoplasmic distribution (Figure 1C, panel c). These data suggest that the nuclear accumulation of PKCδ in response to etoposide does not result from inhibition of nuclear export.
The PKCδ catalytic fragment accumulates in the nucleus during etoposide-induced apoptosis
Caspase cleavage of PKCδ occurs in response to a variety of apoptotic agents and results in the production of a 40 kDa constitutively active catalytic fragment (CFδ). We have shown previously that the CFδ protein can be detected in C5 cells as early as 4 h after treatment with etoposide (Reyland et al., 1999). Since the kinetics of PKCδ nuclear translocation correlated temporally with generation of the CFδ, we sought to determine whether the CFδ protein accumulates in the nucleus of cells treated with etoposide. Figure 2 shows an immunoblot of nuclear and cytosolic fractions from untreated cells or cells treated with etoposide. Although a small amount of the CFδ can be seen in the cytosol at all time points after etoposide treatment, the ratio of nuclear to cytosolic CFδ protein increases dramatically with time after treatment, suggesting that the caspase-cleaved form of PKCδ preferentially accumulates in the nucleus during apoptosis. It should be noted that the abundance of full-length PKCδ in the nuclear fraction also increases, consistent with the possibility that full-length PKCδ is also translocated into the nucleus.

Fig. 2. PKCδ catalytic fragment localizes to the nucleus during etoposide-induced apoptosis. C5 cells were left untreated or treated with etoposide for 4, 6 or 8 h. Nuclear (N) and cytosolic (C) enriched fractions were prepared as described and analyzed by immunoblot for PKCδ (top panel), the nuclear marker lamin B (middle panel) or the cytosolic marker α-tubulin. The positions of the full-length (FL PKCδ) and the catalytic fragment (CF PKCδ) are indicated.
Caspase cleavage of PKCδ promotes its nuclear accumulation in apoptotic cells
The data in Figure 2 suggest that caspase cleavage of PKCδ may facilitate its nuclear localization. To test this hypothesis directly, we mutated the caspase-3 cleavage site and examined the effect of this mutation on the nuclear accumulation of PKCδ in apoptotic cells. In pGFP–PKCδ (GFP–PKCδD→A327), the aspartic acid at the P1 position of the caspase cleavage site (DILD327) was changed to alanine (DILA327), which results in loss of caspase cleavage as shown in Figure 3A. Mutation of this site had no effect on the kinase activity of PKCδ (data not shown). Figure 3B shows a representative field of cells transfected with pGFP–PKCδ (left) or pGFP–PKCδD→A327 (right) and treated with etoposide. As indicated by the arrows, nuclear localization of both GFP–PKCδ and GFP–PKCδD→A327 is observed; however, the percentage of pGFP–PKCδD→A327-transfected cells that exhibit nuclear localization is significantly reduced. To verify this, we counted the number of cells exhibiting predominantly nuclear localization of PKCδ and expressed the data as a percentage of the total number of cells expressing GFP (Figure 3C). As shown, GFP–PKCδ rapidly accumulated in the nucleus of cells treated with etoposide (40% of cells exhibit nuclear localization at 4 h and 54% at 8 h). In contrast, pGFP–PKCδD→A327-transfected cells displayed significant inhibition of nuclear translocation (17% of cells exhibit nuclear localization at 4 h and 26% at 8 h), indicating that caspase cleavage facilitates nuclear accumulation of PKCδ.




Fig. 3. Caspase cleavage of PKCδ facilitates nuclear accumulation in apoptotic cells. (A) Parotid C5 cells were transiently transfected with pGFP–PKCδ or pGFP–PKCδD→A327. After 18 h, cells were left untreated or treated with 50 µM etoposide for 8 h and harvested for immunoblot analysis for GFP expression. Arrows indicate the positions of the full-length and catalytic fragment of GFP–PKCδ. (B) C5 cells were transiently transfected with pGFP–PKCδ or pGFP–PKCδD→A327. After 18 h, cells were left untreated or treated with etoposide for an additional 8 h and viewed by confocal microscopy (magnification, ×20). The white bar represents 10 µm. Arrows indicate transfected cells in which PKCδ is localized to the nucleus. Arrowheads indicate cells with predominantly cytoplasmic localization of PKCδ. (C) Cells were counted by fluorescence microscopy and the number of cells exhibiting nuclear localization of PKCδ after treatment for the indicated time with etoposide was obtained as a percentage of the whole GFP population (∼500 cells counted/variable). Gray bars represent cells transfected with pGFP–PKCδ, while the hatched bars represent cells transfected with pGFP PKCδD→A327. Data are the mean ± SEM from 10 fields of view. The graph represents one of three independent experiments that gave similar results. (D) C5 cells were transiently transfected with pGFP–CFδ. After 15 h, cells were fixed, counterstained with DAPI (right panels) and viewed by confocal microscopy (magnification, ×100). The white bar represents 10 µm.
The reduced nuclear accumulation of GFP–PKCδD→A327 in etoposide-treated cells suggested that CFδ may be imported more efficiently into the nucleus. To address this possibility, we asked if expression of the CFδ results in nuclear accumulation in the absence of an apoptotic stimuli. Cells were transiently transfected with GFP-tagged catalytic fragment PKCδ (pGFP–CFδ, amino acids 325–673) and visualized by confocal microscopy. Figure 3D demonstrates that in contrast to full-length GFP–PKCδ (see Figure 1B), GFP–CFδ co-localizes with the 4′,6-diamidino-2-phenylindole (DAPI)-labeled DNA in untreated cells, indicating accumulation within the nucleus.
