Ozone-Sensitive Arabidopsis rcd1 Mutant Reveals Opposite Roles for Ethylene and Jasmonate Signaling Pathways in Regulating Superoxide-Dependent Cell Death (original) (raw)
Abstract
We have isolated a codominant Arabidopsis mutant, radical-induced cell death1 (rcd1), in which ozone (O3) and extracellular superoxide (O2•−), but not hydrogen peroxide, induce cellular O2•− accumulation and transient spreading lesions. The cellular O2•− accumulation is ethylene dependent, occurs ahead of the expanding lesions before visible symptoms appear, and is required for lesion propagation. Exogenous ethylene increased O2•−-dependent cell death, whereas impairment of ethylene perception by norbornadiene in rcd1 or ethylene insensitivity in the ethylene-insensitive mutant ein2 and in the rcd1 ein2 double mutant blocked O2•− accumulation and lesion propagation. Exogenous methyl jasmonate inhibited propagation of cell death in rcd1. Accordingly, the O3-exposed jasmonate-insensitive mutant jar1 displayed spreading cell death and a prolonged O2•− accumulation pattern. These results suggest that ethylene acts as a promoting factor during the propagation phase of developing oxyradical-dependent lesions, whereas jasmonates have a role in lesion containment. Interaction and balance between these pathways may serve to fine-tune propagation and containment processes, resulting in alternate lesion size and formation kinetics.
INTRODUCTION
Human activities have increased tropospheric ozone (O3) concentration two- to fivefold during the past 40 years (Kley et al., 1999). O3 poses a twofold challenge to plants: photosynthesis and growth can be impaired by increased background concentrations, whereas acutely phytotoxic O3 concentrations are additionally manifested as foliar lesions in sensitive species and cultivars (Kangasjärvi et al., 1994; Kley et al., 1999). Both of these modes of O3 action potentially result in crop losses worth several million dollars annually. Historically, O3 has been regarded as a “wound stress” that causes necrosis by oxidizing and damaging plasma membranes (reviewed in Heath and Taylor, 1997). Recent results, however, indicate that O3 responses resemble components of the hypersensitive response (HR) found in incompatible plant–pathogen interactions (Kangasjärvi et al., 1994; Sharma and Davis, 1997; Sandermann et al., 1998). This similarity is most likely related to the occurrence of reactive oxygen species (ROS), such as superoxide anion radicals (O2•−) and H2O2 in the apoplast. O3-derived ROS apparently trigger, by way of an as yet undescribed mechanism, an oxidative burst in the affected cells (Schraudner et al., 1998; Pellinen et al., 1999; Rao and Davis, 1999). Similarly, an oxidative burst is one of the earliest responses of plants to microbial pathogens and is an integral component in HR-related cell death (Lamb and Dixon, 1997). Accumulation of H2O2 in response to O3 has been reported in tobacco (Schraudner et al., 1998) and birch (Pellinen et al., 1999). In Arabidopsis, O3-induced accumulation of both O2•− and H2O2 has been reported (Rao and Davis, 1999). Sites of ROS accumulation and visible lesions in these plants were of a distinct size, suggesting that ROS production can function as a regulator of cell death not only in the HR, but also under O3 stress.
O3 is known to activate ethylene and salicylate (SA) signal transduction pathways leading to downstream responses, such as antioxidant and antimicrobial defenses (Sharma et al., 1996; Sharma and Davis, 1997; Sandermann et al., 1998). Increased ethylene emission from O3-exposed plants is an early, consistent marker for O3 sensitivity (Tingey et al., 1976; Mehlhorn and Wellburn, 1987; Wellburn and Wellburn, 1996; Tuomainen et al., 1997). O3 has been proposed to exert its toxicity through a chemical reaction with ethylene, yielding toxic products that initiate a self-propagating lipid peroxidation cycle (Elstner et al., 1985; Mehlhorn and Wellburn, 1987). Alternatively, ethylene emission has been postulated as a wounding symptom in O3-exposed plants (Heath and Taylor, 1997). However, ethylene has emerged as a regulator of programmed cell death (pcd)—for example, in pea carpel senescence (Orzáez and Granell, 1997), maize endosperm development (Young et al., 1997), and root aerenchyma formation (Drew et al., 2000). Ethylene also triggers pcd in the accelerated cell death1 (acd1) mutant of Arabidopsis (Greenberg and Ausubel, 1993) and is involved in regulating cell death in plant–pathogen interactions (Bent et al., 1992; Ciardi et al., 2000). These results suggest a potential new role for ethylene as a regulator of cell death in O3 responses.
In O3-exposed plants, ethylene synthesis is a result of the specific activation of 1-aminocyclopropane-1-carbocylic acid (ACC) synthase and ACC oxidase genes (Tuomainen et al., 1997) and is required for O3 damage (Mehlhorn and Wellburn, 1987). Jasmonic acid (JA), on the other hand, can protect tobacco plants from O3 when applied before O3 treatment (Örvar et al., 1997), and the O3 sensitivity of a poplar clone has been proposed to result from JA insensitivity (Koch et al., 1998). Thus, these two signaling pathways, which act synergistically in induced systemic resistance (Pieterse et al., 1998) and in regulating expression of pathogenesis-related (Norman-Setterblad et al., 2000) and wound- induced (O'Donnell et al., 1996) genes, appear to have opposing effects with regard to O3 sensitivity. However, the detailed role or interaction of JA and ethylene signaling in the regulation of ROS accumulation during O3-induced or other forms of ROS-dependent cell death has not been described.
Here, we report the identification and characterization of an Arabidopsis mutant, rcd1 (for radical-induced cell death1), that displays O3-inducible and O2•−-inducible lesion formation. We also show that O3 response mutants can be useful in identifying interacting components of ROS-dependent signaling pathways in plant–pathogen interactions, for example, to elucidate interaction between ROS and other pathways that regulate the HR lesion formation in incompatible plant–pathogen interactions. Our results suggest that a highly regulated induction of ethylene biosynthesis promotes cell death and that functional ethylene perception and signaling are required for O2•− accumulation, which is responsible for the oxyradical-dependent cell death, ultimately leading to lesion propagation. JA signaling, on the other hand, is involved in lesion containment. On the basis of these results, we propose a model for the relative contribution of the different signaling pathways to ROS-driven lesion propagation and the process of lesion containment.
RESULTS
Isolation and Genetic Mapping of a Novel Arabidopsis Mutant, rcd1,That Displays HR-like Lesion Development in Response to O3
Approximately 14,000 individual M2 plants, grown from ethyl methanesulfonate–mutagenized Columbia (Col-0) seed, were exposed for 3 days to 250 nL L−1 O3 (8 hr/day) at a density of 3000 plants m−2. Fifty-six individuals displaying O3-induced lesions on rosette leaves were identified. In the subsequent screenings at low density, four lines representing independent loci and distinct patterns and kinetics of lesion formation were selected from 11 lines displaying consistent O3-sensitive phenotypes. When these four lines were crossed with each other and the O3 sensitivity was assayed in the F1 progeny, no O3-sensitive F1 lines were detected, indicating that these mutants represent independent loci. Detailed characterization of the most sensitive mutant, rcd1, is presented here. To determine its mode of inheritance, rcd1 was crossed with Col-0. In the F2 progeny, three phenotypes, both parentals and an intermediate, segregated as a single codominant Mendelian trait, 1:2:1 (36:66:46, χ2  3.08,
3.08, 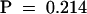 ). The F2 progeny from the rcd1 × Landsberg erecta (L_er_) were used for mapping with polymerase chain reaction–based microsatellite and cleaved amplified polymorphic sequence markers that cover all five chromosomes. Using 32 homozygous, O3-sensitive rcd1 individuals from the F2 progeny, we were able to position the locus in chromosome 1 at 53 ± 2.2 centimorgans in the recombinant inbred map, 2 centimorgans from a microsatellite in bacterial artificial chromosome clone F23M19. No mutants with similar or related phenotypes had previously been assigned to this region.
). The F2 progeny from the rcd1 × Landsberg erecta (L_er_) were used for mapping with polymerase chain reaction–based microsatellite and cleaved amplified polymorphic sequence markers that cover all five chromosomes. Using 32 homozygous, O3-sensitive rcd1 individuals from the F2 progeny, we were able to position the locus in chromosome 1 at 53 ± 2.2 centimorgans in the recombinant inbred map, 2 centimorgans from a microsatellite in bacterial artificial chromosome clone F23M19. No mutants with similar or related phenotypes had previously been assigned to this region.
O3-induced lesions of rcd1 initiate along the leaf margins and spread inward through intervascular tissue. Normally, visible symptoms appear first as dark, water-soaked spots 12 hr after the onset of O3 treatment (250 nL L−1) on middle-aged rosette leaves. When whole tissues are affected by many coalescing lesions, tissue collapse joins medially and progresses toward the leaf base and tip. In this case, damage is already visible at 3 hr as a loss of leaf turgor. By 24 hr, as shown in Figure 1A, lesion progression in rcd1 had halted, and the lesions had developed into well-defined dry patches of dead tissue. Lesion initiation was independent of the duration of O3 exposure (250 nL L−1) between 1 and 8 hr; only the extent of lesion formation increased with time. No visible symptoms were found on Col-0 (Figure 1A). However, microscopic examination of Col-0 tissue revealed individual cells and small clusters of cells that had died as a result of O3 exposure. Lesions in rcd1 were not triggered by application of SA (or its active analog benzo[1,2,3]thiadiazole-7-carbothioic acid [BTH]) or ethylene, high-intensity light, UV-B radiation, wounding, or alterations in daylength (data not shown)—all factors known to trigger lesions in other phenotypically related mutants.
Figure 1.

Progression of Damage and Accumulation of O2•− in O3-Exposed Arabidopsis.
(A) Leaf damage in Col-0 and rcd1 24 hr after the onset of a 6-hr exposure to 250 nL L−1 O3.
(B) O2•− accumulation in O3-exposed leaves. Col-0 and rcd1 were exposed to O3 as above. Plants were removed from the chamber at the times indicated, and detached leaves were infiltrated with nitroblue tetrazolium (NBT). The presence of the purple formazan precipitate indicates the location and extent of O2•− accumulation.
(C) Effect of inhibitors on O2•− accumulation. Leaves of rcd1 were exposed to 4 hr of O3 (250 nL L−1) or 4 hr of O3 (250 nL L−1) followed by norbornadiene (NBD; 30 μL L−1), as indicated (−NBD or +NBD). After O3 exposure, leaves were infiltrated with NBT. The specificity of NBT staining for O2•− and the cellular origin of O2•− were verified by infiltrating O3-exposed (250 nL L−1) leaves with NBT in combination with superoxide disumutase (SOD) or diphenylene iodonium (DPI) (20 μM), an inhibitor of flavin-containing oxidases, as indicated. The role of ethylene insensitivity in O2•− accumulation was elucidated by exposing Col-0 and ein2 to a high concentration (400 nL L−1) of O3. SOD- and DPI-treated leaves were stained at 4 hr, and leaves were treated (or not) with NBD (+/−) and stained at 8 hr. Col-0 and ein2 leaves exposed to 400 nL L−1 O3 were stained immediately after the 2-hr exposure.
O3 Induces O2•− Accumulation in Front of the Spreading Lesion in rcd1
Lesion development in rcd1 is transiently comparable with lesion propagation in lsd1 (for lesion simulating disease resistance1), in which extracellular O2•− production precedes lesion spread (Jabs et al., 1996). This prompted us to assay cellular O2•− accumulation, as shown in Figure 1B, in O3-exposed rcd1 by monitoring the precipitation of purple formazan when reacting nitro blue tetrazolium (NBT) with O2•−. To exclude reactions of NBT with O3-derived O2•− (Runeckles and Vaartnou, 1997), the plants were postcultivated in pollutant-free air for at least 15 min after exposure to O3. No NBT precipitation could be detected in clean air–grown rcd1 or Col-0 (data not shown). However, 2 to 4 hr after the onset of O3 exposure, NBT precipitation was visible in both Col-0 and rcd1, although the distribution in each strain differed. In Col-0, NBT precipitation was scattered throughout the leaf, with greater amounts at the tip and margins, whereas in rcd1, staining was concentrated first (at 2 hr) in the region in which the lesions typically initiated and later (at 4 to 8 hr) just beyond the boundary of the collapsing tissue. No NBT staining was observed in Col-0 at 8 hr or later time points. In rcd1, as illustrated in Figure 2, precipitation of NBT by O2•− continued in front of lesion expansion for as long as 12 hr after the onset of the 6-hr O3 treatment. NBT precipitation clearly was the result of O2•− accumulation because staining was abolished when superoxide dismutase (SOD) was coinfiltrated into the leaves (Figure 1C).
Figure 2.

Spatial Relationship between O2•− Accumulation and Spreading Lesions.
(A) The overall staining pattern of an O3-exposed (6 hr × 250 nL L−1) rcd1 leaf stained for O2•− at 8 hr. The leaf area with collapsed cells is indicated with an asterisk.
(B) Light microscopy showing details of the boxed region in (A).
(C) UV epifluorescence of the same region as in (B), showing autofluorescent phenolic compounds (arrowhead) defining the boundary of lesioned tissue. The O2•− accumulation occurred in the healthy tissue adjacent to the expanding lesion.
We used ion leakage, an indicator of plasma membrane damage, as a quantitative measure of the extent of cell death. When ion leakage was monitored in O3-exposed plants, marked differences were found between Col-0 and rcd1 (Figure 3A). In rcd1, ion leakage increased 10-fold during O3 exposure and showed a further increase, even in the absence of O3, between 6 and 12 hr, when the lesions were spreading. In contrast, O3 induced only a small, transient increase in Col-0 during the exposure period.
Figure 3.

Progression of O3-Induced and O2•−-Induced Leaf Damage in Col-0 and rcd1.
(A) O3-induced cell death. Plants were exposed to O3 (indicated by the black bar) and postcultivated in pollutant-free air. The extent of cell death was measured as relative ion leakage (percentage of total ions ±se).
(B) O2•−-induced cell death. Leaves from clean air–grown plants were detached and vacuum-infiltrated with the O2•−-generating system xanthine and xanthine oxidase (X/XO). The black triangle under the x axis indicates the approximate duration and intensity of O2•− synthesis by X/XO. The time course of cell death, measured as relative ion leakage (±se), was monitored for 24 hr.
(C) Inhibition of cell death by SOD. Leaves of Col-0 and rcd1 were infiltrated with the O2•−-generating system X/XO, MnSOD (440 units mL−1), or infiltration buffer, as indicated. Reagents were included (+) or not (−) in each treatment as indicated. Cell death was measured as ion leakage (±se) after 20 hr.
(D) H2O2 and cell death. Leaves from clean air–grown plants were detached and vacuum-infiltrated with increasing concentrations of H2O2. Cell death in Col-0 and rcd1 was measured as relative ion leakage (±se) after 20 hr.
Extracellular O2•–, but Not H2O2, Is Both Necessary and Sufficient to Trigger Propagation of Cell Death in rcd1
To elucidate the role of O2•− and H2O2 in lesion development, we infiltrated Col-0 and rcd1 leaves with an extracellular O2•−-generating system, xanthine/xanthine oxidase (X/XO); with increasing concentrations of H2O2; and with an H2O2-generating system, glucose/glucose oxidase (G/GO) (Jabs et al., 1996; Alvarez et al., 1998). O2•− production by X/XO lasted for ∼3 hr and induced cell death in Col-0 and rcd1 with different magnitudes and kinetics (Figure 3B). In X/XO-infiltrated Col-0 plants, ion leakage increased to ∼25% during the first 4 to 8 hr and remained at that value during the experiments. Changes in ion leakage were more dramatic in rcd1, which showed a second increase to as much as 65% between 8 and 24 hr (Figure 3B). Thus, O3 could be functionally replaced by O2•− produced with X/XO. When the X/XO-produced O2•− was removed by coinfiltration with MnSOD, cell death in rcd1 was not triggered to any greater extent than in Col-0 (Figure 3C). Therefore, O2•− alone seems sufficient to induce lesion formation. Physiological concentrations of H2O2 did not induce cell death in rcd1. Infiltration of rcd1 leaves with an active H2O2-producing system (G/GO) did not cause increased cell death in either rcd1 or Col-0 in the range of steady state H2O2 production between 5 and 250 μM when compared with buffer alone (data not shown). Furthermore, direct treatment of excised rcd1 or Col-0 leaves with H2O2 solutions up to 10 mM showed no substantial increase in cell death in either strain compared with the buffer control. Greater H2O2 concentrations (50 and 100 mM) resulted in equal increases in cell death in both Col-0 and rcd1 (Figure 3D).
The role of endogenous O2•− accumulation triggered by O3 during the early phases of lesion formation was studied further by inhibition experiments, specifically with diphenylene iodonium (DPI), an inhibitor of ROS accumulation in Arabidopsis (Jabs et al., 1996; Alvarez et al., 1998). Infiltration of rcd1 leaves with DPI after a 2-hr O3 exposure prevented NBT precipitation (Figure 1C) and reduced leaf damage in a concentration-dependent manner (Figure 4). Half-maximal reduction was obtained with 2 μM DPI, maximal reduction (∼45%) with 5 μM. We conclude from these data that both O3 and O2•− from X/XO initiate active cellular O2•− production that continues in rcd1 after exogenous O2•− ceases. Furthermore, this O2•− is both necessary and sufficient to propagate cell death in rcd1.
Figure 4.

Role of Endogenous O2•− Production in O3-Induced Lesion Formation.
Leaves of rcd1 were exposed to 250 nL L−1 O3 for 2 hr and then infiltrated with indicated concentrations of the flavin oxidase inhibitor DPI or with buffer alone. Damaged leaf area was assessed at 24 hr. The effect of DPI is expressed as the percentage of damaged leaf area (±se) of plants infiltrated with buffer.
Incompatible Bacterial Pathogen Induces Extensive Cell Death during the HR in rcd1
An oxidative burst, the active production of apoplastic O2•− and H2O2, is one of the earliest plant responses in incompatible interactions and is integrally involved in regulating hypersensitive cell death (Lamb and Dixon, 1997). To analyze whether incompatible interaction with a bacterial pathogen triggers spreading lesions in rcd1, we infiltrated 3-week-old rosettes of Col-0 and rcd1 with increasing concentrations of Pseudomonas syringae pv tomato, strain DC3000, carrying the avirulence gene avrB on a plasmid (Bent et al., 1992). The extent of tissue collapse caused by the HR was quantified by measuring ion leakage. As seen in Table 1, infiltration of rosette leaves with bacterial concentrations from 104 to 107 colony-forming units (cfu) mL−1 induced substantially higher ion leakage in rcd1, whereas in mock treatments, no difference between Col-0 and rcd1 was visible. The difference between Col-0 and rcd1 regarding ion leakage was of similar magnitude for every concentration used. Thus, we concluded that spreading lesions are triggered in rcd1 also by an incompatible interaction, which involves ROS production in the oxidative burst.
Table 1.
Avirulent Pathogen Triggers Spreading Cell Death in rcd1a
| cfu/mL | Col-0 | rcd1 |
|---|---|---|
| Mock | 9.4 ± 0.5 ab | 9.2 ± 0.7 a |
| 104 | 12.6 ± 0.5 ab | 20.9 ± 2.5 bc |
| 105 | 17.4 ± 1.3 bc | 25.5 ± 2.4 c |
| 106 | 41.1 ± 3.3 d | 53.2 ± 1.7 e |
| 107 | 59.9 ± 3.3 e | 71.9 ± 3.0 f |
Oxygen Radicals Induce Pathogen Defense Genes but Not Antioxidant Genes in rcd1 and Col-0
Expression of antioxidant, antimicrobial, and other stress-related genes was analyzed by using 92 expressed sequence tags (ESTs) and cDNA clones in cDNA macroarrays. A subset of the results is shown in Table 2. No major differences were found between Col-0 and rcd1 cultivated in clean air except for lower amounts of catalase (CAT3) and defensin (PDF1.2) mRNAs in rcd1 (Table 2). O3 induced major changes in the expression of PR-1, two glutathione _S_-transferase genes (GST1 and GST2), and catalases 1 and 3 in both rcd1 and Col-0. O3-induced increases in GST1 and GST2 mRNAs were markedly greater in rcd1 (26-fold) than in Col-0 (nine- to 12-fold). CAT3 transcripts increased in both Col-0 and rcd1 such that by 8 hr, they were approximately equivalent. SOD isoforms showed minor responses, or their transcript levels were decreased (chloroplastic Cu/ZnSOD) in both Col-0 and rcd1. We concluded that because the transcript levels for enzymes detoxifying O2•−, H2O2, and lipid hydroperoxides are approximately equal in Col-0 and rcd1, then the radical-induced cell death in rcd1 is not a result of decreased expression of antioxidative genes.
Table 2.
Stress and Defense Gene Transcript Levelsa
| Geneb | EST Stock/Accession No. | Air | Ozone | ||
|---|---|---|---|---|---|
| Col-0 | rcd1 | Col-0 | rcd1 | ||
| PR1 | M90508 | 0.1c | 0.1 | 2.0⇑d | 2.3⇑ |
| CHIb | 92G1T7 | 0.1 | 0.1 | 0.2 | 0.2 |
| GST1 | 206N21T7 | 0.4 | 0.4 | 5.0⇑ | 11⇑ |
| GST2 | 91H22T7 | 0.6 | 0.5 | 6.5⇑ | 13⇑ |
| AT-ACO2 | G2B6T7 | 0.3 | 0.5 | 0.6 | 1.6↑ |
| MNSOD1 | 109J19T7 | 1.6 | 2.3 | 2.0 | 1.6 |
| Cu/ZnSOD1 | 2G11T7P | 2.5 | 3.3 | 3.0 | 4.2 |
| Cu/ZnSOD2 | 161I22T7 | 4.5 | 5.1 | 2.6 | 1.6 |
| FeSOD1 | 34D9T7 | 1.0 | 2.2 | 0.4 | 0.2 |
| APX1 | 135D24T7 | 1.9 | 2.2 | 1.7 | 2.7 |
| GR | 185P3T7 | 1.1 | 1.7 | 0.3 | 0.8 |
| GPX2 | 190H7T7 | 2.0 | 2.2 | 0.7 | 1.9 |
| catalase3 | 134I1T7 | 5.3 | 2.2 | 12↑ | 10⇑ |
| catalase1 | 118M15T7 | 0.3 | 0.3 | 2.2⇑ | 1.0↑ |
| AOS | 230J8T7 | 0.8 | 0.6 | 0.5 | 0.3 |
| PDF1.2 | 37F10T7 | 1.3 | 0.4 | 1.5 | 0.5 |
Ethylene Signaling Is Required for Accumulation of O2•–, Which Drives Lesion Propagation
Similar concentrations of O3 and O2•− (X/XO treatment) in Col-0 and rcd1 plants led to markedly more cell death in rcd1 (Figures 1B and 2), suggesting that the initial amount of ROS formation from O3 or X/XO is not the only factor involved in the initiation of lesion propagation. Several biochemical variables have been studied as the basis for O3 sensitivity. Ethylene emission has been found to be the most consistent response to O3 (Mehlhorn and Wellburn, 1987; Wellburn and Wellburn, 1996; Tuomainen et al., 1997).
Figure 5 shows results of detailed studies on ethylene biosynthesis in Col-0 and rcd1. Of the five Arabidopsis ACC synthase genes analyzed—AT-ACS1, 2, 4, 5, and _6_—only the isoform AT-ACS6 was responsive to O3. AT-ACS6 was already highly induced in rcd1 at 30 min after the onset of O3 exposure (Figure 5A). Transcript levels were at their maximum at 1 to 2 hr and decreased thereafter. Enhanced activation of AT-ACS6 in rcd1 was also reflected in increased ACC concentrations (Figure 5B). Similarly, the amounts of ACC oxidase transcripts (Table 2) and ethylene evolution (Figure 5C) were also three- to fivefold higher in rcd1 than in Col-0, and in contrast to Col-0, ethylene evolution in rcd1 continued past the period of O3 exposure.
Figure 5.

O3 Induction of Ethylene Biosynthesis.
(A) Time course of O3 induction of the ACC synthase gene AT-ACS6. Transcript levels shown are relative to the hybridization signal of control samples taken at the same times.
(B) Time course of concentrations of the ethylene precursor ACC (±se).
(C) Ethylene evolution (±se) in clean air–grown and O3-exposed Col-0 and rcd1. O3 exposure (250 nL L−1 for 6 hr) was followed by postcultivation in pollutant-free air.
Black bars indicate the duration of exposure. FW, fresh weight.
It has been proposed (Elstner et al., 1985; Mehlhorn and Wellburn, 1987) that a chemical reaction between ethylene and O3 could produce water-soluble, highly reactive radicals that could initiate a self-propagating peroxidative cycle, thus causing ethylene-mediated damage without the action of ethylene signaling. Results of the experiments addressing this question are shown in Figure 6. rcd1 was exposed first to O3, followed by incubation either in clean air or in the presence of an antagonist of ethylene action, norbornadiene (NBD). After rcd1 was treated for 4 hr with O3 (250 nL L−1), ion leakage had increased slightly above that of the clean air–grown control plants (Figure 6A). In plants postcultivated in clean air after the exposure, ion leakage showed an additional fourfold increase. In contrast, blocking of ethylene perception with NBD treatment during postcultivation inhibited any additional increase in ion leakage (Figure 6A) and led to disappearance of the NBT staining-detectable O2•− accumulation (Figure 1C). The effect of ethylene on O2•− accumulation and cell death appeared thus to act by way of ethylene perception, not through a chemical reaction between ethylene and O3.
Figure 6.

Role of Ethylene Perception and Signaling in the Formation of O3-Induced Leaf Damage.
(A) Inhibition of ethylene perception. rcd1 plants were exposed to O3 (250 nL L−1) for 4 hr (indicated by the black bar) and subsequently exposed to clean air or to the ethylene antagonist NBD (30 μL L−1). Cell death was measured as relative ion leakage (percentage of total ions ±se) after 0, 4, and 8 hr.
(B) O3 exposure and external ethylene. Col-0, rcd1, and the ethylene-insensitive mutant ein2 were exposed to clean air, 4 hr of O3 (250 or 400 nL L−1), or 4 hr of O3 followed by 4 hr of ethylene (12 μL L−1). Cell death was measured as ion leakage (±se) after 8 hr. Insert shows ethylene evolution from Col-0 and ein2 in clean air (C) and after 4 hr of 400 nL L−1 O3.
(C) O2•− and exogenous ACC. Leaves of Col-0, rcd1, and ein2 were infiltrated with the O2•−-generating system X/XO, with the ethylene precursor ACC (50 μM), or with an infiltration buffer. Cell death was measured as relative ion leakage (percentage of total ions ±se) after 20 hr.
(+), reagents included; (−), reagents not included.
Ethylene Insensitivity Confers O3 Tolerance
The role of ethylene signaling in O3-induced tissue damage was additionally addressed with the ethylene-insensitive mutant ein2 (Guzman and Ecker, 1990). After exposure to 250 nL L−1 O3 for 4 hr, rcd1 displayed more cell death than did Col-0 and ein2 (Figure 6B). When O3 was followed by 4 hr of ethylene (12 ± 2 μL L−1), a marked increase in cell death was evident in both rcd1 and Col-0 but not in ein2. Ethylene treatment alone did not induce O2•− accumulation or lesion formation in rcd1 (not shown). Furthermore, when the plants were exposed to 400 nL L−1 O3, leaf damage in both Col-0 and rcd1 was extensive (Figure 6B), and predominant accumulation of O2•− was apparent in Col-0 (Figure 1C). Under the same conditions, ein2 showed just minor O2•− accumulation at 2 hr (Figure 1C) and no later leaf damage. Enhanced ethylene synthesis (by two- to fourfold), however, was evident in both Col-0 and ein2 under this regime (Figure 6B, insert), indicating that O3 enters the leaves and elicits a response in both strains. To address whether O2•− in combination with endogenously produced ethylene has similar effects as O3 and exogenous ethylene, leaves of Col-0, rcd1 and ein2 were infiltrated with X/XO in the presence and absence of the ethylene precursor ACC (Figure 6C). X/XO-induced cell death, determined as ion leakage after 20 hr, increased in Col-0 (by twofold), rcd1 (by threefold), and ein2 (by fivefold) in comparison with buffer controls. Although ACC alone had a negligible effect, O2•− and ACC together had synergistic effects over O2•− alone in Col-0 (3.5-fold increase) and rcd1 (4.5-fold increase) but not in ein2.
The position of rcd1 relative to the ethylene pathway was further elucidated by crossing rcd1 with ein2 and identifying the F2 individuals that were homozygous for both mutations. Epistatic relationships between the loci were analyzed by exposing the double mutant to 4 hr of O3 followed by 4 hr of clean air in a setup similar to that used with NBD. Figure 7 shows that no differences were detected in ion leakage in O3-exposed Col-0 or ein2 during the course of the experiments. In O3-exposed rcd1, ion leakage increased from control values to 9% leakage during the 4-hr O3 exposure and continued to increase to 16% during the subsequent 4 hr in clean air. The rcd1 ein2 double mutant showed an increase in ion leakage during the 4-hr O3 exposure similar to that for rcd1. However, the spread of cell death in clean air after O3 was absent, and by 8 hr, ion leakage had decreased back to the control value (Figure 7). Ethylene insensitivity in the double mutant thus prevented the occurrence of spreading cell death after O3 exposure in a manner similar to NBD treatment (Figure 6A) without affecting the increase in cell death during O3 exposure. Thus, we concluded that both a highly regulated increase in ethylene evolution and functional ethylene signaling are required for the amplification of cell death.
Figure 7.

Progression of Cell Death in O3-Exposed Col-0, rcd1, ein2, and rcd1 ein2 Double Mutant.
Three-week-old plants were exposed to O3 (250 nL L−1) for 4 hr (indicated by the black bar) and then kept in clean air. Whole rosettes were collected at 0, 4, and 8 hr, and ion leakage from damaged cells was determined as a measure of cell death. Cell death is expressed as relative ion leakage (percentage of total ions ±se).
JA Signaling Is Involved in Containment of HR-like Lesion Development and Counteracts the Ethylene-Dependent, O2•–-Driven Lesion Propagation
In rcd1, cell death did not spread throughout the leaf; lesion propagation was contained between 12 and 24 hr. We looked at other possible signaling pathways that could be involved in lesion containment. Kangasjärvi et al. (1994) proposed that JA could act as signaling molecule in plant–O3 interactions. Furthermore, Table 2 shows that fewer transcripts of defensin (PDF1.2), a gene that is coregulated by ethylene and JA (Penninckx et al., 1998), were present in both clean air–grown and O3-exposed rcd1. This suggests that JA biosynthesis or signaling, or interaction of JA and ethylene signaling, could be affected in rcd1, because the ethylene biosynthesis and signaling pathways in rcd1 are functional.
Results of the experiments addressing the role of JA in lesion containment are shown in Figure 8. rcd1 was first treated with methyl jasmonate (MeJA) after 4 hr of O3 exposure in an experiment similar to that used with the ethylene antagonist NBD (cf. with Figure 6A). Exposure of rcd1 to 1.4 μM MeJA after O3 exposure inhibited lesion propagation from 4 to 8 hr (Figure 8A). This suggests that JA treatment is counteracting or preventing the lesion propagation executed by the ethylene-dependent O2•− accumulation in rcd1. Furthermore, this suggests that rcd1 is not JA insensitive. Table 3 shows further verification: Roots of rcd1 were sensitive to growth inhibition by MeJA, whereas JA-insensitive jar1 was not affected. In the presence of 10 μM MeJA, root growth inhibition in rcd1 was similar to that in Col-0 (61 and 66%, respectively), whereas in jar1, no substantial inhibition occurred. Thus, based on JA inhibition of root elongation, rcd1 cannot be regarded as JA insensitive. rcd1 is not deficient in JA biosynthesis either, because exposure of rcd1 to O3 led to much greater concentrations of JA than in Col-0 (not shown).
Figure 8.
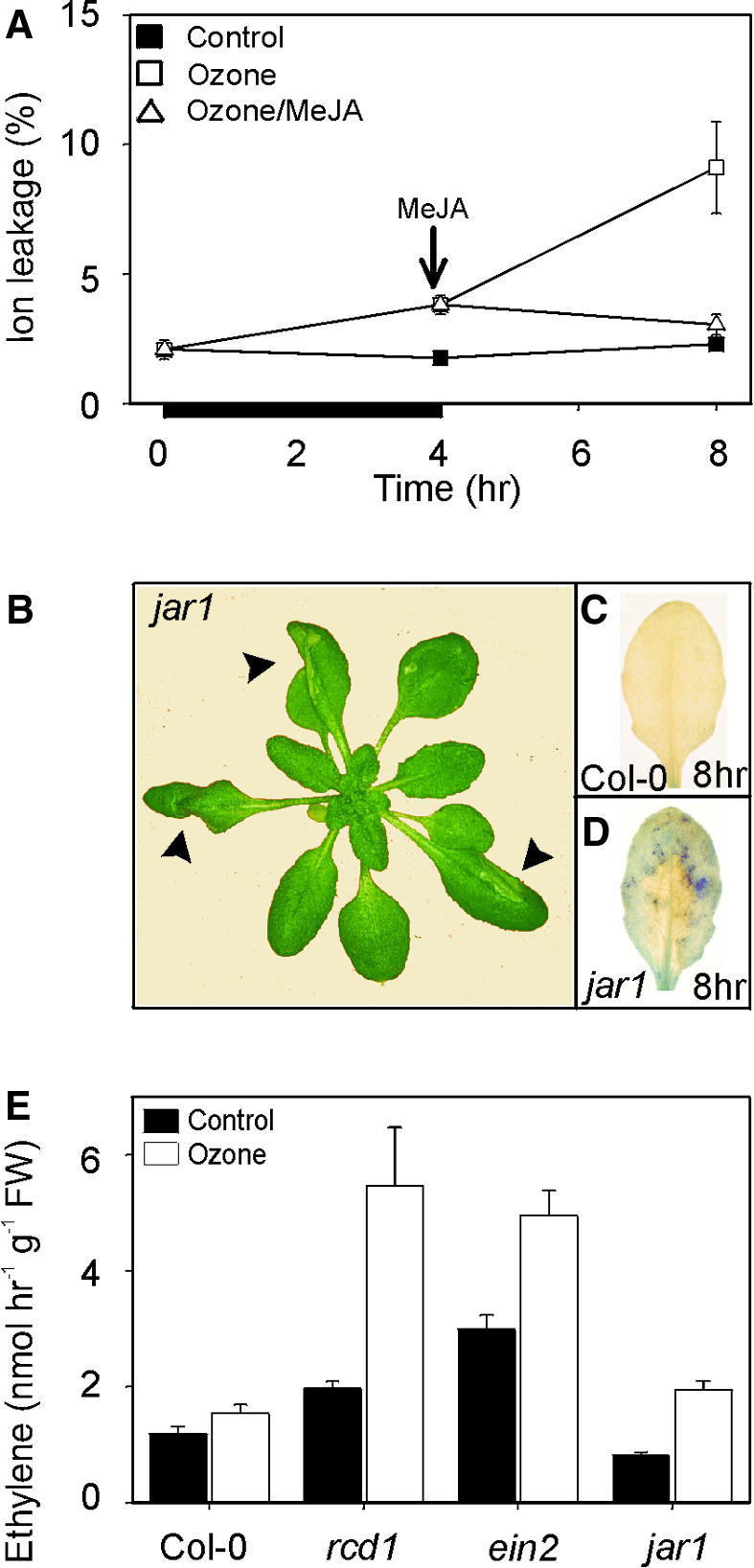
Role of JAs and JA Signaling in Lesion Containment.
(A) Jasmonate protection of O3-induced cell death in the rcd1 mutant. Three-week-old rcd1 plants were exposed to O3 (250 nL L−1) for 4 hr (indicated by the black bar under the x axis) and then exposed to clean air or MeJA (1.4 μM). Cell death was measured as relative ion leakage (percentage of total ions ±se) after 0, 4, and 8 hr.
(B) O3 sensitivity of a JA-insensitive mutant jar1. Shown is the O3 damage in the leaves of jar1 at 24 hr after the onset of a 6-hr exposure to 250 nL L−1 O3.
(C) O3-induced accumulation of O2•− in Col-0. Leaves of O3-exposed Col-0 stained for O2•− accumulation at 8 hr show no NBT precipitation.
(D) O3-induced accumulation of O2•− in a JA-signaling mutant. The JA-insensitive mutant jar1 was exposed to 6 hr of O3 (250 nL L−1). Plants were removed from the chamber at 8 hr, and detached leaves were infiltrated with NBT. The presence of the purple formazan precipitate indicates the location and extent of O2•− accumulation.
(E) Ethylene evolution from Col-0, rcd1, jar1, and ein2 in clean air and after 2 hr of 250 nL L−1 O3. FW, fresh weight. Error bars indicate se.
Table 3.
Inhibition of Root Elongation by MeJa in Col-0, rcd1, and jar1
| MeJa (μM)a | Col-0 | rcd1 | jar1 |
|---|---|---|---|
| 0 | 34.4 ± 1.6 cb | 26.6 ± 0.7 b | 28.3 ± 1.0 b |
| 10 | 11.6 ± 1.1 a | 10.2 ± 0.4 a | 26.4 ± 0.7 b |
To elucidate the role of JA in lesion propagation and containment, we further utilized the JA-insensitive mutant jar1 (Staswick and Howell, 1992). O3-exposed jar1 developed spreading lesions similar to those of rcd1 (Figure 8B). Furthermore, in the margins of the spreading lesion, jar1 exhibited O2•− accumulation (Figure 8D) in a manner similar to rcd1. The same initial O2•− accumulation as in Col-0 at 1 to 2 hr seemed sufficient to initiate lesion propagation in jar1. However, the O3-induced lesion propagation in jar1 was not accompanied by similar high amounts of ethylene as were seen in rcd1 (Figure 8E). This suggests that jar1 is less efficient in lesion containment, in contrast to rcd1, which shows increased lesion propagation. Thus, these results suggest that JA signaling is required in processes that prevent the O2•−-dependent lesion propagation or that contain lesion spread.
DISCUSSION
The novel rcd1 mutant described here is sensitive to extracellular oxygen radicals and shows extensive formation of HR-like lesions and activation of several pathogen defense–related processes in response to ROS. Phenotypically, rcd1 is in part similar to the acd (Greenberg and Ausubel, 1993) and lsd (Dietrich et al., 1994) mutants that have been utilized to identify the components regulating hypersensitive cell death downstream of pathogen infection. In particular, the “runaway” spread of cell death in rcd1 during the first 8 to 12 hr is reminiscent of that in lsd1, in that a front of O2•− accumulation has been found at lesion boundaries in living tissues in both mutants. However, in addition to avirulent Pseudomonas, which induces an oxidative burst in the affected cells, O2•− and O3 are the only treatments known thus far to trigger the spreading lesion formation in rcd1, whereas various other factors, such as SA, BTH, ethylene, or growth conditions, trigger lesion formation in lsd or acd mutants. Thus, rcd1 seems to define a component of the HR related to the role of ROS in cell death regulation. The chromosomal location of rcd1, in addition, has not been described for any acd, lsd, antioxidant, or disease-related mutants. Therefore, rcd1 describes a novel class of ROS-responsive lesion-mimic mutants.
rcd1 Appears to Be Deficient in the Control of Lesion Propagation
The phenotypes described in various lesion-mimic mutants suggest that three separate processes—initiation, propagation, and containment—are involved in lesion formation (Greenberg and Ausubel, 1993; Dietrich et al., 1994; Greenberg et al., 1994; Yu et al., 1998; Rate et al., 1999). For example, lsd1 and acd2 are defective in the control of lesion initiation; that is, several inappropriate stimuli can trigger the HR-like lesions. In addition to quantitative differences in initiation, rcd1, reminiscent of lsd1 and acd1, seems to be deficient in the control of lesion propagation. This deficiency, however, occurs only transiently in rcd1, because after 24 hr, concomitant with the cessation of O2•− accumulation, the propagation of the lesion is brought under control. In contrast, in the dnd1 (for defense, no death1) mutant (Yu et al., 1998), initiation of cell death takes place, but either propagation is deficient or containment is hyperactive. Similarly, lesion initiation but no propagation takes place in O3-treated Col-0 (Figures 1 and 2), tobacco (Schraudner et al., 1998), tomato (Tuomainen et al., 1997), and birch (Pellinen et al., 1999). Related processes can also be seen during systemic acquired resistance to pathogens, in which the systemic leaves show microbursts of H2O2 and subsequent cell death (micro-HR; Alvarez et al., 1998) that resembles lesion initiation.
Increased O3 and O2•–-Induced Lesion Propagation in rcd1 Is Not a Result of Deficient Antioxidative Protection
Propagation of the lesion is most likely to involve generation at the initiation site of a signal that regulates death of the neighboring cells. Both O2•− and H2O2 have been shown to act as signal molecules in various systems (Lamb and Dixon, 1997). O3-exposed tobacco (Schraudner et al., 1998) and birch (Pellinen et al., 1999) showed predominant H2O2 but no O2•− accumulation, whereas O3-exposed Arabidopsis predominately accumulates O2•−. Our results suggest that in Arabidopsis, O2•− can act as a positive regulator in the amplification of a cellular oxidative burst and cell death in the surrounding cells. O3, and O2•− generated by X/XO, induced O2•− accumulation and cell death in rcd1 (Figure 1B), whereas H2O2 concentrations as great as 10 mM were ineffective (Figure 3D). This raises the question of the functionality of O2•− scavenging in rcd1. LSD1 is known to be involved in the induction of CuZnSOD in Arabidopsis (Kliebenstein et al., 1999), and O3 sensitivity of vtc1 is a result of low ascorbate concentrations (Conklin et al., 1996). Similarly, several O3-sensitive mutants that are deficient in Fe and CuZnSOD gene expression have been isolated (D. Kliebenstein and R. Last, unpublished data). However, transcript levels of several antioxidant genes were approximately the same in rcd1 and Col-0 at 8 hr after the beginning of the exposure (Table 2). Furthermore, ascorbate concentrations and total catalase and SOD activities were similar in both clean air–grown and O3-exposed Col-0 and rcd1 (K. Overmyer, H. Tuominen, and J. Kangasjärvi, unpublished data). Thus, at an overall level, deficient antioxidant capacity does not seem to be responsible for lesion propagation in rcd1; other signals and mechanisms must be involved.
Specific Activation of Ethylene Biosynthesis and Functional Ethylene Signaling Are Required for Amplification of O2•– Accumulation and Cell Death
Ethylene synthesis in the O3-exposed rcd1 was a result of fast, specific activation of the ACC synthase gene AT-ACS6 (Figure 5A). A similar O3 responsiveness of only the AT-ACS6 gene has also been seen in Col-0 (Vahala et al., 1998). In Col-0, both O3-induced ethylene evolution and O2•− accumulation lasted approximately the same time as the O3 exposure, whereas in rcd1 they continued beyond the actual exposure period (Figures 1B and 5). Similarly, a correlation between ethylene evolution and O2•− accumulation was evident in a survey of 11 Arabidopsis ecotypes differing in their sensitivity to O3 (K. Mittelstrass, H. Wohlgemuth, and C. Langebartels, unpublished data). A direct role for ethylene in regulating ROS production was shown in further experiments. Both loss-of-function and gain-of-function experiments with O3-sensitive rcd1 and O3-tolerant Col-0 and ein2 revealed a requirement for ethylene synthesis and signaling in cell death. This suggests that the O3-induced ethylene synthesis in Arabidopsis is not a mere indicator of damage (Tingey et al., 1976; Heath and Taylor, 1997) but rather an active component controlling the damage process itself. It also implies that ethylene acts upstream of O2•− accumulation, which is consistent with its rapid induction by O3. However, both 250 and 400 nL L−1 O3 induced ethylene evolution and initial microbursts of O2•− in ein2 without lesion propagation. This suggests that the increase in ethylene synthesis is not under autocatalytic regulation and that these very early responses related to lesion initiation are ethylene independent. The experiments performed with the rcd1 ein2 double mutant also separated functionally O3-induced lesion initiation from propagation. The double mutant showed an initial increase in cell death during O3 exposure, a property of rcd1, but ceased further lesion propagation in clean air because of the ethylene insensitivity conferred by ein2 (Figure 7). Together, these results suggest that ethylene-independent lesion initiation is followed by ethylene-dependent amplification of O2•− accumulation, which is responsible for the execution of spreading cell death. We propose that ethylene primes the cells in the borders of the lesions for hypersensitivity to a message from the O2•−-producing cells, which results in triggering O2•− production or in enhanced O2•− production.
JAs and JA Signaling Are Involved in Lesion Containment
JA signaling appears to be involved in lesion containment. Exposure of rcd1 to MeJA after 4 hr of O3 prevented further lesion propagation (Figure 8A). This suggests that increased JA can override the ethylene and O2•−-dependent propagation of cell death that occurs in rcd1 at that time. Further evidence for this comes from the fact that the JA-insensitive jar1 is O3 sensitive and displays a spreading-lesion phenotype similar to that of rcd1. Initially, at 1 to 2 hr after the beginning of O3 exposure, the pattern of O2•− accumulation in jar1 was similar to that of Col-0 and rcd1. Later, during lesion propagation, O2•− accumulation continued in jar1, but in contrast to rcd1, propagation did not involve ethylene evolution to the same extent as in rcd1. This may seem at first contradictory to the proposed requirement for ethylene as an amplifier of O2•− accumulation and cell death. Actually, however, similar lesion phenotypes would be expected to result from an increase in lesion propagation caused by increased ethylene-dependent O2•− accumulation, as in rcd1, or from a reduced capacity for lesion containment, without a large increase in ethylene, as in jar1.
Does Interaction and Cross-Talk among JA, SA, and Ethylene Signaling Determine the Degree and Extent of O2•–-Driven Lesion Formation?
Recently, an oxidative cell death cycle was postulated from work with pathogen-infected plants undergoing HR (Van Camp et al., 1998). In this model, ROS, SA, and cell death were implicated in a self-amplifying cycle ultimately leading to visible symptom development. Rao and Davis (1999) have shown that NahG Arabidopsis plants failed to develop HR-like lesions after a short exposure to O3. They concluded that SA is involved in processes that contribute to O3-induced cell death by potentiating ROS toxicity. We have also verified this in our own experiments (H. Tuominen, K. Overmyer, and J. Kangasjärvi, unpublished data). Thus, three stress-related signaling pathways, for SA, JA, and ethylene, appear to interact with ROS in HR-like lesion development that takes place in O3-exposed Arabidopsis. Their interactions and roles in ROS-dependent cell death are summarized in Figure 9. During lesion initiation, initial cellular O2•− accumulation in microbursts executes cell death in separate, individual cells, resulting in micro-HR–like microscopic lesions. SA is required for the HR-like cell death, because O3 does not induce HR-like lesions in NahG plants (Rao and Davis, 1999). Activation of the ACC synthase gene AT-ACS6 in rcd1 results in increased ethylene evolution, which promotes the ethylene signaling–dependent O2•− accumulation that drives lesion propagation. A similar role for ethylene was also postulated for the activation of cell death in the acd6 mutant (Rate et al., 1999) and for regulating symptom development in Pseudomonas- and Xanthomonas-infected Arabidopsis (Bent et al., 1992) and in Xanthomonas-infected tomato (Ciardi et al., 2000).
Figure 9.

Roles of Ethylene, JA, and SA in the O3-Induced Oxidative Cell Death Cycle.
Locations of the components of ethylene and JA signaling, for which mutants affecting the extent of cell death were used in this study, are shown in green. Similarly, the sites of action for the inhibitors of O2•− accumulation (SOD and DPI), ethylene perception (NBD), and SA action (NahG) that affect cell death are indicated with red bars. Components addressed in this study are shown in green. The putative location of RCD1 function, which includes negative regulation of ethylene synthesis, is shown.
Exogenously added JA promoted lesion containment in rcd1, also overriding the positive effect of ethylene on cell death. Furthermore, the JA-insensitive jar1 showed a lack of lesion containment and thus formed visible lesions and O2•− accumulation without high ethylene synthesis. A similar role for JA was proposed in an O3-sensitive poplar clone (Koch et al., 1998).
Results from the experiments presented here give some indication as to the possible location of RCD1 function with respect to processes that regulate cell death (Figure 9). Ethylene biosynthesis was activated in rcd1 to a higher degree, or by a lesser stimulus than in the parent ecotype Col-0. RCD1 may be involved in processes related to regulation of ethylene biosynthesis or to communication between the ethylene, JA, and SA pathways, which are known to interact with each other (Reymond and Farmer, 1998). Interaction between these pathways appears to fine-tune the relative contribution of lesion initiation, propagation, and containment, as reflected by different lesion sizes and formation kinetics. It therefore seems obvious to include both ethylene and JA, as presented in Figure 9, as new regulating components in the oxidative cell death cycle.
METHODS
Mutant Screening, Crossing, and Genetic Mapping
Ethyl methanesulfonate–mutagenized Arabidopsis thaliana ecotype Columbia (Col-0) seeds (M2E-1A-4; Lehle Seeds, Round Rock, TX) were grown in a peat/sand mixture at a density of 3000 plants m−2 for 3 weeks in growth chambers (photon flux density 250 μmol m−2 sec−1, 12/12 hr day/night, 22/16°C, 50/75% relative humidity) and exposed for 3 days to 250 nL L−1 ozone (O3) for 8 hr. Details of the screening procedure for O3-induced foliar lesions have been described (Langebartels et al., 2000). Selected mutants were crossed with each other as appropriate to determine allelism with Col-0 to determine the inheritance of the phenotype, and with Landsberg erecta (L_er_) for mapping. F2 populations of rcd1 × L_er_ with an O3-sensitive phenotype were used for linkage analysis by using cleaved amplified polymorphic sequence and microsatellite markers. O3 sensitivity of the F2 lines used was verified in selfed F3 populations. The ethylene- insensitive ein2-1 mutant and the jasmonic acid (JA)-insensitive jar1-1 were obtained from the Arabidopsis Biological Resource Center (Ohio State University, Columbus). For double mutant analysis, ethylene-insensitive F2 individuals from the cross rcd1 × ein2 were selected on plates containing 20 μM 1-aminocylcopropane-1-carboxylic acid (ACC) by screening for the lack of triple response. Double mutant F2 individuals homozygous for rcd1 were identified and confirmed with backcrosses of ethylene-insensitive F2 and F3 plants to rcd1.
Treatments and Biochemical Analyses
O3 exposures were a single 6-hr pulse at 250 nL L−1, except where otherwise indicated. Times of measurement refer to hours after the onset of exposure. Superoxide (O2•−) accumulation in leaves was visualized with 0.1% nitroblue tetrazolium (NBT; Boehringer Mannheim), as described (Jabs et al., 1996), with a 20-min incubation period. Plants were removed from the chambers at least 15 min before staining. Ethylene exposures (12 ± 2 μL L−1) were performed under conditions similar to those described above for O3 by mixing 2% ethylene with the air in the growth chamber. The concentration of ethylene was measured with a photoionizer and regulated with a computer-controlled system. Exposures to high-intensity light (photon flux density 1000 μmol m−2 sec−1, supplied by Philips multimetal lamps; Oy Philips Ab, Espoo, Finland) were for 8 hr in growth chambers at 10°C. UV-B exposure (0.3 kJ m−2 hr−1) was for 7 days and was essentially as described (Landry et al., 1995). Treatments with 30 μL L−1 ethylene antagonist norbornadiene (NBD; Sigma) and with 1.4 μM methyl jasmonate (MeJA; Sigma) were performed in a desiccator jar. Ethylene emission from plants was analyzed from two rosettes incubated for 1 hr at 20°C (Langebartels et al., 2000). ACC was determined as described (Langebartels et al., 2000). Cellular O2•− production was inhibited by diphenylene iodonium (DPI; Sigma), a suicide inhibitor of NADPH oxidase and other flavin-containing oxidases (Cross and Jones, 1986). Leaf halves of rcd1 exposed to 250 nL L−1 O3 for 2 hr were infiltrated with DPI solutions at the indicated concentrations. Leaf damage was measured after 24 hr in at least 10 replicates per treatment. Cell death was quantified by ion leakage from rosette leaves into 5 mL of 18 Mohm water for 1 hr, measured with a Mettler conductivity meter (Mettler Toledo GmbH, Switzerland).
In Vitro Treatments
The O2•−-generating system xanthine/xanthine oxidase (X/XO; 0.5 mM/0.05 unit mL−1; Sigma) or the H2O2-generating system glucose/glucose oxidase (G/GO; 2.5 mM/2.5 to 250 units mL−1; Calbiochem) in 10 mM sodium phosphate buffer, pH 7.0, was vacuum-infiltrated into detached leaves (Jabs et al., 1996; Alvarez et al., 1998). When indicated, 440 units mL−1 MnSOD (Sigma) or 50 μM ACC (Sigma) was included. All treatments were for 20 hr (or as indicated) at 22°C, and only the first three fully expanded leaves were used. The lifetime of O2•− radical production by X/XO and of O2•− production in leaves in each treatment was assessed by NBT staining. H2O2 production was monitored and calibrated against standard dilutions by luminol bioluminescence for concentrations in the micromolar range and by direct spectrophotometric (_A_240) measurement for concentrations in the millimolar range, as described (Chamnongpol et al., 1998). Direct H2O2 treatments were performed in a large volume to prevent H2O2 clearance by detoxification systems (50 mL per 10 leaves); concentration determinations before and after treatments indicated a maximum of 20% decrease in H2O2 over the course of all experiments. For JA root inhibition assays, surface-sterilized seed was grown on vertically oriented plates of 0.5 × Murashige and Skoog (1962) medium in the presence or absence of 10 μM MeJA for 7 days, after which root length was determined as described by Staswick and Howell (1992). Pathogen treatments were performed with detached rosettes that were maintained on wet filter paper during the course of the treatment. Freshly grown Pseudomonas syringae pv tomato DC3000 bacteria bearing the AvrB gene on the plasmid pPSG0002 (Bent et al., 1992) were resuspended, diluted at the indicated densities in 10 mM MgCl2, and delivered by vacuum infiltration. All experiments were repeated at least three times.
RNA Isolation, RNA Gel Blot, and cDNA Macroarray Hybridizations
RNA isolation and hybridizations were essentially as described (Carpenter and Simon, 1998). The expression of 92 selected genes was studied with cDNA macroarrays using cDNA clones and with expressed sequence tag clones from the Arabidopsis Biological Resource Center. Inserts were amplified by polymerase chain reaction, purified, and examined by agarose gel electrophoresis. Amplified polymerase chain reaction products (100 ng) were denatured and blotted onto a Hybond N+ membrane (Amersham) by using a 96-well dot-blot device and then cross-linked with UV light. Used as negative controls were 100 ng of pSPORT vector and 100 ng of oligo-dT21. 32P-labeled cDNA probes were prepared from 1 μg of mRNA by oligo-dT21−primed polymerization with M-MLV reverse transcriptase (Promega) at 42°C for 1 hr, followed by 15 min at 70°C, and then rapid cooling on ice. After addition of 1.5 units of RNase H (Stratagene), the mixture was incubated at 37°C for 30 min and purified on G-50 columns (Amersham). Hybridizations were at 42°C in a 50% formamide buffer with stringent washes at 65°C (0.2 × SSC [1 × SSC is 0.15 M NaCl and 0.015 M sodium citrate] and 0.1% SDS), according to standard protocols. All hybridizations were quantified with a phosphor imager (Bas-1500; Fujifilm, Tokyo, Japan) and an image analysis program.
Acknowledgments
We thank Mr. Timo Oksanen for help with the O3 exposures during the mutant screening, Mr. Jorma Vahala for providing the AT-ACS6 clone, Professor Sheng Yang He for providing the Pseudomonas strain used, and Professor Tapio Palva, Dr. Nigel Kilby, and Dr. Jörg Durner for their comments on the manuscript. This work was supported by Academy of Finland Grant Nos. 43671 and 37995 to J.K. and postdoctoral fellowship No. 41615 to H.T., by the Finnish Centre of Excellence Program (2000-2005), and by grants from EU-FAIR (Brussels, Belgium) and Bundesministerium für Bildung und Forschung (BMBF) and Deutsche Forschungsgemeinschaft (DFG) (Bonn, Germany).
References
- Alvarez, M.E., Pennell, R.I., Meijer, P.-J., Ishikawa, A., Dixon, R.A., and Lamb, C. (1998). Reactive oxygen intermediates mediate a systemic signal network in the establishment of plant immunity. Cell 92**,** 773–784. [DOI] [PubMed] [Google Scholar]
- Bent, A.F., Innes, R.W., Ecker, J.R., and Staskawicz, B.J. (1992). Disease development in ethylene-insensitive Arabidopsis thaliana infected with virulent and avirulent Pseudomonas and Xanthomonas pathogens. Mol. Plant-Microbe Interact. 5**,** 372–378. [DOI] [PubMed] [Google Scholar]
- Carpenter, C.D., and Simon, A.E. (1998). Preparation of RNA. Methods Mol. Biol. 82**,** 85–90. [DOI] [PubMed] [Google Scholar]
- Chamnongpol, S., Willekens, H., Moeder, W., Langebartels, C., Sandermann, H., Jr., Van Montagu, M., Inzé, D., and van Camp, W. (1998). Defense activation and enhanced pathogen tolerance induced by H2O2 in transgenic tobacco. Proc. Natl. Acad. Sci. USA 95**,** 5818–5823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciardi, J.A., