The tyrosine kinase Tyk2 controls IFNAR1 cell surface expression (original) (raw)
Abstract
The four mammalian Jak tyrosine kinases are non-covalently associated with cell surface receptors binding helical bundled cytokines. In the type I interferon receptor, Tyk2 associates with the IFNAR1 receptor subunit and positively influences ligand binding to the receptor complex. Here, we report that Tyk2 is essential for stable cell surface expression of IFNAR1. In the absence of Tyk2, mature IFNAR1 is weakly expressed on the cell surface. Rather, it is localized into a perinuclear endosomal compartment which overlaps with that of recycling transferrin receptors and with early endosomal antigen-1 (EEA1) positive vesicles. Conversely, co-expressed Tyk2 greatly enhances surface IFNAR1 expression. Importantly, we demonstrate that Tyk2 slows down IFNAR1 degradation and that this is due, at least in part, to inhibition of IFNAR1 endocytosis. In addition, Tyk2 induces plasma membrane relocalization of the R2 subunit of the interleukin-10 receptor. These results reveal a novel function of a Jak protein on internalization of a correctly processed cytokine receptor. This function is distinct from the previously reported effect of other Jak proteins on receptor exit from the endoplasmic reticulum.
Keywords: cytokine receptor/endocytosis/IFNAR1/interferon/Tyk2
Introduction
Receptors that bind helical bundled cytokines form a large family of transmembrane proteins characterized by a structurally conserved extracellular cytokine binding module and an intracellular (IC) region of variable length and sequence. These receptors function as homodimeric or heterodimeric ligand binding complexes that are coupled to tyrosine kinases of the Jak family, initiating a phosphorylation cascade (Yeh and Pellegrini, 1999; Kotenko and Pestka, 2000). In most of these receptors, it is unclear how ligand binding to the ectodomain results in the activation of Jak proteins in the cytoplasm. However, the ultimate activation mode of Jak proteins is common to most kinases, and involves _trans_-phosphorylation of tyrosine residues in the regulatory loop of the tyrosine kinase domain with consequent increased substrate accessibility to the active site. A second layer of Jak regulation is exerted by the kinase-like (KL) domain (Yeh et al., 2000).
The type I interferon (IFN) receptor is expressed on all cell types as a heterodimeric complex that possesses a high degree of flexibility, being able to recognize a panoply of IFN subtypes (IFN-α/β) and to induce pleiotropic activities (Stark et al., 1998; Mogensen et al., 1999). In this receptor complex, two members of the cytokine receptor superfamily, IFNAR1 and IFNAR2, contribute to ligand binding and to activation of the associated Tyk2 and Jak1 (Yeh and Pellegrini, 1999). Efforts to understand the dynamics and plasticity of such a complex system include the identification of the interactive surfaces of the components and the study of the conformational changes induced by ligand binding (Lewerenz et al., 1998; Roisman et al., 2001; Runkel et al., 2001). However, little is known about the relative expression of the receptor subunits in different cell types, their intracellular trafficking, their delivery to the cell surface and their basal and ligand-induced endocytic properties. IFNAR1 possesses a large _N_-glycosylated ectodomain, with two cytokine binding modules, and an IC region of 100 residues (Ling et al., 1995). The essential role of this chain in IFN responses is evident from the study of IFNAR1-null mice that are totally unresponsive to all type I IFN subtypes (Muller et al., 1994). Moreover, it is known that the level of surface IFNAR1 influences the sensitivity to IFN-α, as measured in STAT activation and antiviral responses in human cells (Yeh et al., 2000; Dondi et al., 2001). In our previous studies of the specific role of Tyk2 within the IFN receptor complex we found that, in human fibrosarcoma cells, Tyk2 is required for the formation of functional high-affinity receptors and in particular for sustaining the IFNAR1 protein level (Gauzzi et al., 1997).
To investigate the mechanism by which Tyk2 exerts its effect on IFNAR1, we have investigated whether Tyk2 alters the subcellular distribution of the receptor. Our results provide direct evidence that, in the absence of the tyrosine kinase, IFNAR1 is exported to the plasma membrane but then accumulates in endocytic organelles. Tyk2 co-expression prevents intracellular accumulation of IFNAR1 by restraining its constitutive internalization, and thus stabilizing it at the cell surface. These results, combined with studies of the endogenous IFNAR1 and the transfected R2 subunit of the interleukin-10 receptor (IL-10R2), attribute to a Jak family member a novel role in regulating cell surface expression of cognate receptors.
Results
Tyk2 dosage effect on the surface level of endogenous and transfected IFNAR1
The level of IFNAR1 at the surface of three cell lines that differ in their Tyk2 content was analyzed by flow cytometry. The mutant cell line 11,1 (U1A) lacks Tyk2 and is IFN-α unresponsive (Pellegrini et al., 1989). The parental 2fTGH cells express endogenous Tyk2, and WT cell is a reconstituted 11,1-derived clone that expresses Tyk2 at a level four times higher than 2fTGH (Figure 1A, right panel). Whereas surface IFNAR1 was barely detectable in 11,1 cells, an almost 3-fold increase was observed in WT cells compared with 2fTGH cells. The IFNAR1 levels present at the cell surface and in total extracts were analyzed in 11,1 and WT cells by immunoprecipitation (Figure 1B). Both the total and the surface levels of the heavily glycosylated IFNAR1 (110–120 kDa) were increased in WT cells.

Fig. 1. Levels of endogenous and transfected IFNAR1. (A) Surface level of endogenous IFNAR1. The level of IFNAR1 was determined by flow cytometry on the indicated cell lines, using the AA3 mAb. The broken curve represents staining with an isotype matched control mAb. The right panel shows the Tyk2 content in 10 µg of lysate from the indicated cell lines. (B) Surface and total levels of endogenous IFNAR1. WT and 11,1 cells were processed for surface and total IFNAR1 immunoprecipitation (see Materials and methods). The recovered material was separated by SDS–PAGE and analyzed by anti-IFNAR1 immunoblot. The lower panel shows the Tyk2 content in 10 µg of lysate. (C) Surface level of transfected IFNAR1. The level of IFNAR1 was determined by flow cytometry as in (A) on 11,1 cells transiently co-transfected with IFNAR1 and with vector (filled, median fluorescence 11.2), wt Tyk2 (bold, median 48.3) or Δ(1–287) mutant (thin, median 8.6). The broken curve represents IFNAR1 in untransfected 11,1 cells. (D) Surface and total levels of transfected IFNAR1. 11,1 cells were transfected with empty vector (–) or with the plasmids indicated and processed as in (B). (E) Endo H sensitivity of transfected IFNAR1. 11,1 cells were transiently transfected with the plasmids indicated, lysed and subjected to total IFNAR1 immunoprecipitation. Half of the immuno-absorbed material was digested with Endo H and the other half was left undigested. After separation by SDS–PAGE, anti-IFNAR1 immunoblot was performed. The arrow indicates the partly glycosylated form of the receptor (95 kDa) and the arrowhead indicates the Endo H-digested product (64 kDa).
We next investigated whether this effect could be reproduced in a transient co-expression system. For this, 11,1 cells were transfected with IFNAR1 in the presence or absence of Tyk2 and surface IFNAR1 was measured by flow cytometry. Wild-type (wt) Tyk2, but not an N-terminal deleted mutant Δ(1–287), increased the number of cells expressing higher IFNAR1 surface levels (Figure 1C). In immunoprecipitation analyses, the level of IFNAR1 at the cell surface was 5- to 6-fold higher in Tyk2-expressing cells than in control cells (Figure 1D, lanes 2 and 3), whereas the total IFNAR1 content was roughly comparable in cells transfected with or without Tyk2 (Figure 1D, lanes 5 and 6).
In addition to the 110–120 kDa species, a 95 kDa band was present uniquely in immunoprecipitates from whole-cell lysates and was less abundant in Tyk2-expressing cells (arrow in Figure 1D and E). To analyze whether this species represented an intermediate glycosylated form of the receptor, endoglycosidase H (Endo H) digestion was carried out on materials immuno-adsorbed with anti-IFNAR1 Abs from cells transfected with the receptor. As shown in Figure 1E, the 95 kDa band disappeared upon Endo H digestion, giving rise to a new band of ∼64 kDa which is the predicted molecular weight for the core protein. The broad 110–120 kDa smear was Endo H resistant. In parallel, digestion with _N_-glycosidase converted all IFNAR1-specific bands into the unglycosylated 65 kDa band (data not shown). This analysis confirmed the predominant N-linked glycosylation of the mature IFNAR1 (Ling et al., 1995) and identified the 95 kDa species as a minor incompletely processed species which was nearly twice as abundant in the absence of Tyk2. Importantly, however, despite the low level of surface expression, ∼70% of the IFNAR1 molecules were mature (Endo H resistant) in the absence of the kinase. In addition, the endogenous 95 kDa IFNAR1 species in Tyk2-negative cells and in Tyk2-positive derived clones were of similar abundance (Figure 1B) (see also Gauzzi et al., 1997). These observations rule out a major contribution of Tyk2 to the processing of the receptor from the endoplasmic reticulum (ER) through the Golgi.
IFNAR1 localizes in an endosomal compartment when expressed in the absence of Tyk2
The subcellular distribution of the endogenous IFNAR1 protein was difficult to study because of its low abundance. However, transiently transfected IFNAR1 could be visualized by immunofluorescence and confocal microscopy. IFNAR1 was found to be confined in a perinuclear compartment that appeared as a dense multivesicular structure with additional scattered vesicles. Representative profiles are shown in Figure 2A–C (left panels). To define its location better, IFNAR1 was first co-transfected with a Golgi-targeted cyan fluorescent protein (CFP) marker. Simultaneous visualization revealed IFNAR1 in a location intermingled with, but physically distinct from, the Golgi complex (data not shown). This IFNAR1-positive compartment was then compared with that of recycling endosomes (Trowbridge et al., 1993). To this end, cells were transfected with IFNAR1 and 24 h later were allowed to internalize Alexa488-coupled transferrin. Cells were then processed for immunofluorescence. Internalized transferrin was found in a perinuclear region, in a dense cluster of vesicles that in part co-localized with IFNAR1-positive vesicles (Figure 2A). Additionally, a comparison of the localization of IFNAR1 and EEA1, a marker for early recycling endosomes (Mu et al., 1995), revealed a substantial overlap in the scattered vesicles (Figure 2B). The possibility that IFNAR1 accumulated in a late endosomal or a lysosomal compartment was evaluated by co-staining transfected cells with anti-IFNAR1 Abs and with Abs directed to the lysosomal membrane glycoproteins (lgps) Lamp1. No evidence of co-localization of IFNAR1 with the lysosomal marker could be found (Figure 2C).

Fig. 2. In the absence of Tyk2 IFNAR1 accumulates in an endosomal compartment. (A) 11,1 cells were transfected with human IFNAR1 (R1) and 24 h later they were incubated with Alexa488-coupled transferrin for 60 min at 37°C. Cells were fixed, permeabilized and stained with anti-IFNAR1 mAbs, followed by Texas Red-coupled secondary Abs, and analyzed by confocal microscopy as described in Materials and methods. IFNAR1, red; transferrin, green; merging of the two signals is shown in the overlay. (B) IFNAR1-transfected cells were fixed, permeabilized and stained with anti-IFNAR1 and anti-EEA1 Abs, followed by Texas Red- and Alexa488-coupled secondary Abs. IFNAR1, red; EEA1, green. White arrows in the overlay indicate some of the vesicles where IFNAR1-positive and EEA1-positive staining overlapped. (C) IFNAR1-transfected cells were fixed, permeabilized and stained with anti-IFNAR1 and anti-Lamp1 Abs, followed by Texas Red- and Alexa488-coupled secondary Abs. IFNAR1, red; Lamp1, green. (D) 11,1 cells were co-transfected with IFNAR1 and the pECFP-Golgi marker and allowed to internalize the anti-IFNAR1 AA3 mAbs for 60 min at 37°C. Cells were fixed, permeabilized and stained with Texas Red-coupled secondary Abs. IFNAR1, red; CFP fluorescence, green. (E) IFNAR1-transfected cells were allowed to internalize both Alexa488-coupled transferrin and anti-IFNAR1 AA3 mAbs for 60 min at 37°C. Cells were processed as in (D). IFNAR1, red; transferrin, green. The white arrow in the overlay indicates one of several vesicles stained by both the internalized mAbs and transferrin. Scale bar, 5 µm.
These findings raised the question of whether the IFNAR1 protein trafficked through the cell surface before localizing in the endocytic and recycling compartment. To answer this question, cells were transfected with IFNAR1, or co-transfected with IFNAR1 and CFP-Golgi, and then incubated with anti-IFNAR1 mAbs for 60 min at 37°C (see Materials and methods). After washing, cells were processed for confocal microscopy. IFNAR1 mAbs, but not isotype-matched control Abs (data not shown), were efficiently internalized in vesicles that did not ovelap with the CFP-Golgi marker (Figure 2D). In a similar experiment, transfected cells were allowed to internalize labeled transferrin and anti-IFNAR1 mAbs and processed for immunofluorescence. As shown in Figure 2E, internalized IFNAR1 mAbs partially co-localized with transferrin-containing vesicles. Taken together, these results demonstrate that transfected IFNAR1 is able to reach the plasma membrane, from where it is constitutively internalized and localizes in a compartment that is in part common with transferrin-containing early and recycling endosomes.
Tyk2 stabilizes IFNAR1 at the cell surface
To study the impact of Tyk2 on IFNAR1 distribution, we performed indirect immunofluorescence on cells co-expressing IFNAR1 and Tyk2. A remarkable effect was evident, with IFNAR1 strongly decorating the plasma membrane only in Tyk2-expressing cells (Figure 3A, right panel). Despite the variable signal intensity among transfected cells due to heterogenous expression levels, we were able to observe a considerable reduction in intracellular IFNAR1. As previously reported, Tyk2 was distributed throughout the cell, including at the plasma membrane (data not shown) (Ragimbeau et al., 2001). Like its human ortholog, the murine receptor stained the cell surface when co-expressed with either human Tyk2 (Figure 3B) or murine Tyk2 (not shown). To eliminate the possibility of a cell-specific effect, the experiment was performed in other recipient cells. Similar localization patterns were observed in COS-7 cells (Figure 3C), MEF from Tyk2–/– mice (Figure 3D), HT-1080 and IFNAR1–/– MEF (not shown). Immunoprecipitation analyses confirmed the positive effect of Tyk2 on IFNAR1 surface level in co-transfected Tyk2–/– MEF (data not shown). From these data, we conclude that Tyk2 can robustly enhance IFNAR1 surface expression in a variety of cell types.
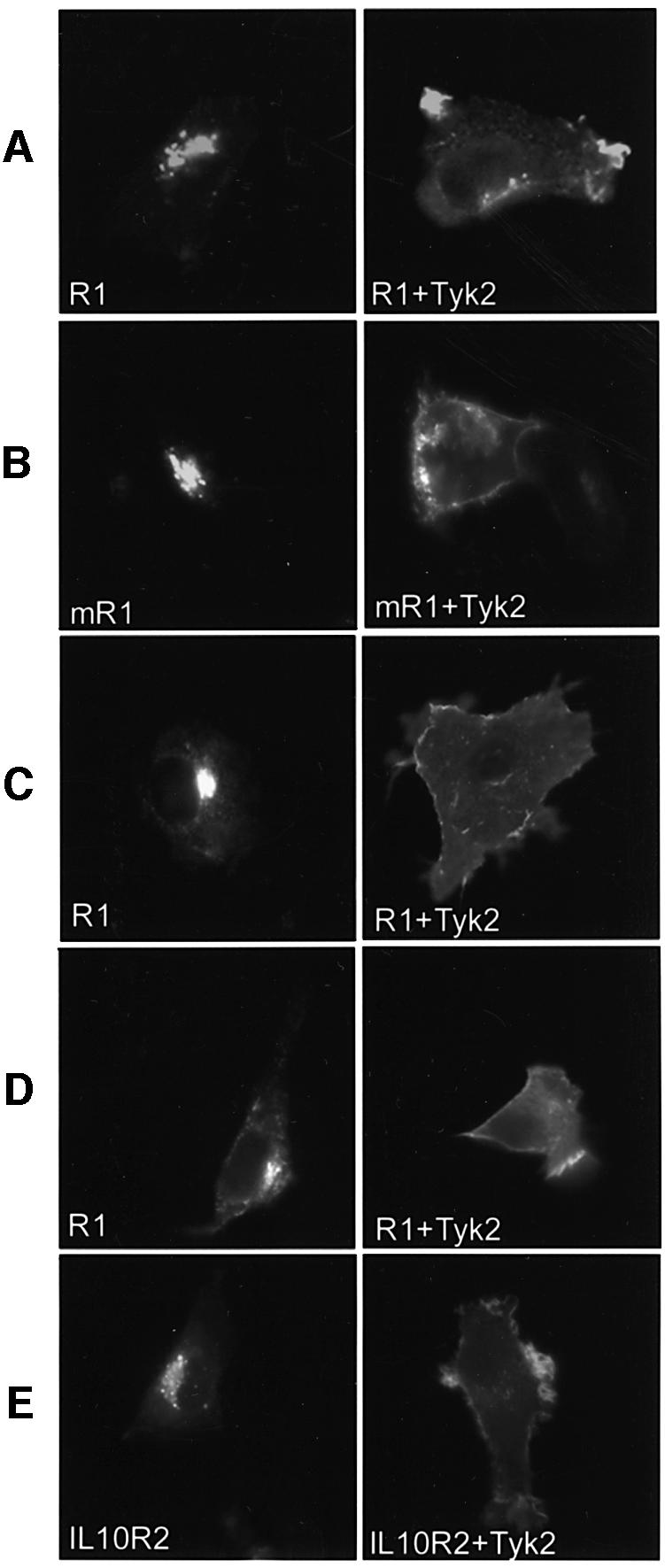
Fig. 3. Tyk2 stabilizes IFNAR1 and IL-10R2 at the plasma membrane. (A) 11,1 cells were co-transfected with human IFNAR1 (R1) and either vector (left) or human (right) Tyk2. Cells were fixed, permeabilized and stained with anti-IFNAR1 and anti-Tyk2 Abs. IFNAR1 staining in Tyk2-positive cells is shown in the right panel. (B) 11,1 cells were co-transfected with the VSV-G epitope tagged murine IFNAR1 (mR1) with either vector (left) or human (right) Tyk2. Staining was with the p5D4 mAb and anti-Tyk2 Abs. IFNAR1 staining in Tyk2-positive cells is shown in the right panel. (C) COS-7 cells were co-transfected with human IFNAR1 (R1) and either vector (left) or human (right) Tyk2. Cells were stained with anti-IFNAR1 and anti-Tyk2 Abs. IFNAR1 staining in Tyk2-positive cells is shown in the right panel. (D) MEF Tyk2–/– cells were co-transfected with human IFNAR1 (R1) and either vector (left) or human (right). Cells were stained with anti-IFNAR1 and anti-Tyk2 Abs. IFNAR1 staining in Tyk2-positive cells is shown in the right panel. (E) 11,1 cells were co-transfected with human IL-10R2 and either vector (left) or human (right). Cells were stained with anti-IL-10R2 and anti-Tyk2 Abs. IL-10R2 staining in Tyk2- positive cells is shown in the right panel. Images were acquired with a CCD camera.
To assess whether Tyk2 could influence the localization of other interacting cytokine receptors, we studied IL-10R2. This receptor chain can pair with IL-10R1 to bind IL-10 and with CRF2-9 to bind IL-22 (Kotenko et al., 1997, 2001). IL-10R2 was transiently expressed in 11,1 cells with or without Tyk2 and stained with a mAb directed to the ectodomain. As shown in Figure 3E, IL-10R2 was confined in an intracellular compartment, closely resembling that of IFNAR1. Conversely, upon Tyk2 co-expression, the intracellular labeling was very weak and IL-10R2 strongly decorated the cell surface. This result was confirmed by flow cytometry (not shown). Thus, the novel Tyk2 function described here is not uniquely exerted towards IFNAR1.
In subsequent studies, we focused on the Tyk2/IFNAR1 pair and further characterized the phenomenon. To identify the minimal Tyk2 determinants required to stabilize IFNAR1 at the cell surface, a panel of Tyk2 mutants was co-transfected with IFNAR1 and cells were analyzed by indirect immunofluorescence. The IFNAR1 profiles obtained upon co-transfection of the most representative Tyk2 mutants are shown in Figure 4A and the overall results are summarized in Figure 4B. Neither of the two kinase domains of Tyk2 was required to stabilize the receptor at the surface. On the other hand, deletion of as few as 51 aa at the N-terminus in mutant Δ(1–51), deletion of 22 aa within the FERM domain in mutant Δ(219–240) or deletion of the SH2-related domain in mutant Δ(385–496) all abrogated the Tyk2 effect. Thus, an intact N region, encompassing the FERM and SH2 domains, is necessary and sufficient to stabilize IFNAR1 at the cell surface.

Fig. 4. Localization of IFNAR1 co-expressed with Tyk2 mutants. (A) 11,1 cells co-transfected with IFNAR1 (R1) and the Tyk2 constructs indicated [schematized in (B)] were stained with anti-IFNAR1 and anti-Tyk2 Abs, as in Figure 3A. Representative IFNAR1 staining profiles in Tyk2-positive cells are shown. Images were acquired with a CCD camera. (B) Schematic representation of the domain organization of Tyk2 and of the various constructs that were tested for their effect on IFNAR1 localization. The N region (590 aa) includes the FERM and the SH2-like domain. The bar over the FERM domain points to the minimal IFNAR1 binding domain (Richter et al., 1998). K930R is a kinase-dead form with a Lys-to-Arg substitution mutation in the ATP-binding site (Gauzzi et al., 1996). KL, kinase-like; TK, tyrosine kinase.
Truncated forms of IFNAR1 are stably expressed at the cell surface
We investigated whether the Tyk2-driven effect on IFNAR1 localization required a specific receptor determinant. The IC region of IFNAR1 contains 100 residues and lacks the box1/2 sequences required by other cytokine receptors for the interaction with Jak proteins (Kotenko and Pestka, 2000). This region can be subdivided into a membrane-proximal half which includes 25 residues (aa 486–511) critical for Tyk2 binding, and a C-terminal portion which includes 16 residues completely conserved in vertebrates (Gibbs et al., 1996; Yan et al., 1996; Basu et al., 1998). From the full-length human IFNAR1, we generated three C-terminal truncation mutants IC-524, IC-480 and IC-463, retaining 67 IC, 23 IC and 6 IC residues, respectively, and one internally deleted mutant, IC-Δ(464–519) (Figure 5A). The four IFNAR1 mutants were transiently transfected in 11,1 cells, with or without Tyk2, and their localization profiles were analyzed. Mutant IC-524 behaved like the wild-type protein, being expressed on the plasma membrane only in Tyk2-positive cells. Conversely, mutants IC-480, IC-463 and IC-Δ(464–519) were localized at the surface irrespective of Tyk2 presence (Figure 5B). The cell surface level of each mutant was also analyzed by surface immunoprecipitation. A 6-fold increase in wild-type IFNAR1 and IC-524 was consistently measured when Tyk2 was co-expressed (Figure 5C). This Tyk2-driven increase is even more remarkable, considering that the transcript level of all receptor forms was 50% reduced when Tyk2, or an unrelated expression vector, was co-transfected (data not shown). This accounts for the finding that the IC-463 and IC-Δ(464–519) proteins were less abundant when co-transfected with Tyk2 than with the vector (Figure 5C). These data demonstrate that the constitutive internalization of IFNAR1 requires an intact 480–520 aa segment, since deletion of this region allows stable surface expression.

Fig. 5. (A) Schematic representation of mutated forms of IFNAR1. Only the IC region is drawn. The unshaded rectangle in wild-type IFNAR1 (WT) corresponds to the Tyk2 binding region (residues 486–511) (Yan et al., 1996). (B) Localization of mutated forms of IFNAR1. 11,1 cells were co-transfected with the IFNAR1 mutants indicated and either vector (upper panels) or Tyk2 (lower panels). Cells were double stained with anti-IFNAR1 and anti-Tyk2 Abs. Representative IFNAR1 staining profiles in Tyk2-positive cells are shown. (C) Surface expression levels of IFNAR1 mutants. 11,1 cells were co-transfected with the IFNAR1 plasmid indicated and vector (–) or Tyk2 (+) and processed for surface immunoprecipitation as in Figure 1B.
Tyk2 protects IFNAR1 from degradation
To determine whether Tyk2 affected the stability of the IFNAR1 protein, we measured the amount of total IFNAR1 at various times after blocking de novo protein synthesis with cycloheximide (CHX). As shown in Figure 6A, when expressed alone, >80% of the mature IFNAR1 band decayed within 4 h of CHX treatment. Pharmacological inhibition of the lysosomal system, using NH4Cl or chloroquine, counteracted the effect of CHX (Figure 6B), suggesting the involvement of lysosomes in targeting IFNAR1 to degradation. A similar result was obtained with the proteasomal inhibitor MG132 (Figure 6B). The effect of MG132 does not necessarily support a role for proteasomal degradation of the receptor, since it may reflect a block in the recycling of ubiquitin, leading to impaired ubiquitin-dependent internalization and lysosomal targeting (van Kerkhof et al., 2000; Rocca et al., 2001). When IFNAR1 was co-transfected with the Δ(1–51) or the Δ(219–240) Tyk2 mutants, which were both unable to stabilize it at the surface, its half-life was unchanged. Conversely, when co-transfected with wild-type Tyk2, only 25% of IFNAR1 was degraded in a 4 h time period (Figure 6A).

Fig. 6. (A) Tyk2 delays IFNAR1 degradation. 11,1 cells were co-transfected with IFNAR1 (R1) and the empty vector or the Tyk2 constructs indicated. The following day, CHX was added for the periods indicated. Total lysates were resolved by SDS–PAGE and blotted with anti-IFNAR1 Abs (top) or anti-Tyk2 Abs (bottom). (B) Pharmacological inhibition of IFNAR1 degradation. 11,1 cells were transfected with IFNAR1 and the following day cells were left untreated or treated for 2 h with CHX alone or in combination with the inhibitor indicated. Total lysates were resolved by SDS–PAGE and blotted with anti-IFNAR1 Abs. (C) Stability of IFNAR1 mutants in CHX-treated cells. Wild-type and three truncated forms of IFNAR1 (scheme in Figure 5A) were transfected and analyzed as in (A). The WT form was also analyzed when co-transfected with Tyk2. The densitometric scan of the signal is shown. For each construct, the total receptor level in cells not treated with the drug was taken as 100%. The data are from one of three experiments yielding comparable results.
Having shown that the degradation of IFNAR1 is slowed down in the presence of Tyk2, we analyzed the lifespan of the three IFNAR1 mutants in a similar setting. As shown in the densitometric scan of Figure 6C, the IC-524 mutant behaved essentially like the wild-type receptor, unlike the IC-463 and the IC-Δ(464–519) mutants which were stable over the 4 h period of CHX treatment. In sum, these results demonstrate that the mature IFNAR1 protein is subject to a degradation process which depends on the presence of the 66 membrane-proximal IFNAR1 residues and is slowed down when Tyk2 is co-expressed.
Tyk2 inhibits basal IFNAR1 internalization
Based on the results described above, we explored the possibility that Tyk2 could exert its function by modifying the constitutive endocytic properties of IFNAR1. To do this, we investigated whether Tyk2 affected the rate of internalization of IFNAR1. The COS-7 cells were utilized for this study, since their transfection efficiency was higher than that of the 11,1 cells. Cell surface IFNAR1 was traced by labeling with the radio-iodinated 64G12 mAb at 4°C. Labeled cells were then warmed to 37°C, allowing endocytosis of the IFNAR1/mAb complexes to proceed for short time intervals. The intracellular and surface labels were quantified after washing with acid. As shown in Figure 7, the rate of endocytosis of IFNAR1, measured within the first 20 min of incubation and estimated by the slope of [125I]64G12 internalization at 37°C, was substantially slowed down by the presence of Tyk2. Consistent with the previous results, at steady state (90 min at 37°C) the partitioning of the receptor between the intracellular compartment and the plasma membrane differed, with less receptor being internalized in cells expressing Tyk2 (see inset to Figure 7). Thus, these results demonstrate that Tyk2 contributes to IFNAR1 surface expression by preventing endocytosis.

Fig. 7. Basal endocytosis of IFNAR1 is slowed down by Tyk2. COS-7 cells were transfected with IFNAR1 (squares) or with IFNAR1 and Tyk2 (circles). Two days later, cells were harvested, incubated with 5 nM of anti-IFNAR1 [125I]64G12 mAb for 1 h at 4°C and then warmed to 37°C to allow endocytosis to proceed. At the times indicated, the internalized material was measured as the cell-associated radioactivity remaining after the acid pH wash (see Materials and methods). Background values obtained from cells transfected with only empty vector were substracted. The plot shows the ratios of internalized (acid-resistant) to surface radioactivity. The inset illustrates the percentage of intracellular (empty bars) and surface (filled bars) radioactivity relative to total radioactivity after 90 min of incubation at 37°C for cells transfected with IFNAR1 (R1) and cells co-transfected with IFNAR1 and Tyk2 (R1+Tyk2). This figure is representative of one of three experiments yielding comparable results.
Discussion
Cells constantly regulate the expression level of proteins reaching the plasma membrane by complex intracellular trafficking processes (Trowbridge et al., 1993; Mellman, 1996). Recent reports have shown that two members of the Jak family of tyrosine kinases play a role in intracellular trafficking of cytokine receptors. One report concerns the short-lived erythropoietin receptor (Epo-R), which is expressed at low level at the cell surface. The vast majority of transfected Epo-R is retained in the ER and undergoes degradation through determinants within the juxtamembrane region without reaching the Golgi apparatus (Neumann et al., 1993; Supino-Rosin et al., 1999). Jak2, and more precisely its N-terminal region, was found to be required for processing and efficient surface expression of transfected Epo-R (Huang et al., 2001). The second report concerns the oncostatin M receptor which, when overexpressed, accumulates mainly in the ER (Radtke et al., 2002). As for the Epo-R, the glycosylation status of the oncostatin M receptor and its localization were greatly modified upon Jak co-expression.
Here, we demonstrate that Tyk2, a third member of the Jak family, has a remarkable influence on the subcellular localization of the IFNAR1 chain of the type I IFN receptor. In the absence of Tyk2, IFNAR1 reaches the cell surface, internalizes and is found in a perinuclear endosomal compartment which overlaps with that of recycling transferrin receptors. Tyk2 modifies such trafficking and profoundly enhances IFNAR1 expression at the cell surface. Taken together, these studies indicate that members of the Jak family are critical regulators of cytokine receptor trafficking and argue for the existence of preformed receptor–Jak complexes. However, the function attributed to Tyk2 is quite distinct, as it is exerted on the internalization rate of a receptor which is intrinsically capable of being processed and reaching the cell surface. This function is strongly reminiscent of that attributed to the cytoplasmic Lck tyrosine kinase which stabilizes CD4 at the cell surface of T lymphocytes (Pelchen-Matthews et al., 1992; Pelchen-Matthews et al., 1998). Whether the other Jak proteins perform a similar function towards some of their cognate receptors needs to be investigated.
A number of studies have addressed the question of the mechanism regulating the surface expression of individual cytokine receptor subunits in the absence of ligand. For instance, the β chain of the IL-2R is efficiently internalized (Hémar et al., 1994; Subtil et al., 1997; Lamaze et al., 2001). Similarly, in the absence of signaling, efficient internalization of gp130 and LIF-R occurs (Thiel et al., 1998, 1999). The IFN-γ R2 appears to recycle between intracellular stores and the cell surface in T lymphocytes (Rigamonti et al., 2000). Whether Jak proteins influence the level or the endocytic properties of these receptor chains is not known. We found that Tyk2 also enhanced the surface expression of IL-10R2. In contrast, a third Tyk2-binding receptor chain, IL-12Rβ1, which is also a subunit of IL-23R (Oppmann et al., 2000), was found highly expressed and localized at the cell surface, irrespective of the presence of Tyk2 (data not shown). These results suggest that properties intrinsic to each receptor will determine their degree of dependency on their tyrosine kinase partner. It is noteworthy that, in contrast with the ubiquitous IFNAR1 and IL-10R2, the expression of IL-12Rβ1 is restricted to specific hematopoietic cell subsets, and its regulation may occur at the transcriptional, rather than the post-translational, level (Gately et al., 1998).
In MEF from Tyk2 knockout mice the signaling amplitude through the type I IFN receptor was shown to be compromised, with a 2-log decreased response to both IFN-α and IFN-β (Karaghiosoff et al., 2000; Shimoda et al., 2000). While this finding is fully compatible with the role attributed to the human Tyk2 in tuning the receptor sensitivity to the ligand, the magnitude of the Tyk2 contribution may be higher in the human system since a residual IFN-β response, and no IFN-α response, can be measured in 11,1 cells (Velazquez et al., 1995). A decreased IFNAR1 surface level in Tyk2–/– murine cells may have gone undetected upon analysis of membrane fractions that are prone to containing intracellular vesicles (Karaghiosoff et al., 2000). The lack of suitable Abs directed to the murine IFNAR1 ectodomain precludes a thorough analysis of its surface level in murine Tyk2–/– cells.
In contrast with the evident, although subordinate, role of Tyk2 in responses to IFN-α/β, Tyk2 was found to be dispensable or redundant for the IL-10-induced downregulation of LPS-stimulated TNFα production in bone marrow macrophages from Tyk2–/– mice (Karaghiosoff et al., 2000; Shimoda et al., 2000). This result does not exclude a structural and/or catalytic role for Tyk2 in other IL-10 induced responses, since IL-10 regulates differentiation and/or growth of a variety of hematopoietic cells and little is known about the role of IL-10R2 and Tyk2 in such responses (Moore et al., 2001). Importantly, IL-10R2 is also a component of the receptor for IL-22 and for a novel family of cytokines exerting antiviral activities (Kindsvogel and Kotenko, 2002). Thus, Tyk2 could exert its structural contribution toward IL-10R2 in the context of some, but not all, IL-10R2-containing receptor complexes.
The entire N portion of Tyk2, comprising the FERM and the SH2-like domains, was found necessary to stabilize IFNAR1 at the cell surface. We know from previous work that the segment encompassing residues 1–221 from the FERM domain interacts in vitro with the IC region of IFNAR1 (Richter et al., 1998). However, when expressed in Tyk2-minus cells, neither this nor a larger segment comprising residues 1–385, can rescue surface IFNAR1. Thus, in addition to this minimal binding interface, other surfaces of the N moiety, including the SH2-like domain, contribute to anchoring IFNAR1 to the plasma membrane. The possibility that the function exerted by Tyk2 on IFNAR1 implicates a reciprocal effect on the conformation and catalytic performance of the enzyme is attractive. Although neither kinase domain is required for the stabilizing function of Tyk2, the N region alone is unable to ensure binding of the ligand to the receptor (Gauzzi et al., 1997). Thus, the N/IFNAR1 interaction may impose a signaling or ‘productive’ conformation on the kinase domains. Related to this, we know that the kinase-like domain itself exerts a positive function on the ligand-binding activity of the IFN receptor, suggesting a complex interplay between the receptor and the various Tyk2 modular domains (Yeh et al., 2000). Importantly, our data exclude the possibility that assembly of the full receptor complex is required for IFNAR1 surface expression, since Jak1 and IFNAR2 would be limiting.
Although the positive effect of Tyk2 on the overall endogenous IFNAR1 level was consistent in 11,1 cells, this effect was less obvious in transient transfections. We believe that some variability is intrinsic to the transient system, where the ratio of IFNAR1 to Tyk2 transfected plasmids is difficult to control and a high biosynthesis may saturate degradation components. Nevertheless, we could clearly reveal a positive effect of Tyk2 on the half-life of IFNAR1 upon blocking de novo protein synthesis in transfected cells. The increased stability of the IC-Δ(464–519) and IC-480 mutants and their surface localization demonstrate that the IC segment between residues 480 and 519 is critical for constitutive internalization and degradation of IFNAR1. Since Tyk2 binds to a region between residues 486 and 511 of IFNAR1 (Yan et al., 1996), it may prevent endocytosis of the receptor by masking endocytic motifs. In doing so, Tyk2 could also sequester the complex away from internalization gates and concomitantly localize it in specific signaling membrane subdomains (Takaoka et al., 2000). Our results do not exclude the possibility that Tyk2 also triggers a conformational change of IFNAR1 towards a more stable folding or diverts the sorting of IFNAR1-containing endocytic vesicles from a degradative pathway to a more efficient recycling. Future work will need to address the question of whether the same or distinct structural determinants are involved in ligand-mediated IFNAR1 internalization and whether Tyk2 contributes to it.
The anchoring function of Tyk2 may provide a mechanism for the rapid modulation of receptor expression. Conformational modification of either protein, induced by ligand binding or intracellular signaling events, could lead to dissociation of the receptor–Jak complex, exposure of endocytic motifs and/or translocation into domains where efficient endocytosis occurs. Our data suggest that Tyk2 could be a limiting factor for IFNAR1 surface expression in certain cell contexts, where Tyk2 is less abundant or is engaged in other signaling complexes. Given the intricate cross-regulation of cytokines and their receptors, the upregulation of receptors that bind and withdraw Tyk2 may reduce IFNAR1 levels and in turn modulate signaling through the IFN receptor (Dondi et al., 2001). From a physiological perspective, our results suggest that Tyk2 may set the sensitivity threshold of the receptor to the various type I IFN subtypes by modulating receptor density (da Silva et al., 2002). This could be particularly relevant in physiological responses to the low constitutively produced type I IFN thought to regulate the homeostasis of cells of the immune system (Gongora et al., 2000; Lee et al., 2000; Takaoka et al., 2000). Inasmuch as Tyk2 is dedicated to signaling pathways of cytokines that perform key functions in early innate responses to pathogens, it is conceivable that its absence will be more or less harmful to the host, depending on the relative impact of these cytokines in the mounting of efficient responses necessary to eradicate a given pathogen (Bogdan, 2000; Karaghiosoff et al., 2000; Shimoda et al., 2000; Biron, 2001).
Materials and methods
Cell culture and transfection
Human fibrosarcoma parental 2fTGH, Tyk2-deficient 11,1 and Tyk2-complemented WT cells were as described (Pellegrini et al., 1989; Yeh et al., 2000). A clone of MEF from Tyk2–/– mice was kindly provided by M.Karaghiosoff (Karaghiosoff et al., 2000). For transient transfection, cells were plated at 5 × 104 in 24-well plates or on glass cover-slips. Next day, 1 µg of IFNAR1 plasmid was mixed with 1 µg of Tyk2 or empty vector and added to the cells as a calcium phosphate precipitate for 6 h. Cells were processed for immunofluorescence or western blot 24 or 48 h later. The protocol was scaled up for immunoprecipitation. Cycloheximide (Sigma) was added at 20 µg/ml 24 h after transfection and left as indicated. MEF Tyk2–/– were transfected for 4 h using the polymer polyethylenimine (Sigma) (Boussif et al., 1995). The lysosomal inhibitors NH4Cl and chloroquinine (Sigma) were used at 50 nM and 100 µM respectively. The proteasomal inhibitor MG132 (Sigma) was used at 50 µM.
Protein analysis
For surface IFNAR1 immunoprecipitation, 4 × 106 cells were washed with phosphate-buffered saline (PBS), detached in PBS and 5 mM EDTA, and incubated with 5 µg of EA12 Abs (Goldman et al., 1999) for 2 h at 4°C. After removal of uncoupled antibodies by extensive washing, cell lysis and immunoprecipitation was as described (Gauzzi et al., 1997). IFNAR1 was detected with 64G12 mAbs (Eid and Tovey, 1995). Tyk2 was detected with T10–2 mAbs (Gauzzi et al., 1996). Endoglycosidase H (Roche) was used at 20 mU per sample according to the manufacturer’s instructions. Bands were revealed with an ECL detection system (Amersham) and quantified with the Kodak Image Station 440CF.
Flow cytometry
Surface IFNAR1 levels were monitored by incubating cells with 10 µg/ml of AA3 mAb (Goldman et al., 1999), followed by incubation with 10 µg/ml of biotinylated anti-mouse IgG Abs and with streptavidin– phycoerythrin (Jackson Immuno Research Laboratories). Cells were analyzed with a FACScan flow cytometer (Becton Dickinson).
Fluorescence microscopy
Cells seeded on cover-slips were transiently transfected and 24 h later fixed with 4% paraformaldehyde and processed as described (Ragimbeau et al., 2001). The IFNAR1 AA3 mAb (Goldman et al., 1999) was used at 5 µg/ml, the anti-IL-10R2 mAb (R&D Systems) was used at 5 µg/ml and the murine IFNAR1 anti-VSV tag p5D4 mAb (Sigma) was used at 10 µg/ml. Appropriate mAbs were revealed with Texas Red-coupled anti-IgG1 secondary Abs (Southern Biotech). Anti-Tyk2 C20 (Santa Cruz) was used to identify Tyk2-positive transfected cells. Goat anti-EEA1 Abs (N19) was obtained from Santa Cruz. Rabbit anti-Lamp1 Abs was a gift from S.Méresse (Centre d’Immunologie de Marseille-Luminy). Rabbit primary Abs was revealed with Alexa488-coupled secondary Abs (Molecular Probes). For transferrin internalization, cells were washed twice and starved for 1 h in serum-free medium. Alexa488-coupled transferrin (150 nM) was added to the culture medium at 37°C for 30–120 min. For internalization, AA3 anti-IFNAR1 mAbs was added to culture medium at 1 µg/ml for 30–120 min. At 30 min, the internalized mAbs was found mostly in scattered vesicles, while from 60 min on it was found in a more compact multivesicular compartment, as shown in Figure 2. Cover-slips were mounted on glass slides with Mowiol containing 2.5% DABCO (Sigma). The confocal microscopy analysis in Figure 2 was performed with a Zeiss LSM-510 microscope; Z-series of optical sections were performed at 0.5 µm increments. Images were acquired with settings allowing the maximum signal detection below the saturation limits. For double staining, sequential aquisitions were performed to avoid fluorescence contamination between the two channels. A three-dimensional reconstruction using 36 projections at an angle of 10° around the Y axis was carried out using the LSM-510 software. A single orientation of the three-dimensional reconstruction is shown. For the other figures, visualization was with a Zeiss Axiovert 135 microscope (40× oil immersion lens). Images were captured with a Hamamatsu Orca II CCD camera and analyzed using AquaCosmos software.
Endocytosis assays
The anti-IFNAR1 mAb 64G12 was iodinated using the chloramine T method (Giron-Michel et al., 2002). The COS-7 cells were transfected by the calcium phosphate protocol. Two days later, 106 cells were harvested with PBS and 10 mM EDTA and were washed twice. After 15 min at 4°C in Dulbecco’s modified Eagle’s medium (DMEM), 10% fetal calf serum and 20 mM HEPES pH 7.2, 5 nM of [125I]64G12 mAb was added for 1 h at 4°C. Internalization was initiated by rapidly warming the cells to 37°C, without removing unbound ligand. At the indicated times, duplicate aliquots were withdrawn, cooled on ice and washed with DMEM and 20 mM HEPES pH 7.2. Surface-bound [125I]mAb was measured by subjecting the cells to three acid washes for 2.5 min at pH 2. This treatment released more than 90% of the surface-bound ligand. Internalized [125I]mAb was determined after lysis of the cell pellet. The steady state distribution of IFNAR1 was measured as follows: 106 transfected COS-7 cells were incubated in the presence of 5 nM [125I]65G12 mAb for 90 min at 37°C, and surface-bound and internalized labeled material was measured as described above.
Plasmids
pRc/CMV–Tyk2 constructs were as described (Gauzzi et al., 1997; Richter et al., 1998; Ragimbeau et al., 2001). Murine Tyk2 cDNA in pcDNA3.1 was a gift from M.Karaghiosoff. The 3′ untranslated region of human IFNAR1 cDNA in pVADN1 was deleted by swapping an _Nde_I–_Bst_XI with a PCR fragment containing a 5′ _Nde_I site and a stop codon after aa 557. Two IFNAR1 mutants were made similarly: in mutant IC-463 the stop codon is after aa 463, and in mutant IC-524 the stop is after aa 524. For mutant IC-Δ(464–519), an _Nde_I–_Bst_XI fragment with a deletion of 168 bp was amplified and swapped into pVADN1-IFNAR1. All fragments derived by PCR were sequenced. Plasmid IC-480 (from G.Lutfalla) contains 23 IC residues followed by 11 unrelated residues. Murine IFNAR1 cDNA in the pVADN1 vector contains a 3′ VSV-G epitope. Human IL-10R2 (Lutfalla et al., 1993) is in pVADN1. The pECFP-Golgi plasmid was from Clontech.
Acknowledgments
Acknowledgements
We thank I.Behrmann, G.Lutfalla, M.Karaghiosoff, S.Méresse, J.Pizarro-Cerda and L.Runkel for reagents; and M.Arpin, A.Dautry-Varsat, M.C.Gauzzi and R.McKendry for discussions and reviewing the manuscript. Work was supported by grants from the Association pour la Recherche sur le Cancer (S.P.), La Ligue contre le Cancer (A.A.) and the Human Frontier Science Program (G.U.). E.D. was supported by the Fondation pour la Recherche Médicale and the Association contre la Sclérose en Plaque.
References
- Basu L., Yang,C.H., Murti,A., Garcia,J.V., Croze,E., Constantinescu, S.N., Mullersman,J.E. and Pfeffer,L.M. (1998) The antiviral action of interferon is potentiated by removal of the conserved IRTAM domain of the IFNAR1 chain of the interferon α/β receptor: effects on JAK-STAT activation and receptor down-regulation. Virology, 242, 14–21. [DOI] [PubMed] [Google Scholar]
- Biron C.A. (2001) Interferons α and β as immune regulators—a new look. Immunity, 14, 661–664. [DOI] [PubMed] [Google Scholar]
- Bogdan C. (2000) The function of type I interferons in antimicrobial immunity. Curr. Opin. Immunol., 12, 419–424. [DOI] [PubMed] [Google Scholar]
- Boussif O., Lezoualch’h,F., Zanta,M.A., Mergny,M.D., Scherman,D., Demeneix,B. and Behr,J.P. (1995) A versatile method for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc. Natl Acad. Sci. USA, 92, 7297–7301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva A.J., Brickelmaier,M., Majeau,G.R., Lukashin,A.V., Peyman,J., Whitty,A. and Hochman,P.S. (2002) Comparison of gene expression patterns induced by treatment of human umbilical vein endothelial cells with IFN-α2b vs. IFN-β1a: understanding the functional relationship between distinct type I Interferons that act through a common receptor. J. Interferon Cytokine Res., 22, 173–188. [DOI] [PubMed] [Google Scholar]
- Dondi E., Pattyn,E., Lutfalla,G., Van Ostade,X., Uze,G., Pellegrini,S. and Tavernier,J. (2001) Down-modulation of type 1 interferon responses by receptor cross-competition for a shared Jak kinase. J. Biol. Chem., 276, 47004–47012. [DOI] [PubMed] [Google Scholar]
- Eid P. and Tovey,M.G. (1995) Characterization of a domain of a human type I interferon receptor protein involved in ligand binding. J. Interferon Cytokine Res., 15, 205–211. [DOI] [PubMed] [Google Scholar]
- Gately M.K., Renzetti,L.M., Magram,J., Stern,A.S., Adorini,L., Gubler,U. and Presky,D.H. (1998) The interleukin-12/interleukin-12-receptor system: role in normal and pathologic immune responses. Annu. Rev. Immunol., 16, 495–521. [DOI] [PubMed] [Google Scholar]
- Gauzzi M.C., Velazquez,L., McKendry,R., Mogensen,K.E., Fellous,M. and Pellegrini,S. (1996) Interferon-α-dependent activation of Tyk2 requires phosphorylation of positive regulatory tyrosines by another kinase. J. Biol. Chem., 271, 20494–20500. [DOI] [PubMed] [Google Scholar]
- Gauzzi M.C., Barbieri,G., Richter,M.F., Uzé,G., Ling,L., Fellous,M. and Pellegrini,S. (1997) The amino-terminal region of Tyk2 sustains the level of interferon α receptor 1, a component of the interferon α/β receptor. Proc. Natl Acad. Sci. USA, 94, 11839–11844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs V.C., Takahashi,M., Aguet,M. and Chuntharapai,A. (1996) A negative regulatory region in the intracellular domain of the human interferon-α receptor. J. Biol. Chem., 271, 28710–28716. [DOI] [PubMed] [Google Scholar]
- Giron-Michel J., Weill,D., Bailly,G., Legras,S., Nardeux,P.C., Azzarone,B., Tovey,M.G. and Eid,P. (2002) Direct signal transduction via functional interferon-αβ receptors in CD34+ hematopoietic stem cells. Leukemia, 16, 1136–1142. [DOI] [PubMed] [Google Scholar]
- Goldman L.A., Zafari,M., Cutrone,E.C., Dang,A., Brickelmeier,M., Runkel,L., Benjamin,C.D., Ling,L.E. and Langer,J.A. (1999) Characterization of antihuman IFNAR-1 monoclonal antibodies: epitope localization and functional analysis. J. Interferon Cytokine Res., 19, 15–26. [DOI] [PubMed] [Google Scholar]
- Gongora R., Stephan,R.P., Schreiber,R.D. and Cooper,M.D. (2000) Stat-1 is not essential for inhibition of B lymphopoiesis by type I IFNs. J. Immunol., 165, 2362–2366. [DOI] [PubMed] [Google Scholar]
- Hémar A., Lieb,M., Subtil,A., DiSanto,J.P. and Dautry-Varsat,A. (1994) Endocytosis of the β chain of interleukin-2 receptor requires neither interleukin-2 nor the γ chain. Eur. J. Immunol., 24, 1951–1955. [DOI] [PubMed] [Google Scholar]
- Huang L.J., Constantinescu,S.N. and Lodish,H.F. (2001) The N-terminal domain of Janus kinase 2 is required for Golgi processing and cell surface expression of erythropoietin receptor. Mol. Cell, 8, 1327–1338. [DOI] [PubMed] [Google Scholar]
- Karaghiosoff M. et al. (2000) Compromised innate and adaptive immunity in Tyk2-deficient mice. Immunity, 13, 549–560. [DOI] [PubMed] [Google Scholar]
- Kindsvogel W. and Kotenko,S. (2002) Meeting on Cytokines and Interferons 2002. J. Interferon Cytokine Res., 22 (suppl. 1), 5–48.11846971 [Google Scholar]
- Kotenko S.V. and Pestka,S. (2000) Jak-Stat signal transduction pathway through the eyes of cytokine class II receptor complexes. Oncogene, 19, 2557–2565. [DOI] [PubMed] [Google Scholar]
- Kotenko S.V., Krause,C.D., Izotova,L.S., Pollack,B.P., Wu,W. and Pestka,S. (1997) Identification and functional characterization of a second chain of the interleukin-10 receptor complex. EMBO J., 16, 5894–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotenko S.V., Izotova,L.S., Mirochnitchenko,O.V., Esterova,E., Dickensheets,H., Donnelly,R.P. and Pestka,S. (2001) Identification of the functional interleukin-22 (IL-22) receptor complex: the IL-10R2 chain (IL-10Rβ) is a common chain of both the IL-10 and IL-22 (IL-10-related T cell-derived inducible factor, IL-TIF) receptor complexes. J. Biol. Chem., 276, 2725–2732. [DOI] [PubMed] [Google Scholar]
- Lamaze C., Dujeancourt,A., Baba,T., Lo,C.G., Benmerah,A. and Dautry-Varsat,A. (2001) Interleukin 2 receptors and detergent-resistant membrane domains define a clathrin-independent endocytic pathway. Mol. Cell, 7, 661–671. [DOI] [PubMed] [Google Scholar]
- Lee C.K., Smith,E., Gimeno,R., Gertner,R. and Levy,D.E. (2000) STAT1 affects lymphocyte survival and proliferation partially independent of its role downstream of IFN-γ. J. Immunol., 164, 1286–1292. [DOI] [PubMed] [Google Scholar]
- Lewerenz M., Mogensen,K.M. and Uzé,G. (1998) Shared receptor components but distinct complexes for α and β interferons. J. Mol. Biol., 282, 585–599. [DOI] [PubMed] [Google Scholar]
- Ling L.E., Zafari,M., Reardon,D., Brickelmeier,M., Goelz,S.E. and Benjamin,C.D. (1995) Human type I interferon receptor, IFNAR, is a heavily glycosylated 120–130 kD membrane protein. J. Interferon Cytokine Res., 15, 55–61. [DOI] [PubMed] [Google Scholar]
- Lutfalla G., Gardiner,K. and Uzé,G. (1993) A new member of the cytokine receptor gene family maps on chromosome 21 at less than 35 kb from IFNAR. Genomics, 16, 366–373. [DOI] [PubMed] [Google Scholar]
- Mellman I. (1996) Endocytosis and molecular sorting. Annu. Rev. Cell. Dev. Biol., 12, 575. [DOI] [PubMed] [Google Scholar]
- Mogensen K.E., Lewerenz,M., Reboul,J., Lutfalla,G. and Uzé,G. (1999) The type I interferon receptor: structure, function and evolution of a family business. J. Interferon Cytokine Res., 19, 1069–1098. [DOI] [PubMed] [Google Scholar]
- Moore K.W., de Waal Malefyt,R., Coffman,R.L. and O’Garra,A. (2001) Interleukin-10 and the interleukin-10 receptor. Annu. Rev. Immunol., 19, 683–765. [DOI] [PubMed] [Google Scholar]
- Mu F.T. et al. (1995) EEA1, an early endosome-associated protein. EEA1 is a conserved alpha-helical peripheral membrane protein flanked by cysteine ‘fingers’ and contains a calmodulin-binding IQ motif. J. Biol. Chem., 270, 13503–13511. [DOI] [PubMed] [Google Scholar]
- Muller U., Steinhoff,U., Reis,L.F., Hemmi,S., Pavlovic,J., Zinkernagel,R.M. and Aguet,M. (1994) Functional role of type I and type II interferons in antiviral defense. Science, 264, 1918–1921. [DOI] [PubMed] [Google Scholar]
- Neumann D., Wikstrom,L., Watowich,S.S. and Lodish,H.F. (1993) Intermediates in degradation of the erythropoietin receptor accumulate and are degraded in lysosomes. J. Biol. Chem., 268, 13639–13649. [PubMed] [Google Scholar]
- Oppmann B. et al. (2000) Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity, 13, 715–725. [DOI] [PubMed] [Google Scholar]
- Pelchen-Matthews A., Boulet,I., Littman,D.R., Fagard,R. and Marsh,M. (1992) The protein tyrosine kinase p56lck inhibits CD4 endocytosis by preventing entry of CD4 into coated pits. J. Cell Biol., 117, 279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelchen-Matthews A., da Silva,R.P., Bijlmakers,M.J., Signoret,N., Gordon,S. and Marsh,M. (1998) Lack of p56lck expression correlates with CD4 endocytosis in primary lymphoid and myeloid cells. Eur. J. Immunol., 28, 3639–3647. [DOI] [PubMed] [Google Scholar]
- Pellegrini S., John,J., Shearer,M., Kerr,I.M. and Stark,G.R. (1989) Use of a selectable marker regulated by α interferon to obtain mutations in the signaling pathway. Mol. Cell. Biol., 9, 4605–4612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radtke S., Hermanns,H.M., Haan,C., Schmitz-Van De Leur,H., Gascan,H., Heinrich,P.C. and Behrmann,I. (2002) Novel role for Janus kinase 1 in the regulation of oncostatin M receptor surface expression. J. Biol. Chem., 277, 11297–11305. [DOI] [PubMed] [Google Scholar]
- Ragimbeau J., Dondi,E., Vasserot,A., Romero,P., Uze,G. and Pellegrini,S. (2001) The receptor interaction region of Tyk2 contains a motif required for its nuclear localization. J. Biol. Chem., 276, 30812–30818. [DOI] [PubMed] [Google Scholar]
- Richter M.F., Duménil,G., Uzé,G., Fellous,M. and Pellegrini,S. (1998) Specific contribution of Tyk2 JH regions to the binding and the expression of the interferon α/β receptor component IFNAR1. J. Biol. Chem., 273, 24723–24729. [DOI] [PubMed] [Google Scholar]
- Rigamonti L., Ariotti,S., Losana,G., Gradini,R., Russo,M.A., Jouanguy,E., Casanova,J.L., Forni,G. and Novelli,F. (2000) Surface expression of the IFN-γ R2 chain is regulated by intracellular trafficking in human T lymphocytes. J. Immunol., 164, 201–207. [DOI] [PubMed] [Google Scholar]
- Rocca A., Lamaze,C., Subtil,A. and Dautry-Varsat,A. (2001) Involvement of the ubiquitin/proteasome system in sorting of the interleukin 2 receptor β chain to late endocytic compartments. Mol. Biol. Cell, 12, 1293–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roisman L.C., Piehler,J., Trosset,J.Y., Scheraga,H.A. and Schreiber,G. (2001) Structure of the interferon-receptor complex determined by distance constraints from double-mutant cycles and flexible docking. Proc. Natl Acad. Sci. USA, 98, 13231–13236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runkel L. et al. (2001) Mapping of IFN-β epitopes important for receptor binding and biologic activation: comparison of results achieved using antibody-based methods and alanine substitution mutagenesis. J. Interferon Cytokine Res., 21, 931–941. [DOI] [PubMed] [Google Scholar]
- Shimoda K. et al. (2000) Tyk2 plays a restricted role in IFNα signaling, although it is required for IL-12-mediated T cell function. Immunity, 13, 561–571. [DOI] [PubMed] [Google Scholar]
- Stark G.R., Kerr,I.M., Williams,B.R.G., Silverman,R.H. and Schreiber,R.D. (1998) How cells respond to interferons. Annu. Rev. Biochem., 67, 227–264. [DOI] [PubMed] [Google Scholar]
- Subtil A., Delepierre,M. and Dautry-Varsat,A. (1997) An α-helical signal in the cytosolic domain of the interleukin 2 receptor β chain mediates sorting towards degradation after endocytosis. J. Cell Biol., 136, 583–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supino-Rosin L., Yoshimura,A., Altaratz,H. and Neumann,D. (1999) A cytosolic domain of the erythropoietin receptor contributes to endoplasmic reticulum-associated degradation. Eur. J. Biochem., 263, 410–419. [DOI] [PubMed] [Google Scholar]
- Takaoka A., Mitani,Y., Suemori,H., Sato,M., Yokochi,T., Noguchi,S., Tanaka,N. and Taniguchi,T. (2000) Cross talk between interferon-γ and -α/β signaling components in caveolar membrane domains. Science, 288, 2357–2360. [DOI] [PubMed] [Google Scholar]
- Thiel S., Behrmann,I., Dittrich,E., Muys,L., Tavernier,J., Wijdenes,J., Heinrich,P.C. and Graeve,L. (1998) Internalization of the interleukin 6 signal transducer gp130 does not require activation of the Jak/STAT pathway. Biochem. J., 330, 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel S. et al. (1999) Identification of a Leu-lle internalization motif within the cytoplasmic domain of the leukaemia inhibitory factor receptor. Biochem. J., 339, 15–19. [PMC free article] [PubMed] [Google Scholar]
- Trowbridge I.S., Collawn,J.F. and Hopkins,C.R. (1993) Signal-dependent membrane protein trafficking in the endocytic pathway. Annu. Rev. Cell. Dev. Biol., 9, 129–161. [DOI] [PubMed] [Google Scholar]
- van Kerkhof P., Govers,R., Alves dos Santos,C.M. and Strous,G.J. (2000) Endocytosis and degradation of the growth hormone receptor are proteasome-dependent. J. Biol. Chem., 275, 1575–1580. [DOI] [PubMed] [Google Scholar]
- Velazquez L., Mogensen,K.E., Barbieri,G., Fellous,M., Uzé,G. and Pellegrini,S. (1995) Distinct domains of the protein tyrosine kinase tyk2 required for binding of interferon-α/β and for signal transduction. J. Biol. Chem., 270, 3327–3334. [DOI] [PubMed] [Google Scholar]
- Yan H., Krishnan,K., Lim,J.T., Contillo,L.G. and Krolewski,J.J. (1996) Molecular characterization of an α interferon receptor 1 subunit (IFNaR1) domain required for TYK2 binding and signal transduction. Mol. Cell. Biol., 16, 2074–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh T.C. and Pellegrini,S. (1999) The Janus kinase family of protein tyrosine kinases and their role in signaling. Cell. Mol. Life Sci., 55, 1523–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh T.C., Dondi,E., Uzé,G. and Pellegrini,S. (2000) A dual role for the kinase-like domain of the tyrosine kinase Tyk2 in interferon-α signaling. Proc. Natl Acad. Sci. USA, 97, 8991–8996. [DOI] [PMC free article] [PubMed] [Google Scholar]