Nuclear accumulation of CFδ requires a putative nuclear localization signal
PKCδ contains a series of basic amino acids in its far C-terminus (amino acids 611–623) that closely resemble the functional bipartite NLS published for Myo-D (Vandromme et al., 1995) (see Figure 4C). This sequence is also conserved between the novel and two conventional PKC isoforms in that each carries at least four of the six basic amino acids (Figure 4C). To determine if this sequence functions in the nuclear import of PKCδ, pGFP–CFδ constructs were generated with various mutations in the putative NLS (Figures 4A and B). pGFP–NLM-CF1δ contains mutations in which the left motif of the NLS sequence was mutated to alanines (K611A/R612A/K613A), while pGFP–NLM-CF4δ contains mutations in which the right motif of the NLS sequence was mutated to alanines (K619A/K621A/K623A). pGFP– NLM-CF8δ harbors mutations in all six amino acids. C5 cells were transiently transfected with pGFP–CFδ or the pGFP–NLM-CFδ mutants and visualized by confocal microscopy. As shown in Figure 5A (panels a and b), while GFP–CFδ localized predominantly to the nucleus, mutation of either the left (NLM-CF1δ; panels c and d) or right motif (NLM-CF4δ; panels e and f) or both (NLM-CF8δ; panels g and h) severely inhibited nuclear accumulation. As seen in Figure 5B, 91% of cells transfected with pGFP–CFδ showed nuclear localization of PKCδ, while nuclear localization was observed in only 14% of cells transfected with pGFP–NLM-CF8δ.




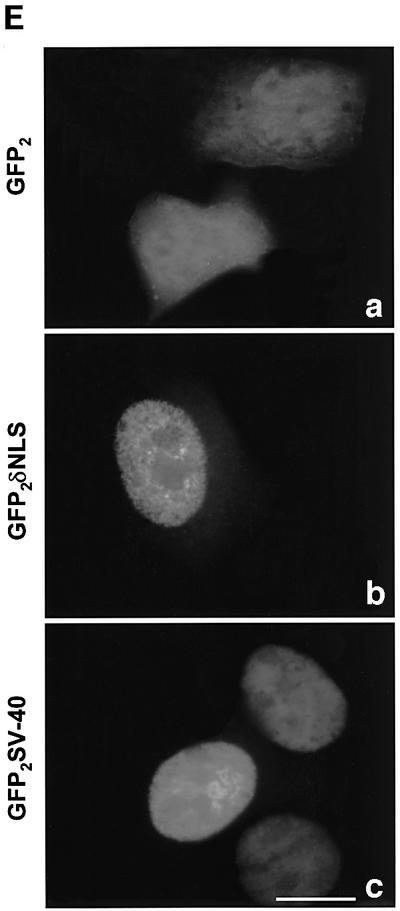
Fig. 5. A bipartite NLS is required for nuclear accumulation of PKCδ. (A) C5 cells were transiently transfected with pGFP–CFδ (panels a and b), pGFP–NLM-CF1δ (panels c and d), pGFP–NLM-CF4δ (panels e and f) or pGFP–NLM-CF8δ (panels g and h). After 15 h, the cells were fixed, counterstained with DAPI and viewed by confocal microscopy for GFP (left panels) or DAPI (right panels) (magnification, ×100). The white bar represents 10 µm. Arrows indicate nuclei of transfected cells. (B) To determine the rate of nuclear accumulation of GFP–CFδ, GFP–NLM-CF1δ, GFP–NLM-CF4δ and GFP–NLM-CF8δ, transfected C5 cells were counted by fluorescence microscopy and the number of cells exhibiting nuclear localization of PKCδ was obtained as a percentage of the whole GFP population (∼100 cells counted/vector). Data are the mean ± SEM from 10 fields of view. The graph represents one of three independent experiments that gave similar results. (C) Cells were transiently transfected with pGFP–PKCδ or pGFP–NLM-FL8δ. After 18 h, cells were left untreated or treated with etoposide for an additional 8 h and viewed by confocal microscopy (magnification, ×100). The white bar represents 10 µm. (D) To determine the rate of nuclear accumulation of PKCδ, transfected cells were counted by fluorescence microscopy and the number of cells exhibiting nuclear localization of PKCδ after treatment with etoposide was obtained as a percentage of the whole GFP population. Gray bars represent untreated cells, while the hatched bars represent etoposide-treated cells (8 h). Data are the mean ± SEM from 10 fields of view and represent one of three independent experiments that gave similar results; >100 cells were counted per variable for each experiment. (E) Cells were transiently transfected with pGFP2 (panel a), pGFP2 δNLS (panel b) or pGFP2 SV-40 (panel c). After 18 h, the cells were analyzed by fluorescence microscopy (magnification, ×100). The white bar represents 10 µm.
To determine if the putative NLS also directs nuclear import of the full-length protein, we generated a pGFP–PKCδ construct in which all six basic amino acids were mutated to alanines. Cells were transiently transfected with pGFP–PKCδ or pGFP–NLM-FL8δ, treated with etoposide and visualized by confocal microscopy. Figure 5C shows the cellular distribution of both constructs in untreated and etoposide-treated cells, while Figure 5D shows the percentage of cells transfected with pGFP–PKCδ or pGFP–NLM-FL8δ in which PKCδ is localized predominantly to the nucleus. As shown here, 55% of etoposide-treated cells showed nuclear accumulation of GFP–PKCδ at 8 h, while the nuclear accumulation of GFP–NLM-FL8δ was completely inhibited in both untreated and etoposide-treated cells. These studies show that the PKCδ NLS is essential for nuclear import of both the full-length and the catalytic fragment of PKCδ. Our observation that the GFP–NLM-FL8δ protein does not accumulate in the nucleus of untreated cells suggests that this sequence is also required for the nuclear accumulation of PKCδ in response to non-apoptotic stimuli.
The PKCδ NLS is sufficient to drive nuclear import
To determine if the PKCδ NLS is sufficient in driving nuclear import of a heterologous protein, we generated a construct in which the 13 amino acids of the PKCδ NLS were cloned downstream of two copies of GFP in tandem (pGFP2–δNLS). As seen in Figure 5E, panel a, in transfected cells the GFP2 protein alone distributed diffusely throughout the cytosol and nucleus. However, as seen in panel b, fusion of the PKCδ NLS to GFP2 resulted in an almost exclusively nuclear pattern of accumulation of GFP, identical to that observed when the SV40 T-antigen NLS (Nigg, 1997) was fused to GFP2 (GFP2–SV40) (panel c). These studies indicate that the PKCδ NLS alone is sufficient to drive nuclear import.
Nuclear localization of CFδ is necessary and sufficient for the induction of apoptosis
Our studies suggest that nuclear localization of the catalytic fragment of PKCδ contributes to the apoptotic pathway. To determine if nuclear localization of CFδ is required for apoptosis, we asked if GFP–NLM-CF1δ, which is excluded from the nucleus, can induce apoptosis. Figure 6A shows that this mutation does not inhibit PKCδ kinase activity. Cells were transfected with pGFP–CFδ, pGFP–NLM-CF1δ or pGFP control vector and apoptosis was assayed by terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) analysis. The number of TUNEL-positive cells was expressed as a percentage of the total number of GFP-positive cells. A parallel experiment was carried out on transfected cells, and cell counts were performed to determine the percentage of cells containing GFP within the nucleus. As seen in Figure 6B, while GFP alone readily accumulates in the nucleus due to its small size, only 7% of cells transfected with pGFP were TUNEL positive. In contrast, 94% of cells transfected with pGFP–CFδ demonstrated nuclear localization of PKCδ and nearly 50% showed positive TUNEL staining, indicating that expression of GFP–CFδ results in nuclear localization and induction of apoptosis. However, only 22% of cells transfected with pGFP–NLM-CF1δ showed nuclear localization, and 16% were TUNEL positive, indicating that nuclear localization of the PKCδ CF is necessary for the induction of apoptosis. The difference in the ability of GFP–CFδ and GFP–NLM-CF1δ to induce cell death was also verified by cell counts, which showed a 43% reduction in GFP-positive cells in the pGFP–CFδ-transfected group, indicating a significant loss of cells, and a 17% reduction in the pGFP–NLM-CF1δ-transfected group relative to cells transfected with the vector alone (data not shown). Taken together, our data demonstrates that nuclear localization of PKCδ is both necessary and sufficient to induce apoptosis.
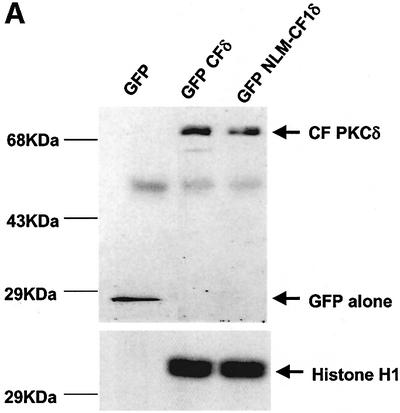

Fig. 6. Nuclear localization of PKCδ CF is necessary and sufficient for the induction of apoptosis. (A) Enzymatic activity of the GFP–CFδ and GFP–NLM-CF1δ proteins and a GFP alone negative control was determined by an immunoprecipitation kinase assay. Top panel: an immunoblot for GFP of the immunoprecipitated GFP fusion proteins. Bottom panel: phosphorylated histone H1 substrate. (B) C5 cells were transfected with pGFP, pGFP–CFδ or pGFP–NLM-CF1δ and after 15 h were prepared for TUNEL analysis. TUNEL-positive cells containing GFP were visualized by immunofluorescence microscopy and counted using a 20× objective. TUNEL-positive, GFP-positive cells were quantitated as the percentage of the total number of GFP-positive cells per field. Parallel experiments were carried out to determine the percentage of cells containing GFP in the nucleus as previously described. Gray bars represent the percentage of GFP-positive cells exhibiting nuclear accumulation of GFP. Hatched bars represent the percentage of GFP-transfected cells that are TUNEL positive. The graph represents one of three independent experiments, which all produced similar results. At least 100 cells were counted for each variable per experiment. Data are the mean ± SEM from 10 fields of view.
Nuclear, but not cytoplasmic, expression of KN-CFδ inhibits etoposide-induced apoptosis
The data presented above indicate that GFP–CFδ preferentially accumulates in the nucleus in the absence of an apoptotic stimulus and that exclusion of PKCδ from the nucleus blocks its ability to initiate apoptosis. To determine whether nuclear PKCδ is also required for apoptosis induced by DNA-damaging agents, we generated a kinase-negative construct of the catalytic fragment of PKCδ fused to GFP (pGFP–KN-CFδ) and the same construct in which the NLS was abolished (pGFP–KN NLM-CF1δ). We have shown previously that a full-length KN PKCδ can suppress apoptosis in response to etoposide (Li et al., 1995; Matassa et al., 2001). Shown in Figure 7A, in the absence of etoposide stimulation, GFP–KN-CFδ, like GFP–CFδ, localized primarily to the nuclei, indicating that kinase activity is not required for nuclear translocation. As expected, mutation of the putative NLS inhibited nuclear accumulation of the catalytic fragment (GFP–KN NLM-CF1δ) (Figure 7A and B). We took advantage of the unique subcellular localization of GFP–KN-CFδ and GFP–KN NLM-CF1δ to ask if expression of KN-CFδ can inhibit etoposide-induced apoptosis, and if nuclear localization of KN-CFδ is required for inhibition. As seen in Figure 7C, expression of GFP–KN-CFδ protected cells from etoposide-induced apoptosis as seen by an almost 65% reduction in TUNEL-positive cells as compared with cells transfected with GFP alone and treated with etoposide. However, in cells transfected with pGFP–KN NLM-CF1δ, no suppression of etoposide-induced apoptosis was observed. These results demonstrate that GFP–KN-CFδ must be imported into the nucleus in order to inhibit etoposide-induced apoptosis, and thus provide conclusive evidence that active nuclear PKCδ is required to initiate the apoptotic process.



Fig. 7. Nuclear, but not cytoplasmic, expression of KN-CFδ inhibits etoposide-induced apoptosis. (A) C5 cells were transiently transfected with pGFP–KN-CFδ (top panels) or pGFP–KN NLM-CF1δ (bottom panels). After 15 h, the cells were fixed, counterstained with DAPI and viewed by confocal microscopy for GFP (left panels) or DAPI (right panels) (magnification, ×100). The white bar represents 10 µm. Arrows indicate nuclei of transfected cells. (B) To determine the rate of nuclear accumulation of GFP–KN-CFδ and GFP–KN NLM-CF1δ, transfected C5 cells were counted by fluorescence microscopy and the number of cells exhibiting nuclear localization of PKCδ was obtained as a percentage of the whole GFP population (∼100 cells counted/ vector). Data are the mean ± SEM from 10 fields of view per experiment and represent the average of three independent experiments. (C) TUNEL analysis was performed on untreated (black bars) and etoposide- (8 h, hatched bars) treated cells transfected with pGFP, pGFP–KN-CFδ or pGFP–KN NLM-CF1δ. After 18 h, TUNEL positive GFP-positive cells were visualized by immunofluorescence microscopy and counted using a 40× objective as described above. Data are the mean ± SEM from at least 10 fields of view per experiment and represent the average of three independent experiments. More than 100 cells were counted per variable for each experiment.
Discussion
Work from our laboratory and others demonstrates an essential role for PKCδ as a regulator of mitochondrial-dependent apoptosis (Ghayur et al., 1996; Mizuno et al., 1997; Bharti et al., 1998). In the present studies, we have shown that nuclear accumulation of PKCδ is both necessary and sufficient for the initiation of apoptosis and have defined a novel and functional NLS sequence in the C-terminus of PKCδ. We show that nuclear PKCδ activity is also required for etoposide-induced apoptosis. Taken together, these findings suggest that PKCδ acts on specific targets within the nucleus that are required directly or indirectly for the induction of apoptosis. Furthermore, they imply that nuclear PKCδ regulates the cytosolic apoptotic machinery, perhaps by controlling expression or activity of a key apoptotic molecule(s).
Endogenous PKCδ is located primarily in the peri-nuclear region in salivary epithelial cells, although diffuse staining is also seen within the cytosol, and in some cells within the nucleus. Our studies clearly demonstrate that cytosolic PKCδ translocates into the nucleus upon treatment of cells with etoposide, and support previous observations that PKCδ localizes to the nucleus during Fas ligand- and cytokine deprivation-induced apoptosis in T cells (Scheel-Toellner et al., 1999), during irradiation-induced apoptosis in MCF-7 cells (Yuan et al., 1998) and in response to etoposide treatment in C6 glioma cells (Blass et al., 2002). It is worth noting that several nuclear proteins have been shown to be substrates of PKCδ in vitro, such as lamin B, DNA-PK and c-Abl tyrosine kinase (Bharti et al., 1998; Cross et al., 2000; Sun et al., 2000). While others have reported that PKCδ translocates to the mitochondria in response to some apoptotic stimuli (Li et al., 1999; Majumder et al., 2000), we find no evidence of PKCδ association with the mitochondria in untreated or in apoptotic salivary epithelial cells.
Our studies demonstrate that the kinetics of nuclear translocation parallel the caspase cleavage of PKCδ, suggesting that these events are linked. Mutation of the caspase cleavage site severely inhibited the ability of PKCδ to translocate to the nucleus following etoposide treatment; however, nuclear translocation was not completely blocked. Likewise, pre-treatment of cells with the caspase inhibitor ZVAD(Ome)-FMK also inhibits etoposide-induced nuclear translocation of PKCδ (T.A.DeVries and M.E.Reyland, unpublished data). This suggests that caspase cleavage facilitates translocation of PKCδ to the nucleus, but is not required for nuclear accumulation per se. This is supported further by our observation that in transfected cells GFP–PKCδ required an apoptotic stimulus to translocate to the nucleus, while GFP–CFδ localized to the nucleus in the absence of such a stimulus. Taken together, our studies indicate that caspase cleavage of PKCδ is an important component of the apoptotic response, since it allows for the nuclear accumulation of an activated form of PKCδ. Notably, apoptotic agents such as 12-_o_-tetradecanoylphorbol-13-acetate (TPA), which do not induce PKCδ cleavage, only weakly induce apoptosis in C5 cells (Reyland et al., 2000).
We show that CFδ is capable of localizing to the nucleus, suggesting that the structural requirement for nuclear localization of PKCδ resides in the catalytic domain and not the regulatory domain. Analysis of the protein sequence of PKCδ demonstrates a putative NLS between amino acids 611 and 623 of PKCδ that resembles the functional NLS published for Myo-D (Vandromme et al., 1995). When mutations were made within this sequence, nuclear localization was significantly inhibited, demonstrating that these basic amino acids are required for efficient import of PKCδ (Figure 5). Additionally, both KRK (611–613) and KKK (619, 621 and 623) are required for nuclear import as neither half can drive nuclear translocation independently. We show that the PKCδ NLS is required for nuclear translocation of the PKCδ catalytic fragment as well as full-length PKCδ. In fact, mutation of this sequence results in near total exclusion of full-length PKCδ from the nucleus of untreated cells, suggesting that this sequence regulates nuclear entry of PKCδ in response to non-apoptotic stimuli. The 13 amino acid PKCδ NLS is also sufficient for driving nuclear import of a tandem GFP protein, indicating that it functions as a canonical NLS. Sequence alignment of the novel, conventional and atypical PKC isoforms shows that the PKCδ NLS is highly conserved in this kinase family (Figure 4C). Interestingly, PKCθ contains five of the six conserved basic residues of the putative PKCδ NLS and has also been shown to translocate to the nucleus in A549 lung carcinoma cells in response to staurosporine (Jones et al., 1997). PKCβII, which translocates to the nucleus during cell proliferation, shares four of the six basic amino acids (Walker et al., 1995; Gokmen-Polar and Fields, 1998). Recently, PKCλ/I and ζ have been shown to contain a functional NLS within the N-terminal regulatory region of the kinase (Perander et al., 2001).
Our studies indicate that even in the absence of caspase cleavage, some PKCδ can localize to the nucleus. This is an important observation since our previous studies, as well as recent studies on cells from PKCδ-null mice (Leitges et al., 2001), indicate that PKCδ is required for early events in the apoptotic pathway which occur at, or prior to, the mitochondria. Since caspase cleavage of PKCδ is a post-mitochondrial event (Emoto et al., 1995), the implication is that the full-length PKCδ protein can also function as a regulator of apoptosis. This is supported by recent studies that showed that tyrosine phosphorylation of the regulatory domain of full-length PKCδ was required for caspase-3 activation and induction of apoptosis. Interestingly, when the tyrosine phosphorylation sites were mutated, they found that full-length PKCδ failed to induce apoptosis, but did, however, translocate to the nucleus (Blass et al., 2002).
A model presented in Figure 8 suggests that full-length PKCδ may be translocated initially into the nucleus where it regulates an early event required for initiation of apoptosis and the subsequent activation of caspase. Activation of caspase resulting in caspase cleavage of PKCδ increases the rate of PKCδ nuclear translocation, and results in amplification of the apoptotic signal. Our studies indicate that a PKCδ-dependent nuclear event regulates initiation of the apoptotic pathway. This scenario presented in Figure 8 predicts that full-length PKCδ and the CFδ may have the same nuclear target(s). Furthermore, since the apoptotic machinery resides in the cytosol, our results imply that PKCδ functions in the nucleus to regulate the cytosolic apoptotic machinery.

Fig. 8. Steps in the regulation of apoptosis by PKCδ. (1) PKCδ is translocated to the nucleus. We have shown previously that expression of KN PKCδ inhibits an early event in the apoptotic pathway that occurs at, or prior to, the mitochondria (Matassa et al., 2001). Our current studies suggest that this PKCδ-dependent event occurs in the nucleus, and that it may regulate entry into the apoptotic pathway. We show that PKCδ is translocated into the nucleus during apoptosis, and that nuclear PKCδ activity is required for apoptosis. (2) PKCδ functions in the nucleus to regulate apoptosis. Expression of GFP–CFδ, which localizes to the nucleus, induces apoptosis, whereas nuclear expression of GFP–KN-CFδ blocks apoptosis. These studies suggest that nuclear PKCδ can regulate the cytosolic apoptotic machinery, resulting in caspase activation and DNA fragmentation. One possibility is that nuclear PKCδ may regulate the expression or activity of key apoptotic molecules which act at, or upstream of the mitochondria. (3) Caspase cleavage of PKCδ facilitates nuclear translocation and amplifies the apoptotic pathway. Our data show that caspase cleavage of PKCδ facilitates its nuclear import, hence caspase cleavage of PKCδ may function to amplify the apoptotic response by enabling rapid import of PKCδ into the nucleus of apoptotic cells.
Materials and methods
Cells and cell culture
The isolation of C5 rat parotid salivary acinar cells was described previously (Quissell et al., 1998). Cells were cultured on Primaria 100 mm culture dishes (Falcon Plastics, Franklin Lakes, NJ) in a 1:1 mixture of Dulbecco’s modified Eagle’s medium/nutrient mixture F-12 supplemented with 2.5% fetal calf serum, 5 µg/ml transferrin, 1.1 µM hydrocortisone, 0.1 µM retinoic acid, 2.0 nM T3, 5 µg/ml insulin, 80 ng/ml epidermal growth factor (Collaborative Biomedical Products, Belford, MA), 5 mM l-glutamine, 50 µg/ml gentamicine sulfate and a trace element mixture (Biofluids, Rockville, MD). Tissue culture reagents were obtained from Gibco-BRL (Gaithersburg, MD). Etoposide (Sigma-Aldrich) was dissolved in dimethylsulfoxide (DMSO) and used at a final concentration of 50 µM in all experiments. LMB (Sigma-Aldrich) was used at a final concentration of 5 ng/ml for 6 h in all experiments.
For immunofluorescence studies, cells were grown on 12 mm coverslips in 12-well polystyrene dishes. For immunoblot analysis, cells were grown on Primeria 100 mm culture dishes. Subconfluent C5 cells were transiently transfected 15–18 h prior to etoposide treat ment using a 6:1 lipid:DNA ratio of FuGene 6 transfection reagent (Roche Molecular Biochemicals, Indianapolis, IN) according to the manufacturer’s protocol.
Construction of EGFP plasmids, and site-directed mutagenesis
Mouse wild-type PKCδ and the dominant-negative PKCδK376R were cloned into pEGFP-N1. The constructs, pPKCδ–EGFP (pGFP–PKCδ) and pPKCδK376R–EGFP, along with pEGFP (pGFP alone; Clontech, Palo Alto, CA) were obtained as a generous gift from Dr Stuart Yuspa (National Institutes of Health) (Li et al., 1999). The pPKCδ–EGFP and pPKCδK376R–EGFP constructs express a protein in which enhanced GFP is fused to the C-terminus of the PKCδ protein. The K376→R mutation in kinase-negative PKCδ resides in the ATP-binding site and the protein has been shown to function as an isoform-specific dominant inhibitory kinase (Li et al., 1995). A mutation in the caspase cleavage site (pGFP–PKCδD→A327) was generated from pPKCδ–EGFP by PCR site-directed mutagenesis using the Quick Change Site-Directed Mutagenesis Kit (Stratagene, LaJolla, CA) according to the manufacturer’s protocol (Clontech, Palo Alto, CA). The primers used were: primer 1, 5′-GTGACATCCTAGCCAACAACGGGACC-3′; and primer 2, 5′-GGTCCCGTTGTTGGCTAGGATGTCAC-3′.
Catalytic fragments of wild-type and kinase-negative PKCδ were generated by PCR using full-length pPKCδ–EGFP and pPKCδK376R– EGFP as a template starting at the caspase cleavage site (amino acid 325) and finishing at the end of GFP. The primers used were: primer 1, 5′-GATGGAATTCGCCACCATGAACAACGGGACCTATGGCAAG-3′; and primer 2, 5′-GGCTGATTATGATCTAGAGTCGCG-3′. Primer 1 incorporated an _Eco_RI site, Kozak sequence and a start codon. Primer 2 contained an endogenous _Xba_I site found downstream of the stop codon in the pPKCδ–EGFP plasmid. The PCR product was double digested with _Eco_RI and _Xba_I, then inserted and ligated into a pEGFP-N1 vector backbone lacking the GFP gene. The resulting constructs were named pPKCδ CF–EGFP (pGFP–CFδ) and pPKCδ CFK376R–EGFP (pGFP– KN-CFδ).
The pPKCδ CFK611A/R612A/K613A–EGFP (pGFP–NLM-CF1δ) and pPKCδ CFK376R/K611A/R612A/K613A–EGFP (pGFP–KN NLM-CF1δ) were generated by site-directed mutagensis according to the manual for the Quick-Change Site-directed Mutagenesis Kit (Stratagene) using pGFP– CFδ or pGFP–KN-CFδ, respectively, as a template with the primers: primer 1, 5′-GGTCCCTCCTGGAGGCGGCGGCGGTGGAGCCGC-3′; and primer 2, 5′-GCGGCTCCACCGCCGCCGCCTCCAGGAGGGACC-3′.
The pPKCδ CFK619A/K621A/K623A–EGFP (pGFP–NLM-CF4δ) and pPKCδ CFK611A/R612A/K613A/K619A/K621A/K623A–EGFP (pGFP–NLM-CF8δ) were generated as described above using pGFP–CFδ or pGFP–NLM-CF1δ, respectively, as a template with the primers: primer 1, 5′-GAG CCGCCCTTTGCGCCCGCAGTGGCATCCCCTTCAGAC-3′; and primer 2, 5′-GTCTGAAGGGGATGCCACTGCGGGCGCAAAGGGCGGCTC-3′.
The pPKCδ FLK611A/R612A/K613A/K619A/K621A/K623A–EGFP (pGFP–NLM-FL8δ) was generated as described above using pGFP–NLM-FL1δ as a template with the above primers.
The pGFP2 and pGFP2 SV40 vectors were obtained as a generous gift from Drs Yihong Wan and Steve Nordeen (University of Colorado Health Sciences Center). pGFP2 δNLS was generated by inserting the following annealed primer pair into a _Bam_HI site downstream of GFP2: primer1, 5′-GATCCTGGAAAAGCGGAAGGTGGAGCCGCCCTTTAAGCCCAAAGTGAAAG-3′; and primer 2, 5′-GATCCTTTCACTTTGGGCTTAAAGGGCGGCTCCACCTTCCGCTTTTCCAG-3′.
Automated sequencing and immunoblot analysis were used to verify the identity and size of all constructs. PCR primers were purchased from either Gibco-BRL (La Jolla, CA) or Macromolecular Resources (Fort Collins, CO).
Immunoblot analysis
Cells were harvested for immunoblots as described previously (Reyland et al., 1999). Enhanced chemiluminescence (NEN) was used to detect the signal. The mouse monoclonal antibody to GFP was obtained from Zymed Laboratories Inc. (San Francisco, CA). Rabbit polyclonal antibodies to PKCδ and lamin B were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), and mouse monoclonal α-tubulin from NeoMarkers (Fremont, CA).
Isolation of cytosolic and nuclear enriched fractions
Adherent cells were scraped onto phosphate-buffered saline (PBS) and collected by centrifugation (1000 g for 5 min). The cells were washed in 1 ml of PBS, centrifuged and the cell pellet resuspended in 300 µl of nuclei isolation buffer [20 mM HEPES-KOH, 100 mM KCl, 1.5 mM MgCl2, 1 mM EGTA, 250 mM sucrose, 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM dithiothreitol (DTT) and 10 µg/ml each of aprotinin, leupeptin and pepstatin]. The cells were incubated on ice for 20 min, and then lysed using a Dounce homogenizer (25×). Efficient cell lysis was verified by Trypan Blue staining. The cell lysate was centrifuged (1000 g for 5 min) and the pellet (nuclei) and supernatant (cytosol) were collected. The nuclei were washed once in 200 µl of nuclei isolation buffer, pelleted by centrifugation and the resulting supernatant was added to the cytosolic extract. To solubilize the proteins in both cytosolic and nuclear enriched fractions, Triton X-100 was added to a final concentration of 1.0%. Protein concentration was quantified using the DC Protein Assay Kit (Bio-Rad).
Immunofluorescence microscopy
Cells were washed with PBS and fixed in 2% paraformaldehyde/PBS for 15 min, followed by permeabilization with 0.5% Triton X-100/PBS for 5 min at room temperature. Cells were washed with PBS and blocked for 1 h in 20 mg/ml of bovine serum albumin/PBS prior to incubation with a rabbit polyclonal PKCδ primary antibody (no. C-17 Santa Cruz Biotechnology) for 1 h. Cells were washed five times with PBS and then incubated with a donkey anti-rabbit fluorescein isothiocyanate (FITC)-conjugated secondary antibody (Jackson Immunoresearch Laboratories Inc., West Grove, PN) containing 5 µg/ml DAPI (Sigma, St Louis, MO) for 1 h. Cells were washed again five times with PBS and coverslips were mounted with _o_-phenylenediamine dihydrochloride (OPDA) mounting medium (Sigma, St Loius, MO). Mounted slides of cells transfected with GFP were prepared in the same way without the antibody staining. In some experiments, cells were incubated with 300 nM Mitotracker red (Molecular Probes, Eugene, OR) for 15 min prior to fixation. In LMB experiments, cells were co-incubated with a mouse cyclin B1 primary antibody (no. sc-245, Santa Cruz Biotechnology) and a donkey anti-mouse Cy3-conjugated secondary antibody as described above. Cells were visualized and images collected using SlideBook software (Intelligent Imaging Innovations Inc., Denver, CO), on a Nikon Diaphot TMD microscope equipped for fluorescence with a xenon lamp and filter wheels (Sutter Instruments, Novato, CA), fluorescent filters (Chroma, Battleboro, VT), cooled CCD camera (Cooke, Tonawanda, NY) and a stepper motor (Intellegent Imaging Innovations Inc., Denver, CO). Multi-fluor images were merged, deconvolved and normalized using SlideBook software.
TUNEL analysis and cell counts
TUNEL analysis was performed using the in situ Cell Death Detection Kit TMR Red (Roche Molecular Biochemicals; Indianapolis, IN) according to the manufacturer’s protocol. GFP-positive cells were visualized by immunofluorescent microscopy and counted using a 20× objective. TUNEL-positive cells containing GFP were identified by co-localization with DAPI and by morphology, and were quantitated as the percentage of the total GFP-positive cells per field. More than 100 cells were counted for each variable per experiment. Experiments quantitating the percentage of cells exhibiting nuclear accumulation of GFP were performed by immunofluorescence microscopy using a 40× objective.
Immunoprecipitation kinase assay
The kinase activity of the PKCδ–GFP fusion proteins was assayed by an immunoprecipitation kinase assay using histone H1 as substrate as previously described. Antibody used in immunoprecipitation was the rabbit polyclonal anti-GFP-ab290 (Abcam) (Reyland et al., 1999).
Acknowledgments
Acknowledgements
The contributions of Kathy Barzen, Linda Sanders, Dr Shinobu Umemura and Linda Hanson are gratefully acknowledged. We thank Drs James DeGregori and Peter Parker for comments on the manuscript. This work is supported by Public Health Service grant PO1DE12798-02 from the National Institute of Dental and Craniofacial Research to M.E.R.
References
- Anderson S., Reyland,M.E., Hunter,S., Deisher,L.M., Barzen,K.A. and Quissell,D.O.(1999) Etoposide-induced activation of c-jun N-terminal kinase (JNK) correlates with drug-induced apoptosis in salivary gland acinar cells. Cell Death Differ., 6, 454–462. [DOI] [PubMed] [Google Scholar]
- Antonsson B. and Martinou,J.C. (2000) The Bcl-2 protein family. Exp. Cell Res., 256, 50–57. [DOI] [PubMed] [Google Scholar]
- Bharti A. et al. (1998) Inactivation of DNA-dependent protein kinase by protein kinase Cδ: implications for apoptosis. Mol. Cell. Biol., 18, 6719–6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blass M., Kronfeld,I., Kazimirsky,G., Blumberg,P.M. and Brodie,C. (2002) Tyrosine phosphorylation of protein kinase Cδ is essential for its apoptotic effect in response to etoposide. Mol. Cell. Biol., 22, 182–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnaiyan A. (1999) The apoptosome: heart and soul of the cell death machinery. Neoplasia, 1, 5–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross T., Griffiths,G., Deacon,E., Sallis,R., Gough,M., Watters,D. and Lord,J.M. (2000) PKCδ is an apoptotic lamin kinase. Oncogene, 4, 2331–2337. [DOI] [PubMed] [Google Scholar]
- Datta R., Kojima,H., Yoshida,K. and Kufe,D. (1997) Caspase-3-mediated cleavage of protein kinase Cθ in induction of apoptosis. J. Biol. Chem., 272, 20317–20. [DOI] [PubMed] [Google Scholar]
- Datta S.R., Dudek,H., Tao,X., Masters,S., Fu,H., Gotoh,Y. and Greenberg,M.E. (1997) Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Science, 91, 231–241. [DOI] [PubMed] [Google Scholar]
- Denning M.F., Wang,Y., Nickoloff,B.J. and Wrone-Smith,T. (1998) Protein kinase Cδ is activated by caspase-dependent proteolysis during ultraviolet radiation-induced apoptosis of human keratinocytes. J. Biol. Chem., 273, 29995–30002. [DOI] [PubMed] [Google Scholar]
- Emoto Y. et al. (1995) Proteolytic activation of protein kinase Cδ by an Ice-like protease in apoptotic cells. EMBO J., 14, 6148–6156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emoto Y., Kisaki,H., Manome,Y., Kharbanda,S. and Kufe,D. (1996) Activation of protein kinase Cδ in human myeloid leukemia cells treated with 1-β-d-arabinofuranosylcytosine. Blood, 87, 1990–1996. [PubMed] [Google Scholar]
- Frasch S., Henson,P.M., Kailey,J.M., Richter,D.A., Janes,M.S., Fadok,V.A. and Bratton,D.L. (2000) Regulation of phopholipid scramblase activity during apoptosis and cell activation by protein kinase Cδ. J. Biol. Chem., 275, 23065–23073. [DOI] [PubMed] [Google Scholar]
- Gama-Carvalho M. and Carmo-Fonseca,M. (2001) The rules and roles of nucleocytoplasmic shuttling proteins. FEBS Lett., 498, 157–163. [DOI] [PubMed] [Google Scholar]
- Ghayur T. et al. (1996) Proteolytic activation of protein kinase Cδ by an ICE/CED 3-like protease induces characteristics of apoptosis. J. Exp. Med., 184, 2399–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokmen-Polar Y. and Fields,A. (1998) Mapping of the molecular determinant for protein kinase C BII isozyme function. J. Biol. Chem., 273, 20261–20266. [DOI] [PubMed] [Google Scholar]
- Gorlich D. (1998) Transport into and out of the nucleus. EMBO J., 17, 2721–2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodel M. et al. (2001) Dissection of a nuclear localization signal. J. Biol. Chem., 276, 1317–1325. [DOI] [PubMed] [Google Scholar]
- James G. and Olson,E. (1992) Deletion of the regulatory domain of protein kinase Cα exposes regions in the hinge and catalytic domains that mediate nuclear targeting. J. Cell Biol., 116, 863–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones T., Courage,C., Hubbard,A. and Gescher,A. (1997) Cellular relocalization of protein kinase Cθ caused by staurosporine and some of its analogues. Biochem. Pharmacol., 53, 1413–1418. [DOI] [PubMed] [Google Scholar]
- Kroemer G. and Reed,J. (2000) Mitochondrial control of cell death. Nat. Med., 6, 513–519. [DOI] [PubMed] [Google Scholar]
- Leitges M. et al. (2001) Exacerbated vein graft arteriosclerosis in protein kinase Cδ-null mice. J. Clin. Invest., 108, 1505–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Lorenzo,P.S., Bogi,K., Blumberg,P.M. and Yuspa,S.H. (1999) Protein kinase Cδ targets mitochondria, alters mitochondrial membrane potential and induces apoptosis in normal and neoplastic keratinocytes when overexpressed by an adenoviral vector. Mol. Cell. Biol., 19, 8547–8558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Yu,J.C., Shin,D.Y. and Pierce,J.H. (1995) Characterization of a protein kinase Cδ ATP binding mutant. J. Biol. Chem., 270, 8311–8318. [DOI] [PubMed] [Google Scholar]
- Majumder P.K., Pandey,P., Sun,X., Cheng,K., Datta,R., Saxena,S., Kharbanda,S. and Kufe,D. (2000) Mitochondrial translocation of protein kinase Cδ in phorbol ester-induced cytochrome c release and apoptosis. J. Biol. Chem., 275, 21793–21796. [DOI] [PubMed] [Google Scholar]
- Matassa A., Carpenter,L., Biden,T.J., Humphries,M.J. and Reyland,M.E. (2001) PKCδ is required for mitochondrial dependent apoptosis in salivary epithelial cells. J. Biol. Chem., 276, 29719–29728. [DOI] [PubMed] [Google Scholar]
- Mizuno K., Noda,K., Araki,T., Imaoka,T., Kobayashi,Y., Akita,Y., Shimonaka.M., Kishi,S. and Ohno,S. (1997) The proteolytic cleavage of protein kinase C isotypes, which generates kinase and regulatory fragments, correlates with Fas-mediated and 12-_O_-tetradecanoyl-phorbol-13-acetate-induced apoptosis. Eur. J. Biochem., 250, 7–18. [DOI] [PubMed] [Google Scholar]
- Nigg E. (1997) Nucleocytoplasmic transport: signals, mechanisms and regulation. Nature, 386, 779–787. [DOI] [PubMed] [Google Scholar]
- Perander M., Bjorkoy,G. and Johansen,T. (2001) Nuclear import and export signals enable rapid nucleocytoplasmic shuttling of the atypical protein kinase Cλ. J. Biol. Chem., 276, 13015–13024. [DOI] [PubMed] [Google Scholar]
- Pongracz J., Webb,P., Wang,K., Deacon,E., Lunn,O.J. and Lord,J.M. (1999) Spontaneous neutrophil apoptosis involves caspase-3 mediated activation of protein kinase Cδ. J. Biol. Chem., 274, 37329–37334. [DOI] [PubMed] [Google Scholar]
- Quissell D.O., Barzen,K.A., Redman,R.S., Camden,J.M. and Turner,J.T. (1998) Development and characterization of SV40 immortalized rat parotid acinar cell lines in vitro. Cell. Dev. Biol., 34, 58–67. [DOI] [PubMed] [Google Scholar]
- Reyland M., Anderson,S.M., Matassa,A.A., Barzen,K.A. and Quissell,D.O. (1999) Protein kinase Cδ is essential for etoposide-induced apoptosis in salivary acinar cells. J. Biol. Chem., 274, 19115–19123. [DOI] [PubMed] [Google Scholar]
- Reyland M., Barzen,K.A., Anderson,S.M., Quissell,D.O. and Matassa,A.A. (2000) Activation of PKC is sufficient to induce an apoptotic program in salivary gland acinar cells. Cell Death Differ., 7, 1200–1209. [DOI] [PubMed] [Google Scholar]
- Scheel-Toellner D., Pilling,D., Akbar,A.N., Hardie,D., Lombardi,G., Salmon,M. and Lord,J.M. (1999) Inhibition of T cell apoptosis by IFN-β rapidly reverses nuclear translocation of protein kinase Cδ. Eur. J. Immunol., 29, 2603–2612. [DOI] [PubMed] [Google Scholar]
- Sun X., Wu,F., Datta,R., Kharbanda,S. and Kufe,D. (2000) Interaction between protein kinase Cδ and the c-Abl tyrosine kinase in the cellular response to oxidative stress. J. Biol. Chem., 275, 7470–7473. [DOI] [PubMed] [Google Scholar]
- Utz P. and Anderson,P. (2000) Life and death decisions: regulation of apoptosis by proteolysis of signaling molecules. Cell Death Differ., 7, 589–602. [DOI] [PubMed] [Google Scholar]
- Vandromme M., Cavadore,J.C., Bonnieu,A., Froeschle,A., Lamb,N., Fernandez,A. (1995) Two nuclear localization signals present in the basic helix 1 domains of Myo D promote its active nuclear translocation and can function independently. Proc. Natl Acad. Sci. USA, 92, 4646–4650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker S., Murray,N.R., Burns,D.J. and Fields,A.P. (1995) Protein kinase C chimeras: catalytic domains of α and βII protein kinase C contain determinants for isotype specific function. Proc. Natl Acad. Sci. USA, 92, 9156–9160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widmann C., Gerwins,P., Johnson,N.L., Jarpe,M.B. and Johnson,G.L. (1997) MEK kinase 1, a substrate for DEVD-directed caspases, is involved in genotoxin-induced apoptosis. Mol. Cell. Biol., 18, 2416–2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf B. and Green,D. (1999) Suicidal tendencies: apoptotic cell death by caspase family proteinases. J. Biol. Chem., 274, 20049–20052. [DOI] [PubMed] [Google Scholar]
- Woo R., McLure,K.G., Lees-Miller,S.P., Rancourt,D.E. and Lee,P.W. (1998) DNA-dependent protein kinase acts upstream of p53 in response to DNA damage. Nature, 394, 700–704. [DOI] [PubMed] [Google Scholar]
- Yang J., Bardes,E.S., Moore,J.D., Brennan,J., Powers,M.A. and Kornbluth,S. (1998) Control of cyclin B1 localization through regulated binding of nuclear export factor CRM1. Genes Dev., 12, 2131–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Z.-M., Utsugisawa,T., Ishiko,T., Nakada,S., Huang,Y., Kharbanda,S., Weichselbaum,R. and Kufe,D. (1998) Activation of protein kinase Cδ by the c-Abl tyrosine kinase in response to ionizing radiation. Oncogene, 16, 1643–1648. [DOI] [PubMed] [Google Scholar]