Relative Influences of Crossing Over and Gene Conversion on the Pattern of Linkage Disequilibrium in Arabidopsis thaliana (original) (raw)
Abstract
In this article we infer the rates of gene conversion and crossing over in Arabidopsis thaliana from population genetic data. Our data set is a genomewide survey consisting of 1347 fragments of length 600 bp sequenced in 96 accessions. It has several orders of magnitude more markers than any previous nonhuman study. This allows for more accurate inference as well as a detailed comparison between theoretical expectations and observations. Our methodology is specifically set to account for deviations such as recurrent mutations or a skewed frequency spectrum. We found that even if some components of the model clearly do not fit, the pattern of LD conforms to theoretical expectations quite well. The ratio of gene conversion to crossing over is estimated to be around one. We also find evidence for fine-scale variations of the crossing-over rate.
SEVERAL recent articles have focused on estimating crossing-over rates from population genetic data (reviewed in Stumpf and McVean 2003). Recently, McVean et al. (2004) presented a methodology based on coalescent theory and Hudson's (2001) pseudo-likelihood, and Crawford et al. (2004) proposed a different approach based on an approximation of the coalescent process for which computations are tractable, originally described in Li and Stephens (2003). Both articles use Markov chain Monte Carlo computations and focus on human data.
Here we present a similar study for Arabidopsis thaliana, using data from a genomewide polymorphism survey (Nordborg et al. 2005). The data set is larger than that in any previous nonhuman study: in addition to making our estimates more reliable, this also allows us to assess the goodness of fit of our statistical model.
Because A. thaliana is highly selfing, it has unique advantages for estimating recombination rates (Nordborg 2000). First, the fact that most individuals are homozygous means that haplotypes can be observed directly (as opposed to being inferred from genotypes). Second, the high degree of inbreeding reduces the efficacy of recombination in breaking up linkage disequilibrium, without also reducing the level of polymorphism (Nordborg 2000). This increases power to detect recombination events, which should make inference more accurate (Hudson and Kaplan 1985).
Previous studies have shown that linkage disequilibrium in A. thaliana typically decays within 50 kb, but that it can be more extensive in particular genomic regions, as well as in some populations (Nordborg et al. 2002, 2005). It has also been argued that A. thaliana has a high rate of gene conversion (Haubold et al. 2002): characterizing the relative importance of crossing over and gene conversion in determining the pattern of linkage disequilibrium is one of the main aims of this article.
Our analysis proceeds in two steps. First, we assess the goodness of fit of different population genetics models and develop a model suitable for analyzing the data. Second, we use this model to estimate rates of crossing over and gene conversion.
MATERIALS AND METHODS
Data:
The data consist of 1347 short (500- to 600-bp) alignments of 96 accessions, generated as part of a genomewide polymorphism survey in A. thaliana (http://walnut.usc.edu/2010). The data differ from those described in Nordborg et al. (2005) in two ways. First, more genomewide fragments have been added. Second, to increase the number of markers at distances of 5–50 kb, data from nine more densely sequenced 500-kb regions were added (M. J. Aranzana, C. Tang, H. Zheng and M. Nordborg, unpublished results). These regions contain ∼20 fragments each, which should be contrasted with a median distance between fragments of 100 kb in the genomewide data. The 1347 fragments analyzed in this article are available as supplemental material at http://www.genetics.org/supplemental/.
For our estimation procedure we used strict criteria regarding the quality of the data and considered as missing sites when the combined forward–reverse base-calling score (Phred) was ≤25. Also two accessions (Ms-0 and Van-0) were removed because many of their loci were heterozygous, indicating recent outcrossing.
Simulations:
All simulations in this article use Hudson's software ms (Hudson 2002). Unless otherwise specified, we simulate sets of 1347 fragments. Each fragment is simulated independently. We first simulate a coalescent tree using appropriate demographic and recombination parameters and then add mutations according to the finite-sites model described below. The mutation rate for each fragment is chosen randomly from a prior that reproduces the variation in the level of polymorphism observed in the data.
Finite sites were incorporated by discretizing the location of the segregating sites produced by ms. Mutations that occurred at the same position after rounding were treated as recurrent mutations and were randomly designated as di- or triallelic with the probabilities estimated below (see model fitting).
Statistical procedures:
Several inference techniques are used in this article. To infer the recombination rate we used Hudson's composite likelihood (Hudson 2002). This procedure incorporates a varying population size model, but assumes no recurrent mutations. Because of this limitation we must be cautious about the interpretation of such estimates, particularly for short-range data where recurrent mutations can easily bias our estimates.
Alternatively, we use our best-fitting model to simulate data sets. Because a full-likelihood approach is not feasible, we measure the level of LD using various summary statistics. Our general approach consists of finding the range of evolutionary parameters that makes the observed statistics plausible.
Because it is difficult to estimate crossing-over and conversion rates simultaneously from the kind of data we have, our inference strategy consists of two steps: we first use the correlations between loci to estimate the recombination rate. Then, given this estimated recombination rate, we infer the amount of gene conversion needed to account for the observed pattern within the loci.
Summary statistics:
For distant markers (that do not belong to the same fragment) we use Lewontin's _D_′,
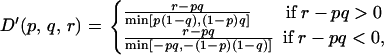
where p and q are the observed frequencies of the minor allele at the two SNPs, and r is the observed frequency of haplotypes with the minor allele at both SNPs.
When measuring linkage disequilibrium (LD) between closely spaced markers we find that _D_′ is very sensitive to recurrent mutations and is therefore not appropriate. We instead look at the number of fragments that show evidence of recombination. A fragment is called incompatible if any two SNPs within the fragment fail the four-gametes test. An equivalent definition is to say that the statistic _R_min (Hudson and Kaplan 1985; Myers and Griffiths 2003) is greater than or equal to one. In the absence of recurrent mutations such a pattern must be caused by a change in the underlying genealogical tree, which indicates either a crossing-over or a conversion event. We observed 330 incompatible fragments in our data set (∼25% of the total number of fragments).
Finally we use a statistic designed to be characteristic of the pattern produced by a single gene conversion event. More precisely, we looked for fragments with the property that a single crossing over cannot explain the data, but a single conversion can. We add the extra property that the conversion tract needs to include at least two SNPs to distinguish conversion from recurrent mutations. We call this pattern a clear conversion. This parsimony analysis was made possible by theoretical tools developed in Gusfield et al. (2004).
MODEL FITTING
The coalescent has become the standard model for analyzing population genetics data (Nordborg 2001; Stephens 2001). As with any model-based inference, a major concern is that the model does not fit the data; however, population geneticists have typically not had enough data to assess model fit. In the present case, it is clear that the standard coalescent does not explain the data (Nordborg et al. 2005). To improve the inference, we therefore begin by modifying the standard coalescent model to fit the data better. Specifically, we find that adding population growth and a finite-sites mutation model improve goodness of fit greatly.
We also observed that even if the average number of pairwise differences is comparable between each pair of accessions (around three differences per kilobase genomewide), two accessions are significantly different (around five differences per kilobase) and were removed (Cvi-0 and Mr-0). Also four pairs (Tamm-2 and Tamm-27, Lov-1 and Lov-5, Got-22 and Got-7, and Bil-5 and Bil-7) and one triplet of accessions (Knox-10, Pna-10, and RRS-10) are very similar (about one difference per kilobase). Bil-5, Got-22, Lov-1, Tamm-2, as well as Knox-10 and Pna-10 were therefore removed. With both heterozygote accessions removed we have thus 86 accessions left. We were unable to improve the fit by incorporating further population structure.
Frequency spectrum:
A useful summary of the data is the allele frequency spectrum. Several of the statistics we use to estimate recombination rates below are highly dependent on the frequency spectrum. For example, Lewontin's _D_′ is always one when a singleton allele is involved. Thus, it is very important that our model reproduces this component of the data as accurately as possible.
The frequency spectrum for our data is highly skewed toward low frequencies, compared to standard expectations (Figure 1). Another view of the same problem is the distribution of Tajima's D: the mean computed across fragments is −0.74, whereas the expectation under a constant population size is close to zero (Tajima 1989).
Figure 1.

The observed frequency spectrum in exons and noncoding regions, compared to coalescent expectations with and without growth.
The simplest explanation for a genomewide excess of rare alleles is demography, in particular some form of recent population growth. However, we note that although the bias is genomewide, it is stronger in exons, indicating that selection must also be responsible (Nordborg et al. 2005). We model the skew in the frequency spectrum using a simple growth model. Because of the evident effect of selection, we use only noncoding regions when fitting the data.
We investigated scenarios of recent exponential growth preceded by a constant population size (cf. Marjoram and Donnelly 1994). This model can be described by two parameters: the growth rate α and the time of the beginning of growth _t_0.
We designed a wide grid for the parameters α and _t_0 and estimated the expected frequency spectrum using simulations at each point of the grid. For each value of the parameters we simulated a large number (105) of coalescent trees. The mutation rate was set such that the average numbers of segregating sites in our simulations and in the data are equal. We assumed no crossing over, no conversion, and an infinitely many sites model (no recurrent mutations).
We compared the observed and simulated frequency distributions at each point in the grid using a χ2-statistic. We binned the SNPs in our data set into 10 bins according to the frequency of the minor allele. Bins were chosen to have a similar number of SNPs in each bin, so that our fitting procedure balances the weight given to rare and frequent alleles.
Including growth dramatically improved the overall goodness of fit. The value of χ2 dropped from 700 without growth to ∼15 for the best fit. On the basis of the frequency spectrum our simulations fit the data well. The best fit was obtained for α = 3 and time _t_0 = 0.22 (ms command line: ms 86 1 -t 5.4 -r 0.5 600 -G 3. -eG 0.22 0.), when normalized _t_0 is slightly less than one-third of the expected time to the most recent common ancestor. With these parameters the ratio r = 2 between the current and the ancestral population.
In the rest of this article we use these estimated growth parameters. When we refer to scaled rates, these are always scaled with respect to the ancestral population size (note that this notation differs from Hudson's ms).
Recurrent mutations:
Because recurrent mutations can lead to patterns of polymorphism similar to those expected under recombination (in particular under gene conversion), it is important to take them into account when estimating rates of recombination. We found clear evidence of recurrent mutations in the data. Consequently, we first estimated the amount of recurrent mutation and then used this estimate to calibrate our coalescent simulations.
Direct evidence for recurrent mutation in our data comes from sites showing more than two alleles. Barring sequencing errors, such sites must have mutated at least twice since the most recent common ancestor (MRCA). Out of 20,888 SNPs we found 411 triallelic and 4 quadriallelic sites.
Because of back mutations, a fraction of sites that have experienced recurrent mutation will be tri- or quadriallelic. This fraction can be estimated using the relative mutation rate between each nucleotide. These rates can be estimated by assuming that the most frequent allele is ancestral, an assumption that is motivated by classical population genetics theory (Watterson and Guess 1977). Using this assumption, we obtain the results presented in Table 1. Although crude, we note that we observe the expected symmetry (due to the A-T and G-C complementarity of both DNA strands) and also a very high rate of cytosine deamination (from C to T).
TABLE 1.
Relative frequencies of mutation
| A | T | G | C | π | |
|---|---|---|---|---|---|
| A | — | 0.34 | 0.43 | 0.23 | 0.301 |
| T | 0.32 | — | 0.21 | 0.46 | 0.301 |
| G | 0.64 | 0.22 | — | 0.14 | 0.199 |
| C | 0.21 | 0.65 | 0.14 | — | 0.199 |
Let r denote the proportion of double-hit mutations that result in a diallelic site. Then 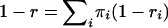 , where
, where 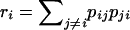 is the probability that a double hit results in a diallelic site given that the ancestral state is i. Thus, from Table 1 and the overall composition in terms of A, T, G, and C, we estimate that ∼60% of the recurrent mutations result in triallelic sites and that, consequently, the number of recurrent sites that are not triallelic is of the magnitude of (100 − 60)/60 × 411 ≈ 274. In other words, recurrent sites (di- and triallelic) represent 3–4% of the total number of segregating sites. Our simulations use a finite-sites model based on these results (see materials and methods).
is the probability that a double hit results in a diallelic site given that the ancestral state is i. Thus, from Table 1 and the overall composition in terms of A, T, G, and C, we estimate that ∼60% of the recurrent mutations result in triallelic sites and that, consequently, the number of recurrent sites that are not triallelic is of the magnitude of (100 − 60)/60 × 411 ≈ 274. In other words, recurrent sites (di- and triallelic) represent 3–4% of the total number of segregating sites. Our simulations use a finite-sites model based on these results (see materials and methods).
In the simulated data we found nonetheless fewer triallelic sites than observed in the data, most likely because of heterogeneity in the mutation rate. To account for this we assumed that a fraction x of the coding region could not mutate. Using the number of triallelic sites as a proxy, this fraction x was estimated at 0.5, which is compatible with the limited redundancy of the genetic code. Specifically, we assumed that if mutations occur in an exon, they must be synonymous. With this supplementary condition the observed amount of recurrent mutation becomes compatible with an otherwise uniform mutation rate within each fragment.
It should be noted that the calculations above assume that almost all triallelic sites result from double mutations. Our results could be biased if there is a large heterogeneity in the mutation rate at the base pair scale. For example, if most triallelic sites are the products of highly mutable sites (mutation hot spots), we would overestimate the total amount of recurrent mutations. However, the very small number of quadriallelic sites indicates that these hot spots cannot be too frequent.
Goodness of fit:
Adding population growth and recurrent mutations dramatically improves the fit of the model; however, the model still does not fit very well. A particularly striking discrepancy lies in the higher-than-expected variance across fragments for several summary statistics (e.g., Tajima's D and the number of segregating sites, see Nordborg et al. 2005).
This discrepancy is largely due to the fact that several fragments appear to have genealogies characterized by an extremely long internal branch, leading to two highly diverged haplotypes. This phenomenon, sometimes referred to as “dimorphism,” has been observed several times in A. thaliana (see, for example, Aguadé 2001), and recent studies have shown that it is not compatible with a standard neutral model (Nordborg et al. 2005; Schmid et al. 2005).
Perhaps the most likely explanation for this pattern is some form of population structure, ancient or current. We attempted to design a simple two-population model that would explain the data, but were not successful (V. Plagnol, M. Nordborg and J. D. Wall, unpublished results). Schmid et al. (2005) reported similar findings. There is at this point no clear explanation for this pattern.
The only way we could improve the fit of the model is by removing outlier fragments. More precisely, we removed a fragment for which we could find a subset of ≥15 segregating sites with the property that the subset of the data restricted to these sites displays only two haplotypes. With our estimated growth parameters, this pattern is very unusual (P = 0.01). We found 105 such loci, for ∼9 expected under the model.
While removing fragments that do not fit the model using arbitrary criteria is not particularly satisfying, we found that the recombination estimates below are largely unaffected by this procedure. In other words, we get the same estimates whether we include the outlier fragments or not (the results below are for the full data). This leads us to believe that our recombination estimates are robust to this particular deviation from the model.
RATE ESTIMATION
Average crossing-over rate:
Since A. thaliana is selfing most individuals are homozygotes. Hence, most of the crossing-over events are undetectable. Thus we are not estimating the actual rate of crossing over but a scaled version that measures its impact on the level of LD. A theoretical description of what this normalization means in a selfing organism is presented in Nordborg (2000).
We estimated the crossing-over rate under models where this rate is uniform across the genome. We go on to discuss how much can be said about the variability of this crossing-over rate, but estimates for the genomewide average are useful when compared to average mutation and conversion rates to assess the relative importance of these phenomena.
Our estimation is based on the observation that the pattern of LD between distant markers depends mostly on the crossing-over rate. Unlike crossing over, the rates of gene conversion and recurrent mutation do not increase with distance (see discussion). More precisely, given the range of parameters that we consider, it is reasonable to assume that when two fragments are >5 kb apart the level of LD is affected almost only by crossing over. We compared estimates obtained from two different methods: Hudson's composite likelihood (Hudson 2001) and a method of least squares using the decay of LD. Note that for both methods we used our estimated growth parameters.
For our least-squares approach we considered all pairs of SNPs separated by at least 5 kb. We estimated the theoretical mean _D_′ for different values of the crossing-over rate. We did so by simulating a large number of two-locus fragments separated by different values of ρ on a grid: 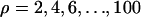 under an infinitely many sites model. We used linear interpolation to obtain the expected _D_′ at precise values. For each pair of fragments we summed the difference between expectation and observation weighted by the number of pairs of SNPs for this pair of fragments. Figure 2 shows how this distance varies with the crossing-over rate. The minimum distance is obtained for a crossing-over rate of 0.3/kb. As shown in Figure 2 we obtained the same estimates using Hudson's pseudo-likelihood. Note that the least-squares approach requires a large amount of data to be efficient and is used mostly to confirm the accuracy of Hudson's composite-likelihood estimator.
under an infinitely many sites model. We used linear interpolation to obtain the expected _D_′ at precise values. For each pair of fragments we summed the difference between expectation and observation weighted by the number of pairs of SNPs for this pair of fragments. Figure 2 shows how this distance varies with the crossing-over rate. The minimum distance is obtained for a crossing-over rate of 0.3/kb. As shown in Figure 2 we obtained the same estimates using Hudson's pseudo-likelihood. Note that the least-squares approach requires a large amount of data to be efficient and is used mostly to confirm the accuracy of Hudson's composite-likelihood estimator.
Figure 2.

Difference between expected and observed decay of _D_′ as a function of the crossing-over rate ρ. The solid line indicates the least-squares distance and the dashed line indicates Hudson's composite likelihood. The minimum of the squared distance and maximum of the composite likelihood (which both estimate the average ρ) are ∼0.3/kb.
Figure 3 shows the decay of _D_′ with distance and compares this curve to what is expected using our genomewide estimate. LD disappears after ∼100 kb, which is comparable to that in humans. The ratio between mutation and crossing-over rates is estimated at ∼10 in exons and 15 elsewhere. These numbers are consistent with theoretical expectations in a selfing organism (Nordborg 2000).
Figure 3.

Decay of _D_′, observed (dashed line) and expected (solid line) for ρ = 0.3/kb, which minimizes distance between expectation and data.
Variation along the genome:
The median distance between fragments is too high to provide reliable estimates of the local variation of the recombination rate. Given our data set it can be done only in the nine regions sequenced with a higher density. Each region consists of ∼20 fragments whose length varies between 450 and 600 bp.
For each region we computed an estimate of ρ using Hudson's composite likelihood (which showed better properties than the least-squares fitting methodology when the amount of data is limited). Results are shown in Table 2, with the location of the regions and the number of fragments in each of them. We also compared these estimates with the genetic distance obtained from recombinant inbred lines. Estimates are based on the data provided in Lister and Dean (1993).
TABLE 2.
Local estimates of recombination rates
| Chr | Start | End | No. frag. | 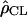 |
Map |
|---|---|---|---|---|---|
| 1 | 28,700 | 29,300 | 18 | 0.31 | 5.3 |
| 2 | 8,300 | 8,800 | 17 | 0.16 | 4.6 |
| 2 | 17,700 | 18,300 | 16 | 0.24 | 3.3 |
| 3 | 6,200 | 6,800 | 20 | 0.32 | 3.8 |
| 4 | 150 | 400 | 17 | 0.43 | 14 |
| 4 | 800 | 1,400 | 16 | 0.47 | 12 |
| 4 | 8,900 | 9,500 | 20 | 0.56 | 5.4 |
| 5 | 2,700 | 3,500 | 23 | 0.19 | 4.5 |
| 5 | 4,000 | 4,600 | 19 | 0.14 | 3.4 |
Both genetic map and population genetics estimates are very noisy. To limit the noise we smoothed the genetic map by fitting a third-degree polynomial on each arm of each chromosome.
For the population genetics estimates we excluded the most polymorphic fragments for two reasons. First, the high number of SNPs gives them a disproportionate weight. Second, high levels of polymorphism might be indicative of an unusually high mutation rate (including recurrent mutations) or perhaps of lower sequencing quality. We set the cutoff at 10% of the fragment (fragments with ≥34 segregating sites were excluded).
We found a weakly significant correlation between the genetic map and our estimates (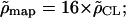 _F_-test _P_-value, 0.09). This correlation is not very strong but this is expected given the noise in the data. Moreover, potential short-range variation of the recombination rate makes the estimation more difficult. Our estimates are for 500-kb regions on average, and the average recombination rate seems to vary up to fourfold between different regions.
_F_-test _P_-value, 0.09). This correlation is not very strong but this is expected given the noise in the data. Moreover, potential short-range variation of the recombination rate makes the estimation more difficult. Our estimates are for 500-kb regions on average, and the average recombination rate seems to vary up to fourfold between different regions.
We then looked for correlations between the variation of ρ and the short-range pattern of LD, which could indicate correlations between rates of gene conversion and crossing over. We found no correlation between the estimate of ρ based on SNP pairs within fragments and the estimates presented above. However, in the regions where the estimate of ρ is high, we found on average higher values of _R_min. It is, however, not clear how to interpret those results because these high _R_min-values are increased only for a few fragments and many factors such as highly mutating loci or alignment issues can cause such a pattern.
Evidence for gene conversion:
Gene conversion is believed to be relatively frequent in A. thaliana (Haubold et al. 2002). In this section, we first demonstrate the evidence of gene conversion in this data set before estimating its rate.
To do so we used the clear conversion statistic (see materials and methods and the appendix). This summary statistic is designed to be characteristic of conversion events. We found four fragments in the data set with this property [chromosome (chr) 1, position (pos) 8,682,419; chr 2, pos 10,617,536; chr 3, pos 22,229,907; and chr 5, pos 18,666,458]. This may seem very few but it is clear that even in the presence of conversion events the required pattern is unlikely to be observed: excess of crossing over, conversion, recurrent mutations, or simply having few segregating sites can prevent us from observing such fragments.
Given the estimated recombination and recurrent mutation parameters we found that in the absence of gene conversion <1% of simulated data sets have four or more clear conversions. Slightly more than half of the simulated data sets have none. Hence our summary statistic provides evidence that there must have been gene conversion in the data. For these four fragments the potential conversion tracts would be ∼190, 400, 55, and 200 bp long (because longer tracts are easier to find these numbers are very biased estimates of the average tract length).
Average conversion rate:
The statistic presented in the previous section demonstrates that gene conversion has indeed occurred. However, this statistic is too stringent to estimate the rate of gene conversion and we now estimate this rate using different statistics.
To measure the level of LD between nearby markers we find that the most appropriate statistic is the number of incompatible fragments (as described earlier). We excluded the most polymorphic fragments (10% of the fragments, with 34 or more segregating sites) because of the likely excess of recurrent mutations. We found 227 incompatible fragments of 1213 remaining fragments in the data set (hence 103 of the 134 fragments with 34 or more segregating sites are incompatible).
It has been observed that there is too much short-range inconsistency in A. thaliana given the amount of crossing over inferred from long-range LD (Haubold et al. 2002). The total number of incompatible fragments is a measure of the amount of short-range inconsistencies. Using coalescent simulations we investigated the distribution of this statistic for different values of f, the ratio between conversion and crossing-over rates.
We used the crossing-over and population growth parameters estimated above. For each simulated data set we computed the number of incompatible fragments. We report the 5 and 95% quantile of the distribution of the number of incompatible fragments for a range of different values of f between 0 and 10 in Figure 4.
Figure 4.

Five percent and 95% quantiles of the distribution of the number of incompatible fragments for different values of f. The horizontal line shows the number observed in the data (227). The dashed line indicates the value of Hudson's pseudo-likelihood.
Our simulation scheme assumed that the mean length of a conversion tract is 100 bp. A ratio of conversion rate to crossing-over rate ∼1 fits our data well, with a 95% confidence interval between 0.5 and 1.2. Thus the conversion rate is estimated at ∼0.3/kb, approximately equal to the crossing-over rate. This implies an average of two recombination events in the history of each fragment, each being as likely to be a crossing over or a gene conversion. This estimate is lower than those found in previous studies (Haubold et al. 2002).
Hudson's pseudo-likelihood estimate is 0.6, at the lower bound of the confidence interval we obtained using the number of incompatible fragments. This value was obtained by setting the crossing-over rate to the value estimated earlier and then estimating the pseudo-likelihood on a grid of values for f. This method does not take into account recurrent mutations or the way the data are divided into different fragments. However, there is a good agreement between both estimates, which indicates that Hudson's pseudo-likelihood seems very robust to such perturbations.
Note that fitting the decay of _D_′ using markers within a fragment was not effective. We first found some very high estimates for f, before we understood that the average _D_′ between markers no more than 500 bp apart depends heavily on the amount of recurrent mutation in the data set. Simulations confirmed that this statistic is greatly affected by recurrent mutations.
Relationship between crossing over and conversion:
We now investigate the relationship between gene conversion and crossing-over rates in our data set. This issue has been addressed previously in Drosophila by Langley et al. (2000) and Andolfatto and Wall (2003). Given the kind of data we have, it is not clear whether we have the power to infer this relationship. Although they are far from being conclusive, the following results show some features of our data set addressing this issue.
We picked 14 large windows of size 2 Mb along chromosome I with nearly 18 fragments on average and tested three different models of rate variation:
- In model I, ρ and γ (the conversion rate) are identical for all windows as well as within windows.
- In model II, ρ and γ vary only between windows but f is a constant.
- In model III, ρ, γ, and f vary between windows but are constant within windows.
To distinguish between these models, we used Hudson's composite-likelihood method. For any two models, we define a summary statistic z as the absolute difference between the total log composite likelihoods under the best-fitting parameter choices for those two models divided by the total number of SNP pairs. A high value of z indicates that the data fit one of the models better than the other. As expected, because model III has more parameters, we find that model III fits the overall data better than models I and II. We computed z for models I and III and for models II and III. The corresponding values are 0.03 and 0.0082, respectively.
To check if these observed summaries are significant, we simulated 100 data sets (of 14 windows each) under the best-fitting parameters choices for models I and II and computed z. Each window was simulated independently and we added mutations only within locations corresponding to each fragment. Values of z as high as the observed values were never seen in any of these simulated data sets. Thus, model III fits our data significantly better than models I and II and this supports the idea that f varies across the Arabidopsis genome. We also note here that estimates of crossing-over and gene conversion rates for 43 large windows (2 Mb) across this genome were not significantly correlated (R = −0.029, _P_-value = 0.8455).
It should be emphasized that our results can be affected easily by fine-scale variation in crossing-over and gene conversion rates within windows as well as by the uncertainty in the levels of recurrent mutations.
DISCUSSION
The scale of decay of LD in our data is comparable to that in humans and is much larger than that observed in Drosophila. A major difference from humans is the greater level of polymorphism for a comparable level of LD. This higher ratio of mutation rate to recombination rate is consistent with theoretical expectations in a selfing organism (Nordborg 2000).
We fitted a coalescent model to the data and used it to show that for the observations to be consistent with the model the ratio between conversion and crossing-over rate must be approximately one. The rates seem to vary on a short scale and we were not able to detect any correlation between them.
Model fitting:
We also found that the data deviate from the standard neutral model. Simple modifications of the coalescent model can improve the fit considerably, but not fully explain the data.
One source of these deviations is the population structure observed by Nordborg et al. (2005). The obvious effects of population structure can be considerably reduced by removing eight outliers (accessions too closely or too distantly related). The reduced sample of 88 accessions shows only relatively weak evidence of population structure. The _F_ST measures presented in Nordborg et al. (2005) show that the level of differentiation between populations is only slightly higher than that in human populations.
However, some features of the data still cannot be explained: for example, the unexpectedly large variance in the level of polymorphism and Tajima's D. It is likely that some of these patterns are also due to the demographic history of A. thaliana; however, it is very difficult to model this. We have no prior knowledge of the demographic history of A. thaliana, with the exception of American accessions for which we have evidence of a very recent expansion (Nordborg et al. 2005). The scenarios we investigated to explain the data involved two populations and different combinations of admixture and migrations between them. The fact that we could not find a simple model fitting the data well suggests that the demography is more complex or that other factors are involved.
Reliability of estimates:
Nonetheless, we believe that our estimates of crossing-over and gene conversion rates are reasonably robust to these problems, mostly because of the very large amount of data and the fact that the estimates appear to be insensitive to removing outliers.
Another source of uncertainty is recurrent mutations. Our study also shows that recurrent mutations are frequent and any estimation procedure must account for this phenomenon. We believe that we have done this successfully.
A greater source of worry is the spacing of the fragments. Given the range over which LD decays, an ideal design would consist of a higher density of fragments, in the range of 10 kb apart (even denser than the nine candidate regions we use in this article). There is little doubt that better estimates would have been obtained with denser data.
Finally, we note that the noise in the estimate of the recombination rate can easily bias our estimate of the ratio of conversion to recombination for two reasons. First, a different level of recombination directly affects how much conversion is needed to explain the data. Second, a bias in the estimate of recombination mechanically affects the estimated ratio.
Acknowledgments
We thank S. Tavaré for helpful comments. The data analyzed in this article were generated with support from the National Science Foundation (DEB-0115062) and the W. M. Keck Foundation to M.N. The analysis was supported by a National Institutes of Health Center for Excellence in Genomic Sciences grant (1 P50 HG002790-01A1) to P.M. and M.N.
APPENDIX: CLEAR CONVERSION STATISTIC
We say that we see a clear conversion event when the genealogy of a fragment is consistent with a unique conversion event, with at least two SNPs in the conversion tract. Also we require that a single crossing over, or conversion event involving only one SNP, cannot explain the genealogy.
The computation of the clear conversion statistic uses ideas presented in Gusfield (2005) and Gusfield et al. (2006). We summarize here a simple way to compute this statistic (D. Gusfield, personal communication).
First, two sites are said to be incompatible if they fail the four-gametes test. Let us consider a fragment in our data set. The first step of the computation consists of clustering SNPs within a fragment such that any pair of incompatible sites belongs to the same set. Gusfield et al. (2006) showed that the number of recombination events or recurrent mutations needed in the history of the sequences is at least the number of sets of SNPs defined in this way. Hence to obtain a clear conversion event it is necessary that there is only one set of incompatible SNPs.
Now we consider the restriction of the haplotypes to that unique set of SNPs. At this step we remove the duplicated haplotypes. If a single recombination event is sufficient to explain the history of these fragments one must be able to enumerate all haplotypes but one so that when one considers a given SNP it mutates only once during the enumeration. Moreover, the haplotype excluded from the enumeration must be the recombinant product of the first and last enumerated haplotypes.
The type of recombination event used to produce the excluded haplotype from both enumerated ones depends on what one is looking for. For our clear conversion statistic we test for crossing over and gene conversion. If a gene conversion can explain the genealogy it is straightforward to see how many SNPs are part of the conversion tract.
References
- Aguadé, M., 2001. Nucleotide sequence variation at two genes of the phenylpropanoid pathway, the FAH1 and F3H genes, in Arabidopsis thaliana. Mol. Biol. Evol. 18**:** 1–9. [DOI] [PubMed] [Google Scholar]
- Andolfatto, P., and J. D. Wall, 2003. Linkage disequilibrium patterns across a recombination gradient in African Drosophila melanogaster. Genetics 165**:** 1289–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford, D. C., T. Bhangale, N. Li, G. Hellenthal, M. J. Rieder et al., 2004. Evidence for substantial fine-scale variation in recombination rates across the human genome. Nat. Genet. 36**:** 700–706. [DOI] [PubMed] [Google Scholar]
- Gusfield, D., 2005. Optimal, efficient reconstruction of root-unknown phylogenetic networks with constrained and structured recombination. J. Comput. Syst. Sci. 70**:** 381–398. [Google Scholar]
- Gusfield, D., S. Eddhu and C. Langley, 2004. Optimal, efficient reconstruction of phylogenetic networks with constrained recombination. J. Bioinform. Comput. Biol. 2**:** 173–213. [DOI] [PubMed] [Google Scholar]
- Gusfield, D., D. Hickerson and S. Eddhu, 2006. An efficiently-computed lower bound on the number of recombinations in phylogenetic networks: theory and empirical study. Special issue on computational biology. Discrete Appl. Math (in press).
- Haubold, B., J. Kroymann, A. Ratzka, T. Mitchell-Olds and T. Wiehe, 2002. Recombination and gene conversion in a 170-kb genomic region of Arabidopsis thaliana. Genetics 161**:** 1269–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R. R., 2001. Two-locus sampling distributions and their applications. Genetics 159**:** 1805–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R. R., 2002. Generating samples under a Wright-Fisher neutral model. Bioinformatics 18**:** 337–338. [DOI] [PubMed] [Google Scholar]
- Hudson, R. R., and N. L. Kaplan, 1985. Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics 111**:** 147–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley, C. H., B. P. Lazarro, W. Phillips, E. Heikinnen and J. M. Braverman, 2000. Linkage disequilibria and the site frequency spectra in the su(s) and su(wa) regions of the Drosophila melanogaster X chromosome. Genetics 156**:** 1837–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, N., and M. Stephens, 2003. Modeling linkage disequilibrium and identifying recombination hotspots using single-nucleotide polymorphism data. Genetics 165**:** 2213–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister, C., and C. Dean, 1993. Recombinant inbred lines for mapping RFLP and phenotypic markers in Arabidopsis thaliana. Plant J. 4**:** 745–750. [DOI] [PubMed] [Google Scholar]
- Marjoram, P., and P. Donnelly, 1994. Pairwise comparisons of mitochondrial DNA sequences in subdivided populations and implications for early human evolution. Genetics 136**:** 673–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVean, G. A. T., S. R. Myers, S. Hunt, P. Deloukas, D. R. Bentley et al., 2004. The fine-scale structure of recombination rate variation in the human genome. Science 304**:** 581–584. [DOI] [PubMed] [Google Scholar]
- Myers, S. R., and R. C. Griffiths, 2003. Bounds on the minimum number of recombination events in a sample history. Genetics 163**:** 375–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordborg, M., 2000. Linkage disequilibrium, gene trees, and selfing: an ancestral recombination graph with partial self-fertilization. Genetics 154**:** 923–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordborg, M., 2001. Coalescent theory, pp. 179–212 in Handbook of Statistical Genetics, edited by D. J. Balding, M. J. Bishop and C. Cannings. John Wiley & Sons, Chichester, UK.
- Nordborg, M., J. O. Borevitz, J. Bergelson, C. C. Berry, J. Chory et al., 2002. The extent of linkage disequilibrium in Arabidopsis thaliana. Nat. Genet. 30**:** 190–193. [DOI] [PubMed] [Google Scholar]
- Nordborg, M., T. T. Hu, Y. Ishino, J. Jhaveri, C. Toomajian et al., 2005. The pattern of polymorphism in Arabidopsis thaliana. PLoS Biol. 3**:** e196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid, K. J., S. Ramos-Onsins, H. Ringys-Beckstein, B. Weisshaar and T. Mitchell-Olds, 2005. A multilocus survey in Arabidopsis thaliana reveals a genomewide departure from a neutral model of DNA sequence polymorphism. Genetics 169**:** 1601–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens, M., 2001. Inference under the coalescent, pp. 213–238 in Handbook of Statistical Genetics, edited by D. J. Balding, M. J. Bishop and C. Cannings. John Wiley & Sons, Chichester, UK.
- Stumpf, M. P. H., and G. A. T. McVean, 2003. Estimating recombination rates from population-genetic data. Nat. Rev. Genet. 4**:** 959–968. [DOI] [PubMed] [Google Scholar]
- Tajima, F., 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123**:** 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson, G. A., and H. A. Guess, 1977. Is the most frequent allele the oldest? Theor. Popul. Biol. 11**:** 141–160. [DOI] [PubMed] [Google Scholar]