Bcl-2 changes conformation to inhibit Bax oligomerization (original) (raw)
Abstract
Bcl-2 inhibits apoptosis by regulating the release of cytochrome c and other proteins from mitochondria. Oligomerization of Bax promotes cell death by permeabilizing the outer mitochondrial membrane. In transfected cells and isolated mitochondria, Bcl-2, but not the inactive point mutants Bcl-2-G145A and Bcl-2-V159D, undergoes a conformation change in the mitochondrial membrane in response to apoptotic agonists such as tBid and Bax. A mutant Bcl-2 with two cysteines introduced at positions predicted to result in a disulfide bond that would inhibit the mobility of α5–α6 helices (Bcl-2-S105C/E152C) was only active in a reducing environment. Thus, Bcl-2 must change the conformation to inhibit tBid-induced oligomerization of integral membrane Bax monomers and small oligomers. The conformationally changed Bcl-2 sequesters the integral membrane form of Bax. If Bax is in excess, apoptosis resumes as Bcl-2 is consumed by the conformational change and in complexes with Bax. Thus, Bcl-2 functions as an inhibitor of mitochondrial permeabilization by changing conformation in the mitochondrial membrane to bind membrane-inserted Bax monomers and prevent productive oligomerization of Bax.
Keywords: apoptosis, Bax, Bcl-2, membrane topology, tBid
Introduction
Bcl-2 inhibits apoptosis by antagonizing proapoptotic family members, including the proteins Bax and Bak that share with it Bcl-2 homology (BH) regions 1–3 as well as proteins containing only a BH3 region. The molecular mechanism by which Bcl-2 antagonizes the actions of the proapoptotic proteins remains uncertain. However, because many proapoptotic proteins share only the BH3 region, the mechanism of inhibition is thought to involve binding interactions mediated by the hydrophobic BH3 binding pocket on the surface of Bcl-2 (Adams and Cory, 1998). Consistent with this model, both Bcl-2 and Bcl-XL, a close relative with similar function, bind to BH3 peptides derived from proapoptotic proteins. Precisely how the binding interactions between pro- and antiapoptotic proteins are regulated is unknown, but based on studies in solution was proposed to involve conformational changes in the proapoptotic Bcl-2 family proteins that expose the BH3 region. The consequences of BH3 region binding are also unclear. There is some evidence that upon binding of BH3 regions, the antiapoptosis proteins are either inactivated or converted to proapoptosis proteins (Letai et al, 2002).
It is now well established that after a cell has been exposed to a cytotoxic stimulus, Bax must undergo multiple conformational changes in order for it to commit the cell to dying by permeabilizing membranes (mitochondria and endoplasmic reticulum) (Sharpe et al, 2004). In most cases, the initial apoptotic stimulus does not activate Bax directly, but rather increases the expression or activity of one or more BH3-only proapoptotic Bcl-2 family members like Bid and Bim. These proteins then trigger a conformation change in Bax causing it to migrate and insert into membranes. Once inserted into membranes, Bax then oligomerizes to permeabilize membranes and release activators of the final effectors of apoptosis such as cytochrome c and SMAC/Diabalo (Sharpe et al, 2004; Annis et al, 2005). We have shown that membrane permeabilization by Bax is preceded by the integration into membranes of helix 9 and two helices (5–6) that in the soluble protein are buried in the interior of Bax. Unlike the better studied bacterial pore-forming proteins that insert a pore-forming domain subsequent to or concomitant with oligomerization, Bax helices 5–6 insert into membranes prior to oligomerization and pore formation (Annis et al, 2005).
We have recently reported that similar to Bax, Bcl-2 undergoes a conformational change after cells are exposed to an apoptotic stimulus (Kim et al, 2004). Unlike Bax, Bcl-2 is constitutively bound to membranes with helix 9 embedded in the bilayer and the rest of the polypeptide, including the putative pore-forming region, located on the cytoplasmic side of the membrane. However, during apoptosis Bcl-2 residue 158, a cysteine located near the base of the putative pore-forming domain of Bcl-2, moves from an aqueous to a hydrophobic environment (Kim et al, 2004). Although this observation clearly indicates that Bcl-2 undergoes a dramatic conformational change during apoptosis, it does not reveal where Bcl-2 is active in the multistep process of apoptosis induction. Some of the candidate steps suggested by recent reports include binding to and sequestering of activated BH3 proteins or prevention of Bax migration, integration, conformation change or oligomerization. Thus, the functional significance of the Bcl-2 conformational change remains unclear.
To understand the significance of the conformational change in Bcl-2, we have examined both Bcl-2 and three functionally impaired mutants, Bcl-2-G145A (Yin et al, 1994; Sedlak et al, 1995; Ottilie et al, 1997), Bcl-2-V159D and Bcl-2-S105C/ E152C in transfected cells and in vitro. By using an in vitro system in which the interactions that control membrane permeabilization by Bax were reconstituted with a minimal number of purified recombinant or transfected components expressed as full-length proteins without tags, it was possible to examine the molecular mechanisms involved at membranes. Our results not only suggest that Bcl-2 and Bax interact directly as was predicted by many previous models, they also strongly suggest an unanticipated molecular mechanism in which both Bcl-2 and Bax must change conformation so that their respective pore-forming domains are inserted into the lipid bilayer before they interact stably. Thus, Bcl-2 prevents oligomerization of membrane-bound Bax only when they are both multispanning transmembrane proteins. Just as insertion of the pore-forming domain of Bax into the membrane is considered an activation step because it is prerequisite for membrane permeabilization, the corresponding conformation change in Bcl-2 also constitutes ‘activation' because it is this form of Bcl-2 that inhibits membrane permeabilization by Bax: mutants that cannot adopt this conformation are inactive with respect to Bax. Conformationally changed Bcl-2 continues to inhibit apoptosis until the concentration of membrane-embedded Bax exceeds that of Bcl-2. Together, these results lead us to propose that the activated form of Bcl-2 is consumed by the reaction and therefore functions similar to a suicide inhibitor of the oligomerization of activated Bax.
Results and discussion
The inactive mutants Bcl-2-G145A and Bcl-2V159D do not change topology at the membrane during apoptosis
Bcl-2 with the mutation G145A (Bcl-2-G145A) does not prevent apoptosis induced by growth factor deprivation in hematopoietic cells. Moreover, Bcl-2-G145A does not co-precipitate with Bax (Yin et al, 1994; Lin et al, 2004), during immunoprecipitation, even using detergent conditions known to induce a conformational change in Bax that allows it to bind to Bcl-2 (Hsu and Youle, 1997). Moreover, we have recently shown that in vitro synthesized Bcl-2G145A does not bind to recombinant Bax (Zhang et al, 2004), and conversely, in vitro synthesized Bax does not bind to recombinant Bcl-2-G145A (Supplementary Figure 1). Thus, one of the reasons that Bcl-2-G145A is nonfunctional is that it cannot bind to Bax. The analogous nonfunctional mutation (G138A) has been generated in Bcl-XL (BclX-G138A) (Sedlak et al, 1995) and it does not bind to Bax lacking the transmembrane domain as measured in yeast two-hybrid and solid-phase binding assays, or to Bax expressed in transfected cells (Jeong et al, 2004). However, BclXL-G138A does bind to a truncated Bax that contains the BH3 domain (Sedlak et al, 1995), and in cells it is still able to bind to and prevent cell death induced by the Bax BH3 domain alone (Ottilie et al, 1997). Moreover, the glycine for alanine substitution at this residue affects homodimerization of Bcl-XL (Jeong et al, 2004), but not of Bcl-2 (Zhang et al, 2004). These conflicting data suggest that some structural constraint other than steric hindrance due to the amino-acid substitution prevents Bcl-2-G145A from binding full-length Bax and inhibiting apoptosis.
As part of our investigations into the binding surfaces in Bcl-2 that mediate homodimerization and hetero-oligomerization with Bax (Zhang et al, 2004), we generated a hypofunctional point mutant Bcl-2-V159D that still binds to Bax in nonionic detergent (Supplementary Figure 1, where Bcl-2 and the mutants are expressed without the α9 helix transmembrane domain). Thus, even though neither Bcl-2-G145A nor Bcl-2-V159D prevents apoptosis, they differ in the extent to which they bind to Bax. To examine the defects in Bcl-2-G145A and Bcl-2-V159D in more detail, we compared the apoptosis-induced conformational changes of these proteins with that observed previously for Bcl-2 when preventing apoptosis (Kim et al, 2004).
In both the human breast cancer cell line MCF-7 and Rat-1myc ERTAM cells (referred to here as Rat-1 cells (Soucie et al, 2001)), Bcl-2 significantly delays apoptosis induced by two different chemotherapy agents compared to Bcl-2-G145A and Bcl-2-V159D (Figure 1A and B). The data for Bcl-2-G145A are not available for 48 h treatment of MCF-7 cells with doxorubicin as no intact cells remained at this time point. Bcl-2-G145A and Bcl-2-V159D were nonfunctional in these assays as the response of expressing cells was not different from vector control cells (data not shown).
Figure 1.
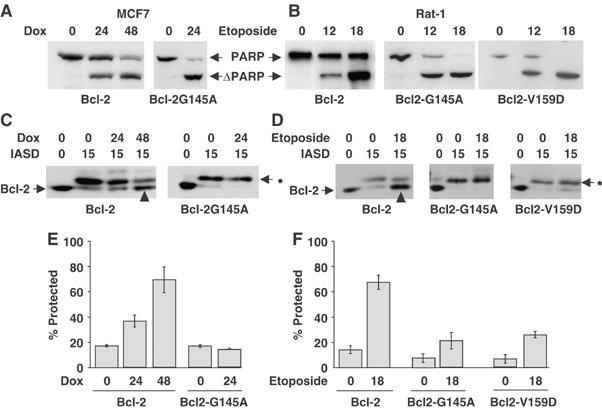
In two different cell lines inactive Bcl-2 mutants do not change topology at the membrane during apoptosis. MCF7 human breast cancer (A, C, E) and Rat-1 fibroblast (B, D, F) cell lines were transfected with vector control (neo), or expression plasmids encoding Bcl-2 or the inactive mutants Bcl-2-G145A or Bcl-2-V159D, as indicated below the panels. (A, B) Bcl-2-G145A and Bcl-2-V159D do not inhibit apoptosis. Cell lines were exposed to 10 μM doxorubicin (Dox) or 6 μM etoposide for the time indicated in hours above the blots. Apoptosis measured by cleavage of the caspase substrate PARP (ΔPARP) was assessed by immunoblotting of cell lysates. (C–F) Cysteine 158 of Bcl-2 but not the inactive mutants is protected from IASD in drug-treated cells. After drug treatment for the time indicated (hours), membrane fractions were incubated with IASD for 0 (control reactions) or 15 min to label proteins with exposed cysteine residues. After separation by SDS–PAGE, labeling was assessed by immunoblotting with antibodies to Bcl-2. (C, D) The migration position of unmodified Bcl-2 is indicated to the left of the panels. Bcl-2 modified by IASD is indicated by an asterisk (*) to the right of the panels. (E, F) Protection of cysteine 158 from IASD labeling for Bcl-2, Bcl-2-G145A and Bcl-2-V159D determined by densitometry from at least three independent experiments for each drug treatment (indicated below the histograms), for samples from (E) MCF7 cells or (F) Rat-1 cells. As reported previously, the limited linear range of the blotting procedure and the small migration difference between labeled and unlabeled proteins limits the range that can be measured to 10–80% protection (Kim et al, 2004). The error bars indicate s.d.
At times when Bcl-2 prevents apoptosis due to doxorubicin (24 and 48 h) and etoposide (12 and 24 h), a substantial fraction of Bcl-2 had undergone a conformational change such that cysteine 158 in helix 5 became embedded in the membrane where it was protected from labeling by the membrane impermeant cysteine-reactive probe IASD (Figure 1C and D, upwards arrowheads), as observed previously (Kim et al, 2004). Protection from IASD is due to insertion into the lipid bilayer as labeling was carried out in sufficient urea (4 M) to unfold Bcl-2 (Kim et al, 2004). As documented previously, the assay ranges from 10% protection for accessible residues to a maximum of 80% protection from labeling for residues embedded in the bilayer (see below). The same residue was efficiently labeled by IASD both when the membrane was solubilized with nonionic detergent (see below, Figure 3) and in membranes from cells not treated with drug (Figure 1). A similar change in accessibility to IASD was found for several other residues within helices 5 and 6, confirming that the protection of cysteine 158 from labeling with IASD is indicative of a major structural change in Bcl-2 that embeds a large fraction of this region in the bilayer (Roberts et al, manuscript in preparation). However, in membranes from similarly drug-treated cells, cysteine 158 in both Bcl-2-G145A and Bcl-2-V159D does not become protected from IASD. Thus, the conformational change observed for Bcl-2 does not occur or is severely inhibited in these inactive mutants in both MCF-7 and Rat-1 cells (Figure 1C–F).
Figure 3.
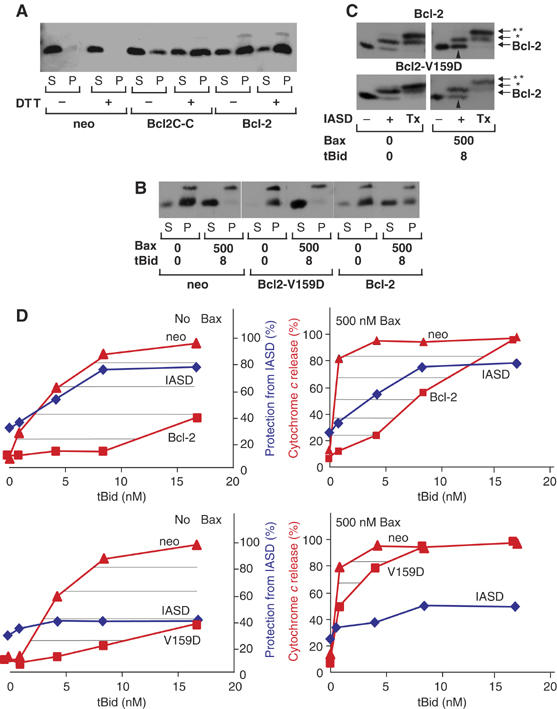
(A) Change in membrane topology of Bcl-2 is required to prevent release of cytochome c from mitochondria treated with recombinant Bax and tBid. The heavy membrane fraction of vector control (neo), Bcl-2-, Bcl-2C-C- or Bcl-2-V159D- (V159D) expressing Rat-1 fibroblasts was exposed to Bax and tBid, as indicated. (A, B) Cytochrome c release was assayed by pelleting the mitochondria and visualizing the cytochrome c (cyt c) in the supernatant (S) and pellet (P) fractions after SDS–PAGE by immunoblotting. X-crossreacting mitochondrial protein. In (A), the heavy membrane fractions were incubated in 1 mM DTT throughout isolation and incubated with tBid (1 nM) and Bax (100 nM). In other panels, the concentration (nM) of tBid and Bax are indicated. (C) Bcl-2 and Bcl-2-V159D were assayed for conformational change in membranes from Rat-1 fibroblasts by IASD labeling as described in Figure 1. In Bcl-2 (upper panel), cysteine 158 was protected; therefore, migration of more than half of the Bcl-2 is not changed after incubation with IASD (upward arrowhead). Much less Bcl-2-V159D (lower panel) was protected from IASD (upward arrowhead). Bands corresponding to IASD labeling of cysteine 158 are indicated by *. Tx indicates an additional control in which Triton X-100 (1%) was added to solubilize the membranes. Labeling of both cysteine 158 and the constitutively membrane embedded cysteine in helix 9 (migration position indicated by **) confirms that protection from labeling was due to integration in the lipid bilayer. (D) Protection (%) of cysteine 158 from IASD labeling (blue diamonds) in heavy membranes from cells expressing Bcl-2 (upper panels) or Bcl-2-V159D (lower panels) compared to the extent of cytochrome c release from mitochondria from the same cells (red, squares indicated Bcl-2 or V159D), and from vector control cells (red, triangles). Immunoblots were quantified by densitometry (_n_>3) and averaged. The area hatched by horizontal lines indicate protection from apoptosis by Bcl-2 (upper panels) or Bcl-2V-159D (lower panels) for reactions without added Bax (left) or containing 500 nM Bax (right). The amount of tBid added to the reactions is indicated. One of three complete data sets for cytochrome c release at different Bax and tBid concentrations is shown in Supplementary Figure 3.
The final stage of the multistep activation of Bax during apoptosis involves oligomerization and membrane permeabilization (Annis et al, 2005). Bcl-2 but not Bcl-2-G145A decreased Bax oligomerization in Rat-1 cells exposed to etoposide (Figure 2, open arrowheads). Furthermore, at a time point during which Bcl-2 was preventing apoptosis, a small fraction of Bcl-2 but not Bcl-2-G145A was found in complexes larger than the Bcl-2 dimers found in untreated cells (Figure 2, solid arrowheads and data not shown).
Figure 2.
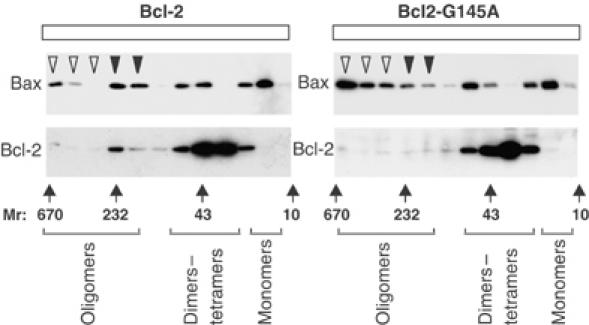
Bcl-2 but not Bcl-2-G145A inhibits oligomerization of Bax. Rat-1 cells expressing either Bcl-2 or Bcl-2-G145A were treated with etoposide for 12 h, and oligomerization of Bax and Bcl-2 were measured by gel filtration chromatography of whole-cell membranes solubilized in CHAPS, followed by immunoblotting of fractions for either Bax or Bcl-2, as indicated. Large oligomers (>250 kDa) are indicated by open arrowheads and are seen primarily in fractions from cells expressing Bcl-2-G145A. A smaller oligomer of approximately 230 kDa seen reproducibly in cells expressing Bcl-2 is indicated by closed arrowheads. Control experiments with untreated cells established elution positions for dimers of Bcl-2 (30–40 kDa fractions) and Bax monomers (15 kDa) as indicated below the panels. The elution positions of molecular weight standards (_M_r in kDa) are shown below the panels.
Thus in intact cells, Bcl-2-mediated prevention of apoptosis is associated with a conformational change in Bcl-2 that moves cysteine 158 (at the base of helix 5) from cytosol to membranes and with inhibition of Bax oligomerization. The Bcl-2 conformation change is a better predictor of function in cellular membranes than Bcl-2 binding to Bax (Supplementary Figure 1). These results suggest that mutation of Bcl-2 at either G145 or V159 prevents a conformation change in Bcl-2 that embeds cysteine 158 in the membrane and is essential for Bcl-2 to inhibit Bax in mammalian cells.
Bcl-2 prevention of cytochome c release and changes in topology are correlated in mitochondria treated with Bax and tBid
To examine the molecular interactions that mediate Bcl-2 inhibition of Bax-mediated membrane permeabilization in more detail, we purified full-length recombinant Bax and tBid and measured cytochrome c release from mitochondria isolated from Rat-1 cells stably transfected with a control plasmid or one encoding Bcl-2, Bcl-2-G145A or Bcl-2-V159D in an assay system described in Supplementary data. Furthermore, we also tested the function of another mutant (Bcl-2C-C) in which we introduced two cysteine residues at locations where an intramolecular S–S bond between helices 2 and 5 (S105C, E152C) is predicted to hinder the conformational change based on the structure of Bcl-2 (Supplementary Figure 2). By changing the oxidative environment in an in vitro system containing mitochondria from Rat-1 cell expressing Bcl-2C-C, we can directly test the role of this conformational change on Bcl-2 function.
Normal neuronal cells contain 0.7–1.1 μg of Bax per milligram of mitochondrial proteins (Polster et al, 2003). However, Bax is markedly upregulated in many conditions, and a 10-fold increase in this concentration is within the range of concentrations in cells subject to apoptotic stress. In the conditions of our assay in which mitochondria were used at a concentration of 1–2 mg protein/ml, we added recombinant Bax in increasing amounts to a maximum of 12 μg of Bax, values consistent with physiologically feasible conditions. The largest amount of Bax corresponded to a concentration of 500 nM, a supraphysiologic concentration at which there was no significant release of cytochrome c from vector control, Bcl-2-V159D, or Bcl-2 expressing mitochondria (Supplementary Figure 3 and data not shown). This result is consistent with observations that Bax has to be activated to permeabilize membranes. Bcl-2 inhibited tBid-dependent permeabilization of membranes by Bax at concentrations similar to those found in normal cells (Polster et al, 2003 and Supplementary Figure 3). However, at the increased concentrations (8 nM tBid and 500 nM Bax), cytochrome c was released even from mitochondria from cells expressing Bcl-2 (Figure 3).
Rat-1 mitochondria also contain endogenous membrane-bound Bak as detected by immunoblotting (data not shown). Experiments with mitochondria from Bax −/− Bak −/− cells indicate that mitochondria are resistant to tBid unless Bax or Bak is present (Zong et al, 2001), and tBid can efficiently induce the oligomerization and activation of Bak, leading to membrane permeabilization (Ruffolo and Shore, 2003). Accordingly, when we exposed mitochondria from vector control Rat-1 cells to tBid, there was a concentration-dependent increase in the amount of cytochrome c release. However, this effect was markedly potentiated by adding increasing concentrations of Bax to this system (e.g. release of 30% of cytochrome c at 1 nM tBid increased to 80% when 500 nM Bax was also added, Supplementary Figure 3A). Thus in experiments containing 500 nM Bax, cytochrome c release is limited by the amount of tBid added.
In cells, the mutant Bcl-2C-C is as functional as Bcl-2 when expressed at equivalent levels (data not shown). This result was expected since the cytoplasm is a reducing environment. However, in mitochondria isolated from cells expressing the mutant Bcl-2C-C, the predicted disulfide bond should inhibit the mobility of α5–α6 helices unless a reducing agent (DTT) is present. Indeed, the release of cytochrome c by tBid-activated Bax was dependent on the addition of DTT (results typical of three experiments are shown in Figure 3A). Bcl-2C-C was as effective as Bcl-2 at preventing tBid/Bax-induced cytochrome c release from mitochondria when DTT was present. However, without DTT cytochrome c release was comparable to vector-transfected controls, an effect we presume is due to the formation of a disulfide bond between the two cysteines as Bcl-2 function was unaffected.
Similar to the results observed in intact cells (Figure 1B), the function of Bcl-2-V159D in mitochondria was severely compromised (Figure 3B and D and Supplementary Figure 3C). At concentrations examined in detail below, membrane permeabilization in mitochondria from cells expressing either Bcl-2 or Bcl-2-V159D was due to the activation of the exogenous Bax, as there was minimal release of cytochrome c above baseline at 1–8 nM tBid unless 500 nM Bax was added (Figure 3D). Presumably, the slight residual activity (and conformation change) detected for Bcl-2-V159D is sufficient to inhibit the endogenous Bak on the mitochondria, but is essentially nonfunctional when excess exogenous Bax is activated by tBid. Thus, an advantage of our system is that it allows us to examine the specific effects of Bcl-2 topology on tBid-induced Bax membrane permeabilization in mitochondria containing a normal complement of the other known and unknown apoptosis regulatory proteins present in Rat-1 cells.
Labeling mitochondrial fractions with IASD revealed that the membrane conformation of Bcl-2 changed substantially after exposure to tBid or tBid and Bax compared to the minor effects noted for Bcl-2-V159D (Figure 3C, upward arrowheads and Figure 3D), similar to our observations in whole cells exposed to chemotherapy drugs (Figure 1C–F). Bcl-2C-C was not assayed with IASD because it contains multiple cysteines and the DTT needed to change conformation would react with IASD. Significantly, the conformational change in wild-type Bcl-2 was strongly evident (∼80 and 60%, respectively) at concentrations of tBid alone (Figure 3D, left panel; tBid 8 nM) or tBid plus Bax (Figure 3D, right panel; tBid 4 nM, Bax 500 nM) at which there was almost complete release of cytochrome c from mitochondria in vector control cells, but not from mitochondria from cells expressing Bcl-2. By contrast, at concentrations of Bax (500 nM) and tBid (4–8 nM) that caused nearly complete release of cytochrome c from mitochondria containing Bcl-2-V159D (Figure 3 B and D), there was a much smaller change from baseline in the amount of protection from IASD labeling of cysteine 158 (Figure 3 C and D). By plotting the extent of cytochrome c release for both the control and Bcl-2-expressing cells on the same graph, it is possible to visualize the ‘effective concentration' for inhibition of cytochrome c release (hatched areas in panels shown in Figure 3D). Comparing this to protection of cysteine 158 from labeling clearly indicates that the conformational change coincides with prevention of cytochrome c release. These data suggest that the conformational change in Bcl-2 induced by BH3-only molecules is required for Bcl-2 to prevent apoptosis. Consistent with these observations, when mitochondria were treated with Bim peptide sufficient to cause release of cytochrome c from mitochondria from control cells, but not from mitochondria from Bcl-2-expressing cells, most of the Bcl-2 had changed conformation as evidenced by protection of cysteine 158 from labeling by IASD (Supplementary Figure 5). These data also confirm that protection of cysteine 158 from IASD labeling in this assay is not due to direct binding of agonist (tBid or Bax) to this residue, as Bim peptide does not bind to Bcl-2 in 4 M urea (Kim et al, 2004). Furthermore, tBid does not remain bound to Bcl-2 in our system (see Figure 5 and Supplementary Figure 4).
Figure 5.
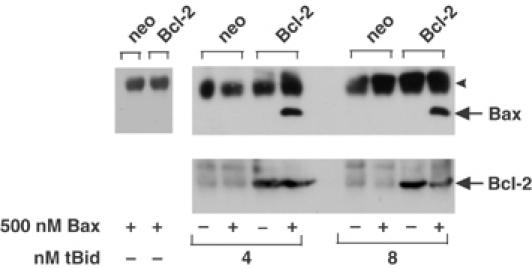
Bcl-2 binds to tBid-activated Bax. Heavy membrane fractions of vector control (neo) or Bcl-2-expressing Rat-1 fibroblasts were incubated with Bax and/or tBid, as indicated. The membranes were solubilized in CHAPS and immunoprecipitated with sheep anti-Bcl-2 antibody, then analyzed by immunoblotting with monoclonal anti-Bax antibody (upper panels) or with sheep anti-Bcl-2 antibody (lower panels). The single arrowhead indicates sheep immunoglobulin light chain, and the migration of Bax and Bcl-2 is indicated at the right.
Taken together, these results suggest a novel mechanism of action in which one way Bcl-2 prevents cytochrome c release from mitochondria is by changing conformation during apoptosis. Since Bcl-2 is constitutively bound to membranes, it can only bind to Bax tightly after Bax migrates to mitochondria (see below), an event coincident with integration of helices 5–6 and 9 of Bax into the membrane (Annis et al, 2005). Thus, it appears that the conformational change in Bcl-2 does not inactivate it as proposed previously (Lin et al, 2004). Instead, our results indicate that the protein continues to inhibit cytochrome c release even when all of the Bcl-2 in the cell has changed conformation (Figure 3D; 4–8 nM tBid), suggesting that this form of Bcl-2 is active in inhibiting cytochrome c release. Furthermore, our data showing that a mutation that is expected to reduce the mobility of helices 5 and 6 (Bcl-2C-C) prevents the protein from inhibiting Bax-induced membrane permeabilization is consistent with the hyperfunction observed for a mutant of Bcl-XL in which point mutations disrupt intramolecular polar interactions (Asoh et al, 2000). In this hyperfunctional mutant, the central α5–α6 helices are more mobile and therefore would be more susceptible to undergoing the conformational change observed here for Bcl-2.
Conformationally changed Bcl-2 inhibits Bax oligomerization, but is consumed by the interaction
Results obtained using many different experimental approaches suggest that stable binding of Bcl-2 to Bax is necessary for Bcl-2 to inhibit cytochrome c release. In our reactions, there is only sufficient Bcl-2 (20 nM) to bind to a small fraction of the added Bax (500 nM). Therefore, we predict that Bcl-2 inhibited cytochrome c release in these experiments by binding stably to only the activated (integral membrane) form of Bax. To test this hypothesis, mitochondria were isolated from both control and Bcl-2 expressing cells and the conformation change was triggered in Bcl-2 by adding tBid (4 nM) and Bax (500 nM). The mitochondrial outer membrane was then solubilized using CHAPS, a detergent that does not alter the conformation or authentic binding interactions of either protein (Hsu and Youle, 1997). The solubilized proteins were analyzed by gel filtration chromatography. In mitochondria from control cells without tBid, the Bax remained largely monomeric (Figure 4, neo, upper panel). However, when both tBid and Bax were added, some of the Bax oligomerized (Figure 4, neo, lower panel). The fraction of the total Bax that oligomerized was relatively small, as expected since the reactions contain more than 100-fold more Bax than tBid.
Figure 4.
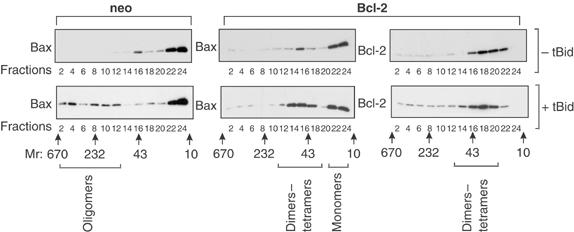
Bcl-2 prevents tBid-induced oligomerization of Bax. The heavy membrane fraction of vector control (neo) or Bcl-2-expressing Rat-1 fibroblasts was incubated with 500 nM Bax without or with tBid, upper and lower panels, respectively. Oligomerization of Bax and Bcl-2 was measured by gel filtration chromatography of the heavy membrane fraction solubilized in 2% CHAPS, followed by immunoblotting of alternate fractions (indicated below the panels) for Bax or Bcl-2 as indicated. In the absence of tBid, Bax elutes primarily as monomers. Although a very small amount of dimers was detected, large oligomers were not observed (upper panels). Oligomerization of Bax induced by tBid resulted in large oligomers (fractions 2–12) in mitochondria from control cells (neo), but was limited to dimers–tetramers in mitochondria from cells expressing Bcl-2 (fractions 14–18, lower middle panel). Bcl-2 (right panels) was found in small complexes (fractions 14–22), regardless of the presence of tBid. However, the small increase in apparent molecular weight of Bcl-2 complexes when tBid was added was very reproducible. The elution positions of molecular weight standards, and the fractions in which different sized complexes of Bax and/or Bcl-2 were detected, are indicated below the blots.
When Bax was added to mitochondria from cells expressing Bcl-2, the Bax remained monomeric consistent with Bcl-2 and Bax not binding to each other in the absence of an apoptotic stimulus (Figure 4, upper panel, compare neo/Bax with Bcl-2/Bax). As seen previously, when analyzed by gel filtration chromatography, Bcl-2 elutes primarily as a monomer or dimer (Figure 4, upper right panel). When both tBid and Bax were added to mitochondria from cells expressing Bcl-2, the elution of Bax revealed both monomers (fractions 22–24) and small oligomers slightly larger than Bcl-2 dimers (Figure 4, compare fractions 14–18 of the lower panels). The precise composition of these oligomers cannot be determined by this method. However, there was also a small but reproducible shift in the elution of Bcl-2 to slightly higher molecular weight fractions (Figure 4, lower right panel), suggesting that some of the small oligomers in fractions 14–18 may contain both Bax and Bcl-2.
The possibility that Bcl-2 binds to Bax only after tBid has caused a conformational change in both proteins was examined by co-precipitation assays using mitochondria from cells expressing Bcl-2. For these experiments, the Bcl-2 in the mitochondria was immunoprecipitated and the bound proteins were separated by SDS–PAGE and visualized by immunoblotting (Figure 5). As a control, mitochondria were pelleted from an aliquot of each reaction prior to detergent solubilization and the amount of Bcl-2 was assayed by immunoblotting. As expected, Bcl-2 was detected only in the cells that expressed the human protein. Importantly, even though Bax was present in one of each of the paired reactions, it co-precipitated with Bcl-2 only from reactions also containing tBid (Figure 5). Furthermore, Bax and Bcl-2 can only be crosslinked on mitochondrial membranes in the presence of tBid (Supplementary Figure 4A). Since sufficient tBid (8 nM) was added to change the conformation of most or all of the Bcl-2 on the membrane (20 nM) (Figures 3C and 6A), the result suggests that the conformationally changed Bcl-2 binds to Bax. Since the only Bax present is in the integral membrane form with helices 5–6 and 9 embedded in the membrane (Annis et al, 2005), we conclude that the conformationally changed Bcl-2 binds to the integral membrane form of Bax. Under the same conditions, Bax eluted from the gel filtration column as a small oligomer of 40–50 kDa instead of the >200 kDa oligomers found without Bcl-2 (Figure 4). Thus, binding of the conformationally changed Bcl-2 to the integral membrane form of Bax inhibits oligomerization of Bax.
Figure 6.
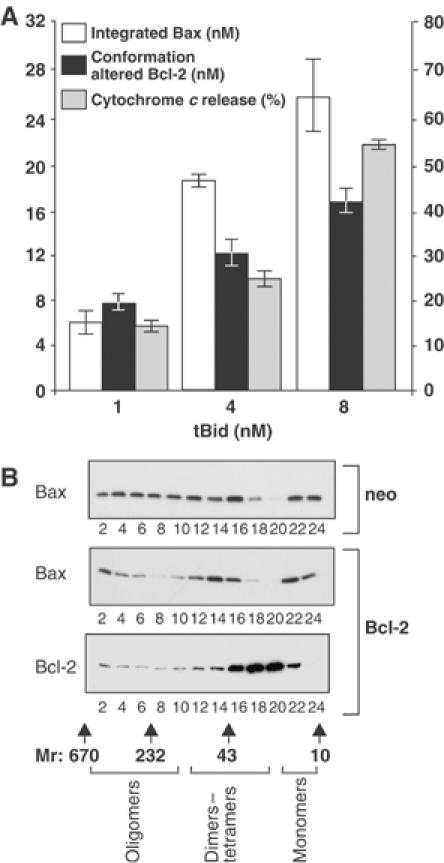
Bcl-2 conformational change correlates with the amount of integrated Bax. (A) Bax (500 nM) and the indicated amount of tBid were added to reactions containing heavy membranes from Bcl-2-expressing cells (final concentration of Bcl-2, 20 nM). Protection of cysteine 158 of Bcl-2 from labeling by IASD measured by immunoblotting indicates the amount of conformationally changed Bcl-2 (solid bars). The amount of Bax integrated into mitochondria was assayed by alkaline extraction (open bars). When the amount of integrated Bax exceeds the amount of conformationally changed Bcl-2, cytochrome c release begins (gray bars) but remains less than that for mitochondria from vector-transfected control cells (>90%). The error bars indicate s.e.m.'s for three independent experiments, each performed in triplicate. (B) Large oligomers of Bax detected by gel filtration chromatography when 8 nM tBid and 500 nM Bax were added to mitochondria from control cells (top panel) were reduced but clearly detectable in Bcl-2-expressing cells (center panel). Bcl-2 is excluded from large oligomers of Bax detected by gel filtration chromatography (bottom panel). The elution position of molecular weight standards on the gel filtration column, and the fractions in which different sized complexes of Bax and/or Bcl-2 are detected, are shown below the blot.
Several observations suggest that tBid only binds transiently to Bcl-2 to activate it, and does not remain sequestered by Bcl-2. First, tBid can only be crosslinked with Bcl-2 at concentrations >20 times higher than we used to elicit the conformational change (Supplementary Figure 4B and C). Secondly, when tBid was added to mitochondria containing Bcl-2 and then mitochondria were pelleted, tBid remained in the supernatant (Supplementary Figure 4D). Finally, Bcl-2 binds to tBid and Bid in CHAPS, but not in a nonionic detergent (Triton X-100) that is known to change the conformation of Bcl-2 family members to their active form (Hsu and Youle, 1997) and likely mimics insertion into a membrane (Supplementary Figure 4E).
It is likely that tBid by itself can drive the conformational change in Bcl-2, because the conformation change in Bcl-2 was induced by tBid even when Bax was not added (Figure 3D). Furthermore, the tBid dependence of the conformational change in Bcl-2 was virtually identical when excess (500 nM) Bax was present (compare protection from IASD in the two upper panels of Figure 3C). Similar results were also observed when Bim peptide was used to induce the conformational change in Bcl-2 (Supplementary Figure 5). However, it is formally possible that activated Bax can also cause this conformational change in Bcl-2.
If the conformationally changed Bcl-2 inhibits the integral membrane form of Bax, then the amount of the two proteins in mitochondria should increase coordinately and the ratio of them would indicate their stoichiometry in inhibited Bax complexes in mitochondrial membranes. When we compared the amount of Bax integrated into heavy membranes with the amount of conformationally altered Bcl-2 after adding different amounts of tBid, there was good concordance with approximately equal amounts of the two species present (Figure 6A). Thus, in reactions containing 500 nM Bax and 20 nM Bcl-2 when tBid was added at 1 nM (a concentration at which 80% of the cytochrome c is released from vector control cells, compared to a value for Bcl-2-expressing cells that does not differ from baseline (15%)), the amount of conformationally changed Bcl-2 and integral membrane Bax was very similar ∼8 and 6 nM each, respectively (Figure 6A). As tBid is increased to 4 or 8 nM, cytochrome c release was detected from mitochondria isolated from Bcl-2-expressing cells (although it remained significantly less than in vector control cells, Figure 3C) and the amount of integrated Bax exceeded the amount of conformationally changed Bcl-2 (18 versus 12 nM at 4 nM tBid, and 26 versus 15 nM at 8 nM tBid, respectively). Although this result suggests that one conformationally changed Bcl-2 inhibits approximately one integral membrane Bax, the gel filtration data in Figure 3 suggest that the mechanism is not as simple one to one binding. While the elution of the smaller Bax complexes partially coincides with the elution of Bcl-2, the gel elution profile is consistent with the complexes containing Bcl-2 dimers, Bcl-2/Bax heterodimers/trimers as well as small Bax oligomers. Thus, at the resolution of these techniques, we propose that one activated Bcl-2 molecule inhibits 1–2 molecules as opposed to 5–10 molecules of integrated Bax.
When sufficient tBid (16 nM) was added to the reactions to activate more than 30 nM Bax, large oligomers of Bax are once again observed by gel filtration chromatography (Figure 6B). The peak corresponding to dimers–tetramers of Bax and Bcl-2 seen in Figure 4 is still seen at 16 nM tBid (Figure 6B). Together, these results are consistent with a mechanism for Bcl-2-mediated inhibition of Bax in which conformationally changed Bcl-2 molecules are consumed. Thus, once the amount of integral membrane Bax is sufficiently greater than the amount of Bcl-2 (20 nM), any further Bax molecules that are activated by tBid can oligomerize and permeabilize mitochondria. Taken together, our data are most consistent with a mechanism in which the conformationally changed Bcl-2 acts as an inhibitor of Bax oligomerization (Figure 7). Conceptually, Bcl-2 behaves like a suicide inhibitor as it is consumed by the reaction; however, nonproductive oligomers rather than a covalently inhibited enzyme end product are generated. Based on the similar structure of Bcl-2 and Bax, we speculate that the conformational change in Bcl-2 results in insertion of helices 5 and 6 of Bcl-2 into the lipid bilayer such that it adopts a transmembrane topology similar to that of the integral membrane form of Bax (Annis et al, 2005). This multispanning transmembrane form of Bcl-2 binds to nascent Bax oligomers in a manner nonproductive for further oligomerization, thereby inhibiting Bax-mediated membrane permeabilization. In these complexes, Bcl-2 remains bound to Bax and therefore it cannot inhibit oligomerization of other Bax proteins. We hypothesize that the selective, rapid binding of the polytopic conformation of Bcl-2 to membrane-associated Bax is due to a marked increase in the affinity of the activated forms (helices 5 and 6 inserted into the membrane) of Bcl-2 and Bax. In this scenario, insertion into membranes may differentially modify the binding surfaces that mediate Bcl-2 homodimerization versus Bax hetero-oligomerization (Zhang et al, 2004). Our model is also consistent with the increased binding affinity of Bcl-2 and Bax in the presence of nonionic detergents (Hsu and Youle, 1997). A rigorous test of this hypothesis awaits the development of a technique that permits accurate measurement of the dissociation constants for the binding of the full-length proteins in membranes.
Figure 7.
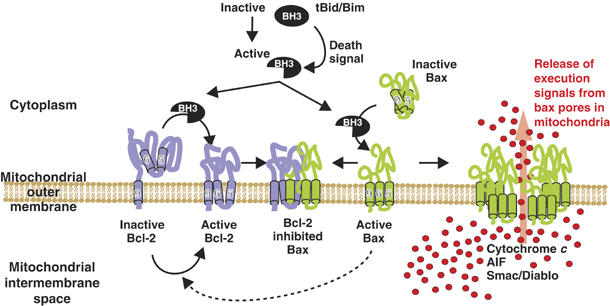
Conformationally changed Bcl-2 inhibits membrane-bound activated Bax. In dividing cells, Bcl-2 is constitutively bound to intracellular membranes by a carboxyl-terminal tail-anchor sequence including α-helix 9. This form of Bcl-2 is inactive for preventing Bax oligomerization, but is probably a homodimer with other antiapoptosis functions, and Bax is an inactive monomer located in the cytoplasm or loosely bound to mitochondria. Death signals activate BH3-only proteins (tBid, Bim), which cause changes in the membrane conformation of both Bax and Bcl-2: Bax translocates and inserts into organellar membrane such that α-helices 5, 6 and 9 are embedded in the bilayer (Annis et al, 2005), and Bcl-2 changes conformation such that cysteine 158 (α-helices 5 and 6) are inserted into the membrane (Figures 1, 3, 6 and Roberts et al, manuscript in preparation). In the absence of conformationally changed Bcl-2, membrane-integrated Bax monomers form large oligomers that permeabilize membranes and release proapoptotic factors. Oligomerization of membrane-embedded Bax (monomers or small oligomers) is inhibited by only those Bcl-2 molecules that have changed conformation. It is possible that when activated by a BH3 protein that does not bind Bcl-2, Bax may induce the conformation change in Bcl-2 (dotted line). The precise stoichiometry of binding of polytopic Bcl-2 to membrane-associated Bax is unknown, but inhibited complexes elute as if they are less than 100 kDa. Binding of Bcl-2 to Bax in nonproductive oligomers appears irreversible, and prevents Bcl-2 from inhibiting other Bax molecules. Thus, when excess Bax molecules are activated, large oligomers form and permeabilize the outer mitochondrial membrane releasing proapoptotic molecules. The number of Bax molecules required to form an oligomer that permeabilizes membranes is not known.
Our results demonstrating that tBid and Bim activate Bcl-2 indicate that activation of Bcl-2 is another role of BH3-only proteins in apoptosis. This function is likely independent of the previously described mechanisms whereby these proteins regulate apoptosis by directly activating Bax or Bak (Letai et al, 2002; Kuwana et al, 2005), by displacing antiapoptotic family members from binding to Bax/Bak (Letai et al, 2002), as an ATM effector in DNA damage response (Kamer et al, 2005), or by triggering membrane integration of antiapoptotic family members to neutralize activity (Wilson-Annan et al, 2003). Finally, others have shown that tBid in excess can overcome the antagonizing influence of Bcl-2 on Bax and Bak, arguing that in this context Bax and Bak have the potential to escape conformationally altered Bcl-2.
Our novel inhibitor model of the mechanism of action of Bcl-2 (Figure 7) has important implications for understanding the function and regulation of Bcl-2 in apoptosis. If the conformationally active Bcl-2 is consumed by binding to membrane-integrated Bax and cells are subjected to low-level tonic apoptotic stress, then measurement of the total Bcl-2 in a cell will overestimate the amount that is available to be activated by BH3 proteins, as it will include Bcl-2 already consumed in dead-end complexes with Bax/Bak. Moreover, the conformational change in Bcl-2 represents an important function that may be inhibited or enhanced not only by mutations within the protein (e.g. the hypofunctional mutants studied here, or ‘super Bcl-XL' (Asoh et al, 2000), respectively) but also may be allosterically modulated by other membrane factors. Moreover, the novel, as yet uncharacterized binding interface between conformationally activated Bcl-2 and Bax is an attractive target for selective drug design. It is tantalizing to speculate that the reason that Bcl-2 and Bax have opposite functions even though they are structurally very similar is that subsequent to the induced conformation change, Bcl-2 functions as an oligomerization-defective version of Bax. Finally, it will be important to determine if a similar conformational change is required to activate other antiapoptotic family members such as Bcl-XL (Jeong et al, 2004) or Mcl-1 (Willis et al, 2005) whose membrane integration is regulated differently than Bcl-2.
Materials and methods
Materials
Chemicals were purchased from Sigma Chemicals or Gibco-Life Science, unless otherwise noted. The BL21-SI strain of Escherichia coli used was from Stratagene. The Bim peptide (Ac-MRPEIWIAQELRRIGDEFNA-amide) corresponding to the BH3 domain was blocked at both ends (Dalton Chemicals). The monoclonal antibodies 1H5, against tBid (used at a dilution of 1:4000), and 2D2, an antibody specific for human Bax (1:10 000), were obtained from Ex-alpha Biologicals, Boston and Richard Youle (Hsu and Youle, 1997), respectively. To monitor cytochrome c release, a purified sheep anticytochrome c primary antibody (1:3000) (Ex Alpha Biologicals) was used. Bcl-2 was detected on immunoblots using St-1 (1:10 000), a rabbit polyclonal antibody specific for Bcl-2. Secondary antibodies conjugated to horseradish peroxidase; donkey anti-mouse, donkey anti-sheep and goat anti-rabbit were used at a dilution of 1:10 000 and purchased from Jackson Immuno Research Laboratories Inc.
Recombinant full-length rat Bid with an N-terminal His6 tag was purified and cleaved with caspase-8 as described previously (Zha et al, 2000) The C-terminal fragment of cleaved Bid (tBid) was purified away from the N-terminal Bid fragment and residual caspase-8 as described (Zha et al, 2000). Bax was purified as described (Yethon et al, 2003). Recombinant Bcl-2 with an amino-terminal histidine tag, and lacking the C-terminal insertion sequence were prepared as described (Zhang et al, 2004). Proteins were judged to be greater than 95% pure based on Coomassie Blue staining of SDS–PAGE gels (Yethon et al, 2003).
The chemical labeling gel shift assay for mapping cysteine residues in Bcl-2 embedded in a bilayer was carried out as described (Kim et al, 2004). Integration of Bax into membranes was assayed by extraction with alkali (Annis et al, 2005).
Standard curves of purified Bcl-2ΔTM or Bax were included on the gels to determine the amount of Bcl-2 in mitochondria of Rat-1 MycERTAM cells expressing human Bcl-2 and of Bax targeted to the mitochondria in vitro, respectively. The immunoblots were analyzed using the Kodak Image Station system (440CF). A linear regression analysis was performed on the net intensities of the standards and blots in which the lines of best fit with _R_2 values of ⩾0.95 were analyzed.
Co-precipitation assays
To determine if Bcl-2 and Bax interact, 100 μg of mitochondrial protein at a concentration of 1 mg/ml was incubated with purified recombinant tBid and/or Bax for 1 h at 30°C as below (cytochrome c release assay). Heavy membranes were pelleted by centrifugation at 13 000 g for 10 min and solubilized in SB (250 mM sucrose, 20 mM HEPES, pH 7.5, 2 mM MgCl2, 1 mM EDTA, 1 mM DTT, 1 mM PMSF, 300 mM NaCl, 2% CHAPS and Complete protease inhibitor cocktail (Roche)) for 30 min on ice. Insoluble material (primarily cytoskeletal components) was removed by centrifugation (90 000 g for 20 min) and then samples were precleared with 5 μl of washed protein G agarose beads (Amersham Biosciences) for 1 h. Sheep anti-Bcl-2 antibodies were precipitated with protein G agarose beads and washed three times with SB followed by two washes of SB without CHAPS. Samples were separated by SDS–PAGE and co-precipitated Bax was visualized by immunoblotting with monoclonal mouse anti-Bax antibody.
Cytochrome c release from mitochondria
Heavy membranes enriched in mitochondria were isolated essentially as in (Eskes et al, 2000) from cells lysed by nitrogen cavitation at 150 psi for 15 min on ice in a 45 ml Nitrogen Bomb (Parr Instrument) as described previously (Annis et al, 2005). The debris was separated from the lysate by centrifugation at 2000 g for 4 min. Mitochondria and other heavy membranes were isolated from the supernatant by centrifugation at 13 000 g for 10 min at 4°C. The heavy membrane pellet was resuspended in MB (10 mM HEPES, pH 7.5, 150 mM KCl, 210 mM mannitol, 70 mM sucrose, 0.5 mM EGTA) and used immediately. The isolated heavy membranes were diluted to a concentration of 1 mg/ml of proteins with MBC buffer (210 mM mannitol, 150 mM KCl, 0.5 mM EGTA, 10 mM HEPES, pH 7.5 4 mM MgCl2, 5 mM Na2PO4, 5 mM succinate and 5 μM rotenone) and incubated with Bim peptide and/or purified proteins for 1 h at 30°C. Mitochondria were pelleted by centrifugation at 13 000 g for 10 min and the amount of cytochrome c in the supernatants and pellets was analyzed by SDS–PAGE and immunoblotting using nitrocellulose membranes (Gelman).
Bax oligomerization
The oligomeric state of Bcl-2 and Bax on the mitochondrial membrane was analyzed by gel filtration chromatography. Mitochondrial protein (250 μg at a concentration of 1 mg/ml) was incubated with purified recombinant tBid and/or Bax for 1 h at 30°C as described above. Heavy membranes were pelleted by centrifugation (13 000 g for 10 min) and solubilized in SB buffer (30 min on ice). Insoluble material was removed by centrifugation at 90 000 g for 20 min and the supernatant was applied to a Superdex 200 HR 10/30 column (Amersham Biosciences). The column was equilibrated and eluted in 20 mM HEPES, pH 7.5, 300 mM NaCl, 0.2 mM DTT and 2% CHAPS, and 400 μl fractions were collected. Proteins in every other fraction were precipitated with trichloroacetic acid and analyzed by immunoblotting.
Supplementary Material
Supplementary Information
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Acknowledgments
This work was supported by CIHR Grant FRN 12517 (to DWA and BL) and NIH Grant GM062964 (to JL). Suzanne M Lapolla, G Jane Roberts and Steve Primorac provided technical assistance. DWA holds the Canada Research Chair in Membrane Biogenesis.
References
- Adams JM, Cory S (1998) The Bcl-2 protein family: arbiters of cell survival. Science 281: 1322–1326 [DOI] [PubMed] [Google Scholar]
- Annis MG, Soucie EL, Dlogosz PJ, Cruz-Acado JA, Penn LZ, Leber B, Andrews DW (2005) Bax forms multispanning momomers that oligomerize to permeabilize membranes during apoptosis. EMBO J 24: 2096–2103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asoh S, Ohtsu T, Ohta S (2000) The super anti-apoptotic factor Bcl-xFNK constructed by disturbing intramolecular polar interactions in rat Bcl-xL. J Biol Chem 275: 37240–37425 [DOI] [PubMed] [Google Scholar]
- Eskes R, Desagher S, Antonsson B, Martinou JC (2000) Bid induces the oligomerization and insertion of Bax into the outer mitochondrial membrane. Mol Cell Biol 20: 929–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu YT, Youle RJ (1997) Nonionic detergents induce dimerization among members of the Bcl-2 family. J Biol Chem 272: 13829–132834 [DOI] [PubMed] [Google Scholar]
- Jeong SY, Gaume B, Lee YJ, Ryu SW, Yoon SH, Youle RJ (2004) Bcl-x(L) sequesters its C-terminal membrane anchor in soluble, cytosolic homodimers. EMBO J 23: 2146–2155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamer I, Sarig R, Zaltsman Y, Niv H, Oberkovitz G, Regev L, Haimovich G, Lerenthal Y, Marcellus RC, Gross A (2005) Proapoptotic BID is an ATM effector in the DNA-damage response. Cell 26: 593–603 [DOI] [PubMed] [Google Scholar]
- Kaufmann T, Sclipf S, Sanz J, Neubert K, Stein R, Borner C (2003) Characterization of the signal that directs Bcl-x(L), but not Bcl-2, to the mitochondrial outer membrane. J Cell Biol 160: 53–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim PK, Annis MG, Dlugosz PJ, Leber B, Andrews DW (2004) During apoptosis bcl-2 changes membrane topology at both the endoplasmic reticulum and mitochondria. Mol Cell 14: 523–529 [DOI] [PubMed] [Google Scholar]
- Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR, Mewmeyer DD (2005) BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell 17: 525–535 [DOI] [PubMed] [Google Scholar]
- Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ (2002) Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2: 183–192 [DOI] [PubMed] [Google Scholar]
- Lin B, Kolluri SK, Lin F, Liu W, Han YH, Cao X, Dawson MI, Reed JC, Zhang XK (2004) Conversion of Bcl-2 from protector to killer by interaction with nuclear orphan receptor Nur77/TR3. Cell 116: 527–540 [DOI] [PubMed] [Google Scholar]
- Ottilie S, Diaz JL, Chang J, Wilson G, Tuffo KM, Weeks S, McConnell M, Wang Y, Oltersdorf T, Fritz LC (1997) Structural and functional complementation of an inactive Bcl-2 mutant by Bax truncation. J Biol Chem 272: 16955–16961 [DOI] [PubMed] [Google Scholar]
- Polster BM, Basanez G, Young M, Suzuki M, Fiskum G (2003) Inhibition of Bax-induced cytochrome c release from neural cell and brain mitochondria by dibucaine and propranolol. J Neurosci 23: 2735–2743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffolo SC, Shore GC (2003) BCL-2 selectively interacts with the BID-induced open conformer of BAK, inhibiting BAK auto-oligomerization. J Biol Chem 278: 25039–25045 [DOI] [PubMed] [Google Scholar]
- Sedlak TW, Oltvai ZN, Yang E, Wang K, Boise LH, Thompson CB, Korsmeyer SJ (1995) Multiple Bcl-2 family members demonstrate selective dimerizations with Bax. Proc Natl Acad Sci USA 92: 7834–7838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe JC, Arnoult D, Youle RJ (2004) Control of mitochondrial permeability by Bcl-2 family members. Biochim Biophys Acta 1644: 107–113 [DOI] [PubMed] [Google Scholar]
- Soucie EL, Annis MG, Sedivy J, Filmus J, Leber B, Andrews DW, Penn LZ (2001) Myc potentiates apoptosis by stimulating Bax activity at the mitochondria. Mol Cell Biol 21: 4725–4736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, Adams JM, Huang DCS (2005) Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev 19: 1294–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson-Annan J, O'Reilly LA, Crawford SA, Hausmann G, Beaumont JG, Parma LP, Chen L, Lackmann M, Lithgow T, Hinds MG, Day CL, Adams JM, Huang DCS (2003) Proapoptotic BH3-only proteins trigger membrane integration of prosurvival Bcl-w and neutralize its activity. J Cell Biol 162: 877–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yethon JA, Epand RF, Leber B, Epand RM, Andrews DW (2003) Interaction with a membrane surface triggers a reversible conformational change in Bax normally associated with induction of apoptosis. J Biol Chem 278: 48935–48941 [DOI] [PubMed] [Google Scholar]
- Yin XM, Oltvai ZN, Korsmeyer SJ (1994) BH1 and BH2 domains of Bcl-2 are required for inhibition of apoptosis and heterodimerization with Bax. Nature 369: 321–323 [DOI] [PubMed] [Google Scholar]
- Zha J, Weiler S, Oh KJ, Wei MC, Korsmeyer SJ (2000) Posttranslational _N_-myristoylation of BID as a molecular switch for targeting mitochondria and apoptosis. Science 290: 1761–1765 [DOI] [PubMed] [Google Scholar]
- Zhang Z, Lapolla SM, Annis MG, Truscott M, Roberts GJ, Miao Y, Shao Y, Tan C, Peng J, Johnson AE, Zhang XC, Andrews DW, Lin J (2004) Bcl-2 homodimerization involves two distinct binding surfaces, a topographic arrangement that provides an effective mechanism for Bcl-2 to capture activated Bax. J Biol Chem 279: 43920–43928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong WX, Lindsten T, Ross AJ, MacGregor GR, Thompson CB (2001) BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes Dev 15: 1481–1486 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5