Phosphorylation of Nucleotide Excision Repair Factor Xeroderma Pigmentosum Group A by Ataxia Telangiectasia Mutated and Rad3-Related-Dependent Checkpoint Pathway Promotes Cell Survival in Response to UV Irradiation (original) (raw)
. Author manuscript; available in PMC: 2007 Mar 15.
Abstract
DNA damage triggers complex cellular responses in eukaryotic cells, including initiation of DNA repair and activation of cell cycle checkpoints. In addition to inducing cell cycle arrest, checkpoint also has been suggested to modulate a variety of other cellular processes in response to DNA damage. In this study, we present evidence showing that the cellular function of xeroderma pigmentosum group A (XPA), a major nucleotide excision repair (NER) factor, could be modulated by checkpoint kinase ataxia-telangiectasia mutated and Rad3-related (ATR) in response to UV irradiation. We observed the apparent interaction and colocalization of XPA with ATR in response to UV irradiation. We showed that XPA was a substrate for in vitro phosphorylation by phosphatidylinositol-3-kinase-related kinase family kinases whereas in cells XPA was phosphorylated in an ATR-dependent manner and stimulated by UV irradiation. The Ser196 of XPA was identified as a biologically significant residue to be phosphorylated in vivo. The XPA-deficient cells complemented with XPA-S196A mutant, in which Ser196 was substituted with an alanine, displayed significantly higher UV sensitivity compared with the XPA cells complemented with wild-type XPA. Moreover, substitution of Ser196 with aspartic acid for mimicking the phosphorylation of XPA increased the cell survival to UV irradiation. Taken together, our results revealed a potential physical and functional link between NER and the ATR-dependent checkpoint pathway in human cells and suggested that the ATR checkpoint pathway could modulate the cellular activity of NER through phosphorylation of XPA at Ser196 on UV irradiation.
Introduction
DNA damage triggers a variety of cellular responses in eukaryotes, including the removal of DNA damage via repair mechanisms, arrest of cell cycle progression if DNA lesions are not removed in a timely fashion, and elimination of severely impaired cells by apoptosis (1-4). Among the repair mechanisms that cells use to cope with the genotoxic insults, nucleotide excision repair (NER) represents the most versatile and flexible DNA repair pathway in cells as it deals with a wide range of structurally unrelated bulky DNA lesions (5). In the mechanism of NER pathway, xeroderma pigmentosum group A (XPA) plays an indispensable role and is required for both global genome repair and transcription-coupled repair (6-9). It is generally believed that XPA is involved in DNA damage recognition and also in the recruitment of other NER factors to the DNA damage site to form dual incision complexes through protein-protein interactions (8, 10-12).
DNA damage checkpoints are signal transduction pathways that monitor the structure of chromosomes and coordinate DNA repair with cell cycle progression (4). The checkpoint signaling cascades, conceptually, consist of three major biochemical components: damage sensors, signal mediators/transducers, and effector molecules. The ataxia-telangiectasia mutated and Rad3-related (ATR) and ataxia-telangiectasia mutated (ATM) checkpoint proteins and the Rad9-Rad1-Hus1/Rad17-Rfc2-5 checkpoint complex have been suggested to be involved in DNA damage recognition and signaling in human cells (2, 4, 9, 13-15). ATR and ATM belong to the phosphatidylinositol-3-kinase-related kinase (PIKK) family of proteins. The PIKK family also includes the catalytic subunit of DNA-dependent protein kinase (DNA-PK), which is an essential factor for nonhomologous end-joining pathway of DNA double-strand break repair (14, 16). ATM kinase seems to be activated primarily in response to the induction of double-strand breaks whereas ATR is critical for cellular checkpoint responses to stalled replication forks and to many types of DNA damage, such as those induced by UV light and cisplatin (16, 17). When activated, these kinases display a serine/threonine kinase activity toward the so-called checkpoint mediators (e.g., MDC1, 53BP1, and BRCA1), transducers (Chk1/Chk2), and a multitude of effectors (e.g., p53, CDC25A/C, E2F, and SMC1) in cells to spread the initial signal and to induce the global cellular response to DNA damage (1, 2, 4, 18-20). Recent analyses using an oriented peptide library have revealed that the S/TQ sequence is a minimal requirement for phosphorylation by all three PIKK family kinases (21, 22). Whereas most of the S/TQ-containing substrates identified to date are involved in checkpoints, proteins with the S/TQ motif in other pathways that are physiologically and functionally relevant to checkpoints could also be the target of ATR/ATM.
DNA damage checkpoints were originally defined as the molecular signaling pathways that promote cell cycle delay or arrest in response to DNA damage to allow repair of the damage. Recent evidence, however, indicated that in addition to controlling cell cycle transitions, checkpoint activation is also important for optimal DNA repair activity, activation of transcription, and, in some instances, induction of apoptosis (4). The regulation of DNA repair activity by the checkpoint mechanism has been relatively well studied for the irradiation-induced double-strand break repair pathway. Both homologous recombination and nonhomologous end-joining pathways of double-strand break repair have been shown to be modulated by the ATM- and Chk2-dependent checkpoint (23-25). Moreover, in ATM/ATR-deficient cells, the radiosensitive phenotype has been ascribed to the double-strand break repair defects (26, 27). Strikingly, even haploinsufficiency of checkpoint factors (ATM/Rad9) leads to different double-strand break repair dynamics, implying that checkpoint pathways may control DNA repair activity quantitatively (28). Similarly, such a checkpoint-dependent regulation of double-strand break repair has been observed in yeast where deletion of the MEC1 and Rad53 checkpoint kinases or downstream effector kinases led to a dramatic decrease in double-strand break repair efficiency (29, 30).
In the case of NER, however, little is known about the cellular relationship between NER and the ATR/ATM-dependent checkpoints with regard to DNA damage response. Several studies have suggested that NER may function in the upstream of cellular checkpoint response to UV irradiation as NER processing of UV damage was necessary for checkpoint activation in nonreplicating cells (31-33). On the other hand, two checkpoint genes in budding yeast (Rad9 and Rad24, the human homologues of BRCA1 and Rad17, respectively) have been shown to be required for the inducible NER (34). And in mammalian system, the checkpoint abrogator UCN-01 inhibited the NER activity against cisplatin (35). Given the central role of ATR/ATM kinases in the entire DNA damage response network, we were particularly interested on whether these kinases could directly regulate the cellular NER activity in response to UV damage. In the present study, we have identified the NER factor XPA as an in vitro and in vivo substrate for phosphorylation by checkpoint kinase ATR. Such phosphorylation was UV-inducible and the phosphorylated XPA predominantly resided in the nuclei. We also showed that ATR and XPA could physically interact and apparently colocalize to form nuclear foci on UV irradiation. Strikingly, it was found that phosphorylation of residue Ser196 of XPA was required for the optimal cell survival in response to UV treatment. Taken together, our results revealed a functional link between NER and the ATR-dependent checkpoint pathway and suggested a potential mechanism for regulation of the cellular NER activity by checkpoint pathway following UV irradiation.
Materials and Methods
Cell culture and treatments. Human lung adenocarcinoma cells A549 were obtained from American Type Culture Collection (Manassas, VA) and maintained at 37°c and 5% CO2 in DMEM supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin. Three mutant human fibroblast cell lines, GM18366, GM09607, and GM04429 (ATR-, ATM-, and XPA-deficient cells, respectively), were purchased from Coriell Cell Repositories (Camden, NJ). For UV exposure, cells were irradiated with various doses of UV using a UV cross-linker at a dose rate of 0.5 J/m2/s and further incubated for 4 hours at 37°C before harvesting.
Generation of the XPA and XPA-mutant constructs. Human XPA cDNA was isolated from the pTYB-XPA plasmid by PCR amplification with Pfu Turbo (Stratagene, La Jolla, CA) and then incubated with Taq polymerase (Promega, Madison, WI) for addition of 3VA-overhangs. The cDNA was then inserted into the pcDNA3.1/V5-His TOPO plasmid (Invitrogen, Carlsbad, CA) and transformed into XL10-Gold competent E. coli (Stratagene) resulting in the pcDNA3.1-XPA-WT vector. Ser→Ala and Ser→Asp mutations were generated by site-directed mutagenesis of the pcDNA3.1-XPA-WT plasmid at Ser173 and Ser196 using the QuickChange XL Site-Directed Mutagenesis Kit (Stratagene). The pcDNA3.1-XPA-S173/196A double mutant plasmid was generated by site-directed mutagenesis of the pcDNA3.1-XPA-S173A plasmid as described above. All plasmids were verified by DNA sequencing.
Transfection. Human XPA-deficient fibroblast, grown to 50% to 80% confluence, were transfected with XPA wild-type (XPA-WT) and XPA-mutant constructs using LipofectAMINE reagent according to the instructions of the manufacturer (Invitrogen, Life Technologies). The stably transfected cells were screened through neomycin selection (500 Ag/mL) until the resistant colonies appeared. The cells were then maintained in DMEM medium containing 250 Ag/mL neomycin for all experiments.
Cell viability assays. The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assays were done as previously described (9). For colony formation assays, cells (500 per dish) were plated in triplicate. After overnight attachment, cells were irradiated with indicated doses of UV and then allowed to grow for f2 weeks. The formed colonies were counted under microscope (the colonies having >50 cells were defined as positive colonies).
Coimmunoprecipitation assays. Coimmunoprecipitation assays were conducted as previously described (13, 15). Four micrograms of rabbit anti-ATR (Oncogene, Cambridge, MA) or anti-XPA (Santa Cruz Biotechnology) were used for each coimmunoprecipitation assay. For pull-down assays using purified His-XPA, recombinant His-XPA was purified as described (36). For each assay, 2 μg of His-XPA were mixed with whole-cell extracts prepared from 2 × 106 cells. The reaction mixtures were rotated at 4°c for 10 to 14 hours and then 50 AL of Nickel-NTA resin (Qiagen, Valencia, CA) were added, followed by an additional incubation at 4°c for 30 minutes. The bound proteins were extracted from the agarose/nickel beads by boiling in 1 × SDS gel loading buffer.
Western blotting. Whole-cell lysates and nuclear extracts were prepared as previously described (13, 15). Proteins were separated on 8% SDS-polyacrylamide gels and transferred to polyvinylidene difluoride membrane. The membranes were blocked with TBST buffer containing 5% powered milk and probed using the following primary antibodies: anti-XPA, replication protein A 32-kDa subunit (RPA32; Kamiya, Seattle, WA), anti-ATR, anti-ATM (GeneTex, Inc., San Antonio, TX), anti-Rad51, and anti-Hus1 (Santa Cruz Biotechnology). The membranes were then incubated with horseradish peroxidase-linked secondary antimouse antibodies and bound antibodies were visualized using the enhanced chemiluminescence method.
In vitro kinase assay. The in vitro kinase assays with ATM and ATR were done as described earlier (21, 22). Briefly, endogenous ATM or ATR was immunoprecipitated from 40 J/m2 UV-irradiated A549 cells. The beads were washed thrice with PBS containing 0.05% NP40, followed by one wash with kinase buffer [50 mmol/L HEPES (pH 7.5), 150 mmol/L NaCl, 10 mmol/L MgCl2, 10 mmol/L MnCl2, 2 mmol/L DTT, 10% glycerol, protease and phosphatase inhibitors, 0.5 Amol/L cold ATP]. Then the immunoprecipitates were resuspended in 20 AL of kinase buffer containing 10 ACi of [g-32P]ATP and 1 Ag of purified His-XPA proteins or peptides. The kinase reaction was conducted at 30°c for 10 to 30 minutes and stopped by the addition of SDS loading buffer. Proteins were separated by SDS-PAGE and the radiolabeled proteins were visualized by gel scanning using a PhosphorImage scanner (Fuji, Stamford, CT). Immunoprecipitated endogenous ATM or ATR was confirmed by Western blotting. The in vitro kinase assay with DNA-PK for phosphorylation of purified recombinant XPA or plasmid-expressed XPA and mutants in transfected cells was done in the presence of calf thymus DNA as previously described (21, 22, 37). The DNA-PK isolated from HeLa nuclear extracts as a complex consisting of the catalytic subunit DNA-PKcs and Ku70/80 heterodimer was purchased from Promega.
In vivo phosphorylation. A549 cells were grown on 6-cm dishes overnight in phosphate-depleted medium (DMEM without phosphate; Invitrogen) at 37°c. Thereafter, cells were treated with 20 J/m2 UV irradiation and 32P-labeled orthophosphoric acid was added at 0.2 mCi/mL medium. Cells were incubated for an additional 8 hours at 37°c and harvested for immunoprecipitation with anti-XPA antibody. The immunoprecipitates were separated by SDS-PAGE and the radiolabeled proteins were visualized using a PhosphorImager (Fuji). Immunoprecipitated endogenous XPA was confirmed by Western blotting.
Immunofluorescence. Immunofluorescent staining was carried out as previously described (13, 15). Primary antibody dilutions used are as follows:rabbit anti-XPA (Santa Cruz Biotechnology), 1:500; mouse anti-ATR, 1:2,000 (GeneTex). Secondary antibody dilutions are as follows: antirabbit Alexa Fluor 488, 1:250; antimouse Alexa Fluor 568, 1:250 (Molecular Probes, Carlsbad, CA).
Small interfering RNA transfections. The small interfering RNA (siRNA) transfection experiments were carried out using TransIT-TKO Transfection Reagent (Mirus) essentially following the instructions of the manufacturer. Transfection-ready siRNA duplexes were purchased from Dharmacon (Chicago, IL) and Invitrogen. The siRNA sequences used in this study were ATR, 5V-CCUCCGUGAUGUUGCUUGA; ATM, SMART pool siRNA; and DNA-PK, 5V-AAAGGGCCAAGCUGUCACUCU. Transfection of ATR or ATM siRNA was conducted with A549 cells whereas transfection of DNA-PK siRNA was carried out with HeLa cells.
Results
XPA was phosphorylated in vivo in response to UV irradiation. The NER factor XPA is a zinc-finger protein with a molecular weight of 32 kDa but migrates as two distinct bands in SDS-PAGE at the positions of f38 and 40 kDa, probably due to the different reduction status of XPA related to the formation of disulfide bonds within the protein (12, 38, 39). During our studies of cellular XPA function with A549 cells, we consistently observed a third band of XPA when the gel was run for an unusually long time (Fig. 1A, top, band 3). Interestingly, this band was susceptible to treatment with calf intestinal alkaline phosphatase (CIAP; Fig. 1A (a), lanes 3 and 4) whereas addition of a CIAP inhibitor, glycerophosphate, preserved the mobility shift of this band (Fig. 1A (a), lanes 5 and 6). These data suggest that band 3 could represent a phosphorylated form of XPA. The XPA phosphorylation seems to be UV-inducible (Fig. 1A (a), lanes 1 and 2) although a marginal amount of the phosphorylated XPA band was also observed in the absence of UV irradiation. As a control, the mobility shift of hyperphosphorylated 32-kDa subunit of replication protein A (RPA32) was reversed by CIAP treatment and preserved by CIAP inhibitor (Fig. 1A (b)). It has been well known that RPA32 undergoes extensive phosphoryl modification (hyperphosphorylation) in cells on UV irradiation (40). We also treated XPA immunoprecipitates with CIAP and, as shown in Fig. 1A (c), the treatment removed the third band of XPA, as expected. The significant increase of total XPA in the nuclear extracts following UV treatment was due to an UV-induced cytoplasm-to-nucleus translocation of XPA (data not shown). To further examine if XPA was phosphorylated in vivo, cells were UV irradiated, immediately followed by culture in the presence of radiolabeled orthophosphate. Immunoprecipitation of XPA from cell lysates, SDS-PAGE analysis, and Western blotting revealed a radioactively labeled band corresponding to the third form of XPA (Fig. 1B). Based on these results, we concluded that the occurrence of the third form of XPA, mainly due to UV irradiation of cells, represented a phosphoryl modification of the protein.
Figure 1.
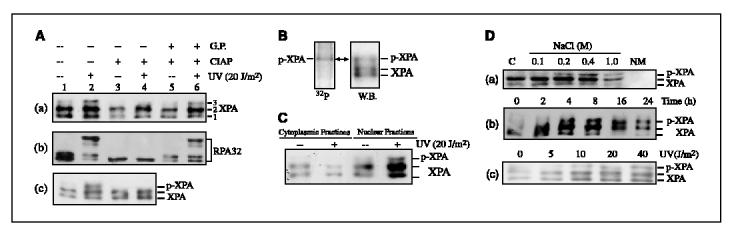
XPA is phosphorylated in cells on UV irradiation. A, A549 cells were treated with 20 J/m2 UV or mock treated. Total cell lysates were harvested at 4 hours after UV irradiation and then treated with 200 units of CIAP (Promega) for 1 hour at 37°c in the absence (lanes 3 and 4) or presence (lanes 5 and 6) of 50 mmol/L glycerophosphate (G.P.) or mock treated (lanes 1 and 2). The treated cell lysates were then subjected to Western blotting and probed with anti-XPA (a) and anti-RPA32 (b), respectively. c, cells were treated with 20 J/m2 UV or mock treated and total cell lysates were harvested for immunoprecipitation assays with anti-XPA antibody. The immunoprecipitated XPA was treated with CIAP or mock treated and then analyzed by Western blotting with anti-XPA antibody. B, A549 cells were grown overnight in phosphate-depleted medium before irradiation with 20 J/m2 UV. Then, 32P-labeled orthophosphoric acid was added and cells were further incubated for 8 hours before harvest. Immunoprecipitation assay was done with anti-XPA antibody. Immunoprecipitates were separated on SDS-PAGE and radiolabeled proteins were detected (left). Immunoprecipitated endogenous XPA was probed by Western blotting (right). C, cells were UV irradiated or mock treated and then cytoplasmic and nuclear extractions were separated on SDS-PAGE for Western blot analysis using anti-XPA antibody. D, a, cells were irradiated with 20 J/m2 of UV and the cytoplasmic fraction (lane 1) was isolated. The nuclear pellet was then sequentially extracted with buffer of increasing salt concentration (lanes 2-5). NM, nuclear matrix (lane 6). b, nuclear extracts were prepared at indicated times after 20 J/m2 UV treatment of cells. c, cells were irradiated with indicated doses of UV and the nuclear extracts were prepared at 4 hours after UV treatment.
We next examined the cellular localization of the phosphorylated XPA. As shown in Fig. 1C, phosphorylated XPA was overwhelmingly located in the nucleus, likely as the chromatin-bound protein, whereas the cytoplasmic fraction contained little, if any, phosphorylated XPA. Consistently, phosphorylated XPA seemed to be more resistant to the salt extraction of nuclei compared with native XPA as more phosphorylated XPA was extracted with higher concentration of salt in nuclear lysates (Fig. 1D (a)). In time-course experiments, it was found that whereas limited fraction of phosphorylated XPA was observed at 2 hours after UV exposure (20 J/m2), phosphorylation of XPA was much more apparent at 4 hours and after (Fig. 1D (b)). The phosphorylation increased gradually and then reached a maximum after 16 hours and persisted until the 24th hours. The XPA phosphorylation also occurred in an UV dose-dependent manner (5 to 40 J/m2) as shown in Fig. 1D (c). In addition, experiments with two other cell lines (HeLa and MCF7) exhibited similar XPA phosphorylation induced by UV irradiation (data not shown).
Phosphorylation of XPA by PIKK family kinases in vitro. The above observations prompted us to identify the kinase(s) responsible for phosphorylation of XPA. Because XPA phosphorylation was UV-inducible and the protein contained two S/TQ sequences, the consensus target for PIKK family kinases (21, 22), we explored the possibility of XPA phosphorylation by checkpoint kinases ATR/ATM and another PIKK family member, DNA-PK. Thus, in vitro kinase assays were done for recombinant XPA with DNA-PK isolated from HeLa cells (Promega) and ATR/ATM immunoprecipitated from cell lysates. As shown in Fig. 2A, DNA-PK did phosphorylate XPA in the presence of [g-32P]ATP under in vitro experimental conditions (lanes 2-4), albeit the phosphorylation efficiency was relatively low in comparison with that of RPA32, which contains multiple S/TQ motifs and undergoes hyperphosphorylation in response to DNA damage (Fig. 2A, lane 1; ref. 40). The in vitro phosphorylation of XPA by DNA-PK also altered the mobility of XPA on SDS-PAGE in the same way as the endogenously phosphorylated XPA (Fig. 2B).
Figure 2.
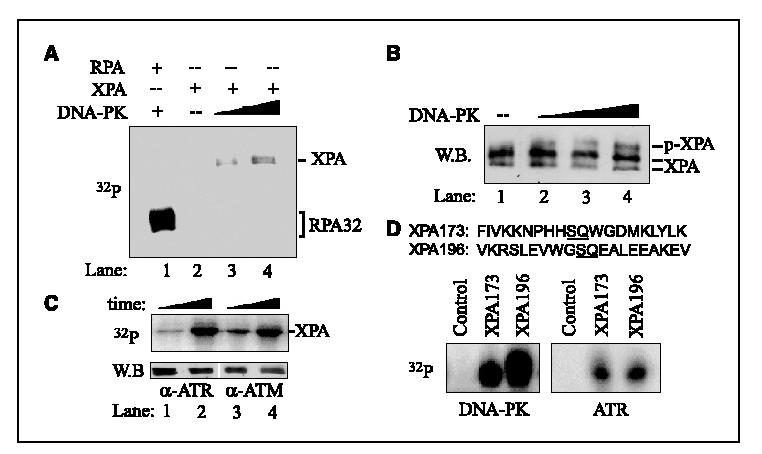
Phosphorylation of XPA in vitro. A, equal amount (40 pmol) of RPA (lane 1) and XPA (lanes 2-4) were incubated in a 20-μL reaction, respectively, with 50 units (lanes 1 and 3) or 100 units (lane 4) of DNA-PK for 30 minutes at 30°C in the presence of [γ32P]ATP. The radiolabeled proteins were visualized using PhosphorImager following SDS-PAGE. B, the kinase assays were done in 20-μL reaction system containing 200 nmol/L XPA, 100 Amol/L cold ATP, and 50 to 200 units of DNA-PK for 30 minutes at 30°c. The phosphorylated XPA was visualized by Western blotting (W.B.) with anti-XPA antibody. C, the immunoprecipitation kinase assays were done as described in Materials and Methods. Cells were first treated with 40 J/m2 UV irradiation, and then endogenous ATR and ATM were immunoprecipitated by anti-ATR and ATM antibodies, respectively. The ATR and ATM immunoprecipitates were then resuspended in 20 μL of kinase buffer containing 10 μCi of [γ-32P]ATP and 1 μg of purified His-XPA protein and incubated at 30°C for 10 to 30 minutes. The radiolabeled XPA was visualized following SDS-PAGE (top). Immunoprecipitated endogenous ATM or ATR was confirmed by Western blotting (bottom). D, the kinase assays were done as above except that the XPA peptides, XPA173 and XPA196, were used. The control peptide was the XPA173 peptide with a substitution of the serine to alanine.
We next employed the immunoprecipitation kinase assay to assess the phosphorylation of XPA by checkpoint kinases ATR and ATM. The endogenous ATR/ATM immunoprecipitated from cell lysates were incubated with purified XPA in the presence of [g-32P]ATP, followed by analysis on SDS-PAGE. As shown in Fig. 2C (top), significant phosphorylation of XPA by endogenous ATR and ATM was observed. The presence of ATR/ATM in immunoprecipitates was confirmed by Western blotting using antibodies against ATR and ATM, respectively (Fig. 2C, bottom).
Given that XPA protein contains two SQ motifs, S173Q and S196Q, both being potential phosphorylation sites for PIKK kinases, it was of interest to determine if both sites were phosphorylatable. Therefore, kinase phosphorylation assays were done with synthesized short peptides (20-mer) with the sequences at Ser173 and Ser196 of XPA, respectively. Both peptides seemed to be good substrates for DNA-PK, ATR (Fig. 2D), and ATM (data not shown). The much higher efficiency of phosphorylation by DNA-PK than by ATR was due to the use of purified DNA-PK in the assays in comparison with the ATR obtained from immunoprecipitation, which usually produced only limited amount of the targeted protein.
ATR was the major kinase responsible for the cellular phosphorylation of XPA following UV irradiation. We rationalized that the kinase(s) responsible for the cellular phosphorylation of XPA should be biologically and functionally relevant to NER pathway. Although ATR/ATM and DNA-PK exhibit similar substrate preferences in vitro, they play distinct biological roles in cells. Because the primary functions of DNA-PK are in double-strand break repair and V(D)J recombination, we next made efforts to explore the potential involvement of ATR and ATM kinases in the cellular phosphorylation of XPA following UV treatment. For this purpose, wortmannin and caffeine, two widely used checkpoint kinase inhibitors, were first employed in our experiments (41, 42). Figure 3A (lanes 3 and 4) showed that both treatments significantly compromised the UV-induced XPA phosphorylation, indicating that cellular XPA phosphorylation was indeed carried out by checkpoint kinase(s).
Figure 3.
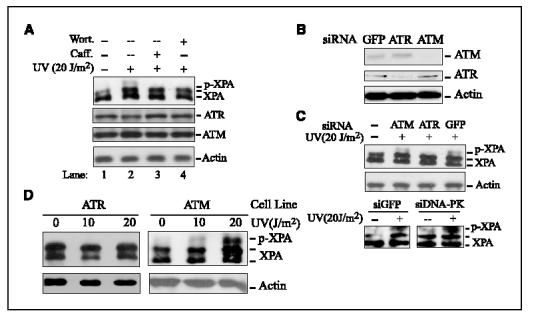
ATR is the major kinase responsible for cellular XPA phosphorylation after UV irradiation. A, A549 cells were mock treated (lane 1) or treated with 20 J/m2 UV irradiation, and then further incubated for 4 hours in the presence of 100 Amol/L wortmannin (Wort; lane 4) or 10 mmol/L caffeine (Caff; lane 3) before harvesting. Total cell lysates were used for Western blot analysis with anti-XPA, anti-ATR, and anti-ATM, respectively. B, A549 cells were transfected with ATR, ATM, or green fluorescent protein (GFP) siRNA as described in Materials and Methods. Total cell lysates were harvested 72 hours after transfection and probed with the indicated antibodies, respectively. C, A549 or HeLa cells were transfected with indicated siRNA and then treated with 20 J/m2 UV irradiation at 72 hours after transfection. Total cell lysates were immunoblotted with anti-XPA and antiactin antibodies, respectively. D, ATR- and ATM-deficient cells were treated with the indicated doses of UV and total cell lysates were prepared at 4 hours after treatment for Western blotting with anti-XPA and antiactin, respectively.
To further distinguish the roles of ATR and ATM in the XPA phosphorylation event, we adopted the technique of siRNA-mediated gene repression to specifically knock down the expression of ATR or ATM in cells. Transfection of cells with siRNA oligonucleotides complementary to ATR or ATM mRNAs resulted in >85% reduction of cellular ATR and ATM expression levels, respectively, as compared with cells transfected with siRNA targeting green fluorescent protein (Fig. 3B). The transfected cells were then treated with UV irradiation. As shown in Fig. 3C, XPA phosphorylation was only marginally affected by ATM knockdown whereas ATR siRNA transfection almost completely eliminated the UV-induced phosphorylation of XPA. However, it should be noted that the difference in XPA phosphorylation with siRNAs for ATR and ATM could be affected by the different knockdown efficacy of the siRNAs. We also did the same experiment with DNA-PK siRNA. Result same as that with ATM siRNA was obtained, indicating that the knockdown of DNA-PK had no apparent effect on the phosphorylation of XPA in vivo (Fig. 3C). We then went further to confirm the dependence of UV-induced cellular XPA phosphorylation on the expression of ATR by using ATR- and ATM-deficient cell lines. As revealed in Fig. 3D, the XPA was phosphorylated in UV-irradiated ATM cells as in normal cells but no phosphorylation was observed for XPA in ATR cells after UV exposure.
ATR and XPA interacted and colocalized in cells after UV treatment. To examine the possible cellular interaction of XPA with ATR, coimmunoprecipitation assays were done using total cell lysates prepared from mock-treated and 20 J/m2 UV-irradiated cells. As shown in Fig. 4A, the ATR antibody could efficiently immunoprecipitate XPA from cell lysates whereas rabbit immunoglobulin G could not, indicating that the coimmunoprecipitation of ATR with XPA was not the result of nonspecific antibody binding. Notably, UV irradiation seemed to moderately enhance the interaction between ATR and XPA. In the reciprocal experiments, XPA antibody was able to immunoprecipitate ATR (Fig. 4B, top) and, when recombinant His-XPA was added into cell lysates, endogenous ATR also was pulled down by the exogenous protein (Fig. 4B, bottom). These data suggest that ATR and XPA may interact with each other in cells. However, it remains possible that the interaction could be mediated by other proteins or the independent binding of both proteins to chromatin. To test these possibilities, we carried out the following experiments (13, 15). After immunoprecipitation with anti-ATR antibodies, the immunoprecipitates were washed with buffer of high salt concentration (0.6 mol/L NaCl) to disrupt protein-protein and protein-DNA interactions (Fig. 4C, lane 2, shows the loss of XPA from the complex following the high salt wash). Then recombinant His-XPA was supplied to allow for interaction with the endogenous ATR in normal buffer. Under these conditions, the interaction of His-XPA with ATR was clearly observed (Fig. 4C). These results suggest that XPA and ATR may interact directly with one another. In addition, coimmunoprecipitation of XPA with the Rad9/Rad1/Hus1 checkpoint complex was not detected under our experimental conditions (data not shown) although their counterparts in budding yeast (Rad14 and Rad17-Mec3-Ddc1, respectively) have been shown to interact with one another (31).
Figure 4.
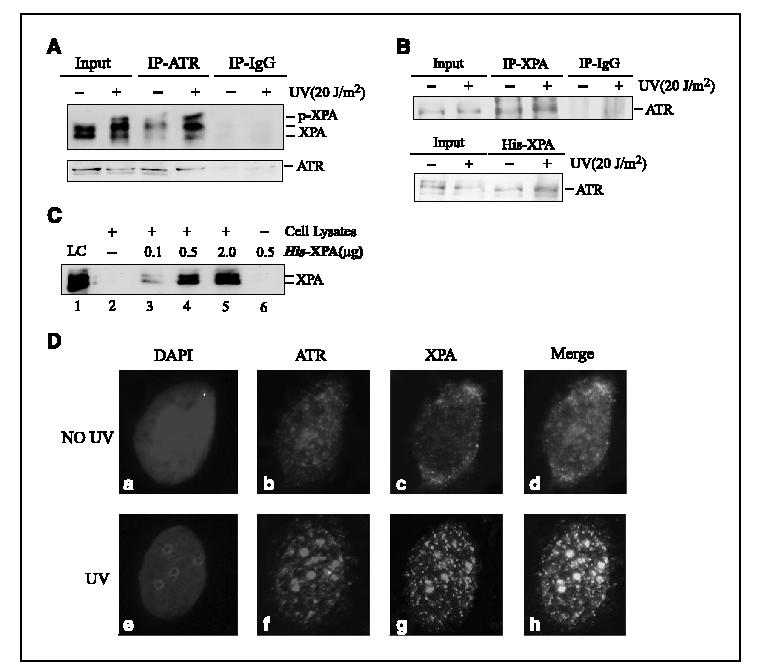
ATR interaction and colocalization with XPA in cells after UV irradiation. A, total cell lysates prepared from UV-treated or mock-treated A549 cells were used for coimmunoprecipitation assays with anti-ATR antibody. Proteins from the immunoprecipitates were detected by Western blotting using anti-XPA and anti-ATR antibodies. As controls, 10% of the total volumes of the whole cellular lysates used for the coimmunoprecipitation were also included. B, top, total cell lysates prepared from A549 cells were used for coimmunoprecipitation assays with anti-XPA antibody; bottom, whole-cell extracts prepared from 2 × 106 cells were mixed with 2 μg of His-XPA and incubated at 4°c for 10 to 14 hours. The XPA-bound proteins were probed by anti-ATR antibody. C, total cellular lysates were incubated with anti-ATR antibodies for 4 to 6 hours, followed by 1-hour incubation with protein A/G-agarose beads. The immunoprecipitates were washed thrice with PBS containing 0.5% NP40 and further incubated with the buffer of high concentration of salt [15 mmol/L Tris-Cl (pH 7.5), 0.6 mol/L NaCl, 0.1% NP40] for 30 minutes at 4°c. After centrifugation and washing, purified His-XPA was added and further incubated in 500 AL of XPA binding buffer [40 mmol/L HEPES-KOH (pH 7.5), 75 mmol/L KCl, 8 mmol/L MgCl2, 1 mmol/L DTT, 5% glycerol and 100 Ag/mL bovine serum albumin, 0.1% NP40] for 4 to 6 hours. The bound proteins were detected by Western blotting with anti-XPA antibody. LC, loading control (20 ng purified His-XPA). D, cells were treated with 20 J/m2 UV or mock treated, followed by 4-hour incubation. After extraction of cytoplasmic proteins with PBS containing 0.5% NP40, cells were fixed and incubated with anti-XPA and anti-ATR antibodies. Cells were then stained with fluorescent dye-linked secondary antibodies and visualized by fluorescence microscopy. b and f, red, anti-ATR stained cells; c and g, green, anti-XPA stained cells; d and h, merged images of the anti-XPA and anti-ATR stained cells; a and e, 4‘,6-diamidino-2-phenylindole-stained nuclei.
We next characterized the cellular association of ATR with XPA using immunofluorescent staining assays. In the cells without UV irradiation, immunostaining with ATR antibody revealed a few bright nuclear spots [Fig. 4D (B)], probably reflecting the proposed role of ATR in monitoring genome integrity during normal cell cycle progression (43). In contrast, XPA seemed to be homogeneously distributed throughout the nucleus without formation of apparent bright foci [Fig. 4D (C)]. On exposure to UV, a clear redistribution of ATR to nuclei occurred and brighter nuclear foci were formed [Fig. 4D (F)]. In the case of XPA, UV irradiation induced significant nuclear focus formation [Fig. 4D (G)]. When overlapped, ATR and XPA foci displayed extensive colocalization [Fig. 4D (H)], suggesting that both proteins could reside in the same complex after UV irradiation.
Defects in ATR-dependent XPA phosphorylation increases the cell sensitivity to UV irradiation. We then went further to examine the biological significances of XPA phosphorylation in vivo. Several expression plasmids for producing XPA-mutant proteins in human cells were constructed. In these constructs, either Ser173 or Ser196 was substituted with alanine, or both were replaced, to generate XPA-S173A, XPA-S196A, and XPA-S173A.S196A mutants, respectively. These XPA-mutant constructs, together with the one for expression of XPA-WT, were transfected into XPA-deficient cells. Stably transfected cell lines were selected with neomycin and Western blotting showed the comparable level of XPA expression in these cell lines (data not shown).
Our in vitro kinase assays with XPA peptides suggested that Ser173 and Ser196 were the potential phosphorylation sites (Fig. 2D). To unambiguously define the phosphorylation residues in XPA, the expressed exogenous XPA was precipitated from total cellular lysates of stably transfected cells with Nickel-NTA resin (Qiagen; all expressed XPA contain a His tag), and then purified DNA-PK was supplied and incubated in the kinase buffer. Under theses conditions, we found that XPA-S173A, XPA-S196A, and XPA-WT all could be phosphorylated efficiently by DNA-PK in vitro, confirming that both residues are phosphorylable in vitro (Fig. 5A, top, lane 1-3). However, double mutation of Ser173 and Ser196 to alanine completely eliminated the phosphorylation of XPA by DNA-PK (Fig. 5A, top, lane 4), suggesting that phosphorylation of XPA exclusively occurred at these two sites. To determine the in vivo phosphorylation site(s) of XPA after UV irradiation, cells were cultured in the presence of 32P-radiolabeled orthophosphate after UV exposure. The XPA was precipitated from cell lysates and separated by SDS-PAGE. As shown in Fig. 5B, mutation of Ser196 alone was sufficient to cause the loss of the radioactive band (top) and the up-shifted band (bottom) of XPA induced by UV irradiation. On the other hand, Ser173A mutation had little, if any, effect on the phosphorylation of XPA. Based on these results, we concluded that phosphorylation of XPA primarily occurred at Ser196 residue in vivo although both Ser173 and Ser196 could be phosphorylated in vitro.
Figure 5.
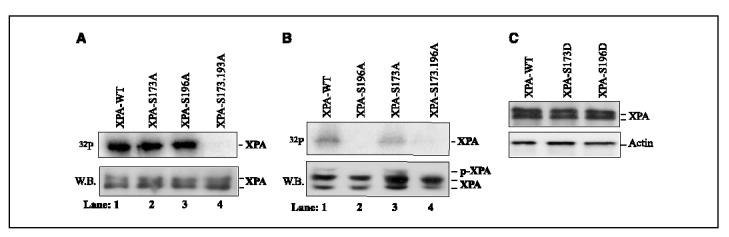
Identification of XPA phosphorylation sites. A, XPA-deficient cells were transfected with expression constructs of XPA-WT, XPA-S173A, XPA-S196A, and XPA-S173.196A, respectively. The stably transfected cells were selected as described in Materials and Methods. Total cell lysates were prepared and incubated with Nickel-NTA resin (Qiagen) for 1 hour at 4°c. After centrifugation, the precipitates were washed thrice with PBS containing 0.05% NP40 and once with kinase buffer. The precipitates were resuspended in 20 AL kinase buffer and the kinase reactions were done for 30 minutes at 30°c in the presence of 100 units DNA-PK and [γ-32P]ATP. The radiolabeled proteins were visualized following SDS-PAGE (top). The precipitated His-XPA was confirmed by Western blotting with anti-XPA antibody (bottom). B, transfected XPA cells were grown overnight in phosphate-depleted medium before irradiation with 20 J/m2 UV. Then, 32P-labeled orthophosphoric acid was added and cells were further incubated for 8 hours before harvest. Total cell lysates were prepared and precipitated with Nickel-NTA resin at 4°c. After extensive washing, the precipitates were separated on SDS-PAGE and radiolabeled proteins were detected (top). Precipitated exogenous XPA was probed by Western blotting (bottom). C, XPA-deficient cells were stably transfected with XPA-WT, XPA-S173D, and XPA-S196D constructs, respectively. Total cell lysates were prepared and probed with anti-XPA and antiactin antibodies, respectively.
To investigate if the phosphorylation of XPA could alter the cellular response to UV irradiation, an MTT assay was first applied to determine the cell viability after UV treatment. Strikingly, replacement of Ser196 of XPA with alanine increased the UV sensitivity of the cells whereas Ser173A mutation resulted in little effects on cell viability (Fig. 6A). Double mutation of Ser173 and Ser196 to alanine displayed the similar effects as the single mutation of Ser196A, confirming that the phosphorylation of Ser196 of XPA is physiologically important for cell survival. Interestingly, all XPA mutants and WT displayed the similar sensitivity to camptothecin, a topoisomerase inhibitor that induces DNA double-strand breaks (Fig. 6B). This strongly suggests that the phosphorylation of XPA is specifically in response to NER-related DNA damage, which is fully consistent with the unique role of XPA in NER. We also determined the colony formation of the cells expressing XPA and its mutantsafter UV treatment. As shown in Fig. 6C, the XPA-S173A and XPA-WT cells showed the similar efficiency in forming colonies whereas, for both XPA-S196A and XPA-S196.173A cells, the number of colonies was significantly reduced after the same dose of UV treatment. In contrast to the moderate effect of S196A mutation on the cell viability against UV in MTT assay, the much more dramatic effect with colony formation assay was due to the fact that this assay took a much longer period (2 weeks) to complete, which was a measurement of accumulated effect on cell survival rate after a long-term incubation of irradiated cells.
Figure 6.

UV and camptothecin (CPT) sensitivities of cells with XPA mutants. A, transfected XPA cells were seeded in 96-well plates and allowed to attach overnight. The cells were then treated with indicated doses of UV irradiation. After treatment, cells were washed once with PBS and further incubated for 72 hours. Aliquots of 10 μL of MTT (5 mg/mL) were then added to each well. After 4 hours of incubation, intensity of the formed color was quantitated by a spectrophotometric plate reader at 490 nm after solubilization in 150 AL of DMSO. Data were normalized to the controls (as the value of 1). Points, mean of four different measurements; bars, SD. B, cells were incubated with indicated doses of camptothecin for 8 hours, and then washed with PBS and further cultured for 24 hours. The MTT assays were done as above. C, colony formation assays of cells with phosphorylation-deficient XPA mutants following UV irradiation. XPA-deficient cells were transfected with constructs for cellular expression of XPA-WT, XPA-S173A, XPA-S196A, and XPA-S173.196A, respectively. The stably transfected cells were obtained as described in Materials and Methods. Five hundred cells were then plated to each dish. After overnight attachment, cells were irradiated with indicated doses of UV and then allowed to grow for 2 weeks. The colonies formed were counted under microscope. The data were normalized to the controls (as the value of 1). Points, mean of three different measurements; bars, SD. D, colony formation assays were done as described in (C).
To further characterize the biological functions of XPA phosphorylation, we constructed two plasmids for cellular expression of XPA phosphorylation-mimicking mutants, in which Ser173 and Ser196 were substituted with Aspartic acid, respectively. After stable transfection of the constructs into XPA-deficient cells, total cellular lysates were prepared to probe the expression level of XPA. We observed similar expression levels of XPA-S173D, XPA-S196D, and XPA-WT (Fig. 5C). Then colony formation assays were done. As shown in Fig. 6D, on UV exposure, the XPA-S196D cells were more likely to form cell colonies than XPA-WT although the aspartic acid could not completely mimic the phosphoryl group in cells. This enhancement in cellular resistance to UV due to the S196D mutation was significant as determined by the t test (P = 0.011 <0.05 at 5 J/m2). In contrast, the increase in colony formation for XPA-S176D mutant, as compared with XPA-WT, was not significant (P = 0.075 >0.05 at 5 J/m2). Taken together, these results suggest that the ATR-dependent phosphorylation of XPA at Ser196 is important for cell survival in response to UV damage, probably by fine-tuning the NER activity in cells.
Discussion
Despite recent significant advances in understanding the molecular mechanisms of NER and ATR/ATM checkpoints, the relationship between the two major DNA damage response pathways in cells remains elusive. In particular, the cellular regulation of NER in response to DNA damage is poorly understood. The unphosphorylated form of XPA is a unique factor in NER and is indispensable for both subpathways (global genome repair and transcription-coupled repair) of NER in cells (6-8). Also importantly, the predominant cellular function of XPA is its role in NER. It is generally believed that XPA, likely in dimer (36), is involved in DNA damage recognition and also in the recruitment of other NER factors, such as RPA, XPAG, and XPF/ERCC1, to the DNA damage site to form dual incision complexes through protein-protein interactions (8, 10). Therefore, it is conceivable that modulation of XPA function may exclusively affect the cellular NER activity. The results from our study, for the first time, indicated that the checkpoint kinase ATR was able to interact with XPA and to actively regulate the phosphorylation of XPA in response to DNA damage, which seemed to be important for cell viability following UV irradiation. Our results suggest that the phosphorylation of XPA at residue Ser196 may play an important role in the DNA damage responses and in modulation of the cellular NER activity.
Several recent studies have reported that protein phosphorylation occurring in cells may modify the cellular NER activity, either positively or negatively. Specifically, phosphorylation of xeroderma pigmentosum group B helicase by casein kinase II has been suggested to impair the NER function of transcription factor IIH (44) whereas overexpression of protein kinase C or activation of extracellular signal-regulated kinase 1/2 signaling stimulated NER activity (45, 46). Here we show that XPA was phosphorylated by checkpoint kinase ATR in cells after UV irradiation although the possibility cannot be completely excluded that XPA could be phosphorylated by the downstream kinase(s) of ATR (e.g., Chk1). Although the observed fraction of XPA under phosphorylation was relatively small for the reason described below, the phosphorylation occurred in an UV dose-dependent manner (Fig. 1D (c)). Interestingly, the same phosphorylation could not be induced by camptothecin, a topoisomerase inhibitor causing double-strand break production in cells (data not shown). This confirmed that XPA phosphorylation was a NER-related unique event. It is also worth noting that a relatively low level of phosphorylated XPA was present in cells even without UV treatment. This could be possibly due to a low level activation of ATR in S phase with the recruitment of ATR to chromatin for monitoring genome integrity during normal S-phase progression in unperturbed cells (43, 47).
The observation that phosphorylated XPA was predominantly located in nucleus as the chromatin-bound protein indicated that either the XPA became phosphorylated by ATR after localization to nucleus or the phosphorylated XPA preferentially translocated to nucleus. The former was likely true as ATR was recruited to chromatin even in the absence of DNA damage (43) and XPA phosphorylation was induced by DNA damage. Phosphorylation also converted XPA into a more extraction-resistant form, suggesting a preferred engagement of phosphorylated XPA in NER reaction. It should be noted, however, that the phosphorylation was not significant in the early post-UV exposure hours (<4 hours), during which the cellular NER function was believed to be very active (Fig. 1D (b)). This suggests that the phosphorylation of XPA may not be required for NER in the early cellular response to UV irradiation. Nevertheless, a substantial portion of XPA became phosphorylated at the later periods. A possible explanation for this is that when DNA lesions become persistent in UV-irradiated cells (6), checkpoint kinase ATR could be translocated to the sites of DNA damage where the replication forks stalled. Thus, ATR was activated and subsequently phosphorylated DNA damage-associated proteins such as XPA. The apparent interaction and colocalization of ATR with XPA following UV irradiation (Fig. 4) and the preferential binding of ATR to UV-damaged DNA support this notion (48). Moreover, the phosphorylated XPA interacted with RPA more efficiently as analyzed by coimmunoprecipitation assays (data not shown). Based on these observations, we propose that phosphorylation of XPA may play a role in facilitating the cellular function of NER in the late stage for removal of persistent DNA lesions (6), which is critical for cell survival. Because most of XPA molecules in cells are involved in normal NER, which does not require the phosphorylated form of XPA (8), the XPA subjected to phosphorylation for repair of persistent DNA damage is probably only a small fraction of total XPA in cells.
In support of our model, substitution of Ser196 of XPA with alanine resulted in substantial decrease in the cell survival in response to UV irradiation. In contrast, phosphorylation of Ser173 of XPA was absent in cells, and thus the mutation of Ser173 to alanine did not alter the cellular UV resistance (Fig. 6A and C). In addition, replacement of Ser196 with aspartic acid to mimic the phosphorylated XPA did enhance the cell survival after UV treatment as compared with XPA-WT (Fig. 6D). The backup or fine-tuning role of XPA phosphorylation in NER seems to be supported by the moderate effect of the S196A of XPA mutation on the cell survival of UV determined by the MTT assay. The much more dramatic decrease in cellular UV sensitivity due to the S196A mutation in colony formation assay was likely attributed to the measurement of a long-term (2 weeks) accumulated effect of the mutation on the cell viability by the assay. Taken together, we propose that the phosphorylation of XPA by ATR checkpoint may positively regulate NER activity and thus may facilitate the cells to recover from NER-related DNA damages.
The radiosensitive phenotype of ATR kinase-dead cells has been ascribed to the double-strand break repair defects (27). Interestingly, such cells and the ATR-deficient cells both displayed an UV-sensitive phenotype (33, 49). Because the loss of cell cycle checkpoints themselves did not result in decreased cell viability after DNA damage (50), our results herein suggest that the XPA phosphorylation defect in ATR-deficient cells may contribute to its UV-sensitive phenotype.
Acknowledgments
Grant support: National Cancer Institute grant CA86927 (Y. Zou). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact. We thank Dr. Margie Tucker for critical reading of the manuscript.
REFERENCES
- 1.Bartek J, Lukas C, Lukas J. Checking on DNA damage in S phase. Nat Rev Mol Cell Biol. 2004;5:792–804. doi: 10.1038/nrm1493. [DOI] [PubMed] [Google Scholar]
- 2.Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432:316–23. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- 3.Wood RD, Mitchell M, Sgouros J, Lindahl T. Human DNA repair genes. Science. 2001;291:1284–9. doi: 10.1126/science.1056154. [DOI] [PubMed] [Google Scholar]
- 4.Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–9. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 5.Cline SD, Hanawalt PC. Who’s on first in the cellular response to DNA damage? Nat Rev Mol Cell Biol. 2003;4:361–72. doi: 10.1038/nrm1101. [DOI] [PubMed] [Google Scholar]
- 6.Costa RM, Chigancas V, Galhardo Rda S, Carvalho H, Menck CF. The eukaryotic nucleotide excision repair pathway. Biochimie. 2003;85:1083–99. doi: 10.1016/j.biochi.2003.10.017. [DOI] [PubMed] [Google Scholar]
- 7.Thoma BS, Vasquez KM. Critical DNA damage recognition functions of XPC-hHR23B and XPARPA in nucleotide excision repair. Mol Carcinog. 2003;38:1–13. doi: 10.1002/mc.10143. [DOI] [PubMed] [Google Scholar]
- 8.Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 9.Wu X, Fan W, Xu S, Zhou Y. Sensitization to the cytotoxicity of cisplatin by transfection with nucleotide excision repair gene xeroderma pigmentosun group A antisense RNA in human lung adenocarcinoma cells. Clin Cancer Res. 2003;9:5874–9. [PubMed] [Google Scholar]
- 10.Wood RD. DNA damage recognition during nucleotide excision repair in mammalian cells. Biochimie. 1999;81:39–44. doi: 10.1016/s0300-9084(99)80036-4. [DOI] [PubMed] [Google Scholar]
- 11.Patrick SM, Turchi JJ. Xeroderma pigmentosum complementation group A protein (XPA) modulates RPA-DNA interactions via enhanced complex stability and inhibition of strand separation activity. J Biol Chem. 2002;277:16096–101. doi: 10.1074/jbc.M200816200. [DOI] [PubMed] [Google Scholar]
- 12.Liu Y, Liu Y, Yang Z, et al. Cooperative interaction of human XPA stabilizes and enhances specific binding of XPA to DNA damage. Biochemistry. 2005;44:7361–8. doi: 10.1021/bi047598y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu X, Yang Z, Liu Y, Zou Y. Preferential localization of hyperphosphorylated replication protein A to double-strand break repair and checkpoint complexes upon DNA damage. Biochem J. 2005;391:473–80. doi: 10.1042/BJ20050379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rouse J, Jackson SP. Interfaces between the detection, signaling, and repair of DNA damage. Science. 2002;297:547–51. doi: 10.1126/science.1074740. [DOI] [PubMed] [Google Scholar]
- 15.Wu X, Shell SM, Zou Y. Interaction and colocalization of Rad9/Rad1/Hus1 checkpoint complex with replication protein A in human cells. Oncogene. 2005;24:4728–35. doi: 10.1038/sj.onc.1208674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15:2177–96. doi: 10.1101/gad.914401. [DOI] [PubMed] [Google Scholar]
- 17.Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22:7265–79. doi: 10.1038/sj.onc.1206933. [DOI] [PubMed] [Google Scholar]
- 18.Kastan MB, Lim DS. The many substrates and functions of ATM. Nat Rev Mol Cell Biol. 2000;1:179–86. doi: 10.1038/35043058. [DOI] [PubMed] [Google Scholar]
- 19.Anderson L, Henderson C, Adachi Y. Phosphorylation and rapid relocalization of 53BP1 to nuclear foci upon DNA damage. Mol Cell Biol. 2001;21:1719–29. doi: 10.1128/MCB.21.5.1719-1729.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lou Z, Minter-Dykhouse K, Wu X, Chen J. MDC1 is coupled to activated CHK2 in mammalian DNA damage response pathways. Nature. 2003;421:957–61. doi: 10.1038/nature01447. [DOI] [PubMed] [Google Scholar]
- 21.Kim ST, Lim DS, Canman CE, Kastan MB. Substrate specificities and identification of putative substrates of ATM kinase family members. J Biol Chem. 1999;274:37538–43. doi: 10.1074/jbc.274.53.37538. [DOI] [PubMed] [Google Scholar]
- 22.O’Neill T, Dwyer AJ, Ziv Y, et al. Utilization of oriented peptide libraries to identify substrate motifs selected by ATM. J Biol Chem. 2000;275:22719–27. doi: 10.1074/jbc.M001002200. [DOI] [PubMed] [Google Scholar]
- 23.Chen G, Yuan SS, Liu W, et al. Radiation-induced assembly of Rad51 and Rad52 recombination complex requires ATM and c-Abl. J Biol Chem. 1999;274:12748–52. doi: 10.1074/jbc.274.18.12748. [DOI] [PubMed] [Google Scholar]
- 24.Morrison C, Sonoda E, Takao N, Shinohara A, Yamamoto K, Takeda S. The controlling role of ATM in homologous recombinational repair of DNA damage. EMBO J. 2000;19:463–71. doi: 10.1093/emboj/19.3.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang J, Willers H, Feng Z, et al. Chk2 phosphorylation of BRCA1 regulates DNA double-strand break repair. Mol Cell Biol. 2004;24:708–18. doi: 10.1128/MCB.24.2.708-718.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuhne M, Riballo E, Rief N, Rothkamm K, Jeggo PA, Lobrich M. A double-strand break repair defect in ATM-deficient cells contributes to radiosensitivity. Cancer Res. 2004;64:500–8. doi: 10.1158/0008-5472.can-03-2384. [DOI] [PubMed] [Google Scholar]
- 27.Wang H, Wang H, Powell SN, Iliakis G, Wang Y. ATR affecting cell radiosensitivity is dependent on homologous recombination repair but independent of nonhomologous end joining. Cancer Res. 2004;64:7139–43. doi: 10.1158/0008-5472.CAN-04-1289. [DOI] [PubMed] [Google Scholar]
- 28.Smilenov LB, Lieberman HB, Mitchell SA, Baker RA, Hopkins KM, Hall EJ. Combined haploinsufficiency for ATM and RAD9 as a factor in cell transformation, apoptosis, and DNA lesion repair dynamics. Cancer Res. 2005;65:933–8. [PubMed] [Google Scholar]
- 29.DeMase D, Zeng L, Cera C, Fasullo M. The Saccharomyces cerevisiae PDS1 and RAD9 checkpoint genes control different DNA double-strand break repair pathways. DNA Repair (Amst) 2005;4:59–69. doi: 10.1016/j.dnarep.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 30.Haghnazari E, Heyer WD. The DNA damage checkpoint pathways exert multiple controls on the efficiency and outcome of the repair of a double-stranded DNA gap. Nucleic Acids Res. 2004;32:4257–68. doi: 10.1093/nar/gkh717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Giannattasio M, Lazzaro F, Longhese MP, Plevani P, Muzi-Falconi M. Physical and functional interactions between nucleotide excision repair and DNA damage checkpoint. EMBO J. 2004;23:429–38. doi: 10.1038/sj.emboj.7600051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Neecke H, Lucchini G, Longhese MP. Cell cycle progression in the presence of irreparable DNA damage is controlled by a Mec1- and Rad53-dependent checkpoint in budding yeast. EMBO J. 1999;18:4485–97. doi: 10.1093/emboj/18.16.4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O’Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, Goodship JA. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat Genet. 2003;33:497–501. doi: 10.1038/ng1129. [DOI] [PubMed] [Google Scholar]
- 34.Yu S, Teng Y, Lowndes NF, Waters R. RAD9, RAD24, RAD16 and RAD26 are required for the inducible nucleotide excision repair of UV-induced cyclobutane pyrimidine dimers from the transcribed and non-transcribed regions of the Saccharomyces cerevisiae MFA2 gene. Mutat Res. 2001;485:229–36. doi: 10.1016/s0921-8777(01)00061-1. [DOI] [PubMed] [Google Scholar]
- 35.Jiang H, Yang LY. Cell cycle checkpoint abrogator UCN-01 inhibits DNA repair: association with attenuation of the interaction of XPA and ERCC1 nucleotide excision repair proteins. Cancer Res. 1999;59:4529–34. [PubMed] [Google Scholar]
- 36.Yang ZG, Liu Y, Mao LY, Zhang JT, Zou Y. Dimerization of human XPA and formation of XPA2-RPA protein complex. Biochemistry. 2002;41:13012–20. doi: 10.1021/bi026064z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Y, Kvaratskhelia M, Hess S, Qu Y, Zou Y. Modulation of replication protein A function by its hyperphosphorylation-induced conformational change involving DNA binding domain B. J Biol Chem. 2005;280:32775–83. doi: 10.1074/jbc.M505705200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Iakoucheva LM, Kimzey AL, Masselon CD, Smith RD, Dunker AK, Ackerman EJ. Aberrant mobility phenomena of the DNA repair protein XPA. Protein Sci. 2001;10:1353–62. doi: 10.1110/ps.ps.40101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miura N, Miyamoto I, Asahina H, Satokata I, Tanaka K, Okada Y. Identification and characterization of xpac protein, the gene product of the human XPAC (xeroderma pigmentosum group A complementing) gene. J Biol Chem. 1991;266:19786–9. [PubMed] [Google Scholar]
- 40.Binz SK, Sheehan AM, Wold MS. Replication protein A phosphorylation and the cellular response to DNA damage. DNA Repair (Amst) 2004;3:1015–24. doi: 10.1016/j.dnarep.2004.03.028. [DOI] [PubMed] [Google Scholar]
- 41.Sarkaria JN, Tibbetts RS, Busby EC, Kennedy AP, Hill DE, Abraham RT. Inhibition of phosphoinositide 3-kinase related kinases by the radiosensitizing agent wortmannin. Cancer Res. 1998;58:4375–82. [PubMed] [Google Scholar]
- 42.Sarkaria JN, Busby EC, Tibbetts RS, et al. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res. 1999;59:4375–82. [PubMed] [Google Scholar]
- 43.Dart DA, Adams KE, Akerman I, Lakin ND. Recruitment of the cell cycle checkpoint kinase ATR to chromatin during S-phase. J Biol Chem. 2004;279:16433–40. doi: 10.1074/jbc.M314212200. [DOI] [PubMed] [Google Scholar]
- 44.Coin F, Auriol J, Tapias A, Clivio P, Vermeulen W, Egly JM. Phosphorylation of XPB helicase regulates TFIIH nucleotide excision repair activity. EMBO J. 2004;23:4835–46. doi: 10.1038/sj.emboj.7600480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin YW, Chuang SM, Yang JL. Persistent activation of ERK1/2 by lead acetate increases nucleotide excision repair synthesis and confers anti-cytotoxicity and anti-mutagenicity. Carcinogenesis. 2003;24:53–61. doi: 10.1093/carcin/24.1.53. [DOI] [PubMed] [Google Scholar]
- 46.Louat T, Canitrot Y, Jousseaume S, et al. Atypical protein kinase C stimulates nucleotide excision repair activity. FEBS Lett. 2004;574:121–5. doi: 10.1016/j.febslet.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 47.Shechter D, Gautier J. ATM and ATR check in on origins: a dynamic model for origin selection and activation. Cell Cycle. 2005;4:235–8. [PubMed] [Google Scholar]
- 48.Unsal-Kacmaz K, Makhov AM, Griffith JD, Sancar A. Preferential binding of ATR protein to UV-damaged DNA. Proc Natl Acad Sci U S A. 2002;99:6673–8. doi: 10.1073/pnas.102167799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wright JA, Keegan KS, Herendeen DR, et al. Protein kinase mutants of human ATR increase sensitivity to UV and ionizing radiation and abrogate cell cycle checkpoint control. Proc Natl Acad Sci U S A. 1998;95:7445–50. doi: 10.1073/pnas.95.13.7445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Slichenmyer WJ, Nelson WG, Slebos RJ, Kastan MB. Loss of a p53-associated G1 checkpoint does not decrease cell survival following DNA damage. Cancer Res. 1993;53:4164–8. [PubMed] [Google Scholar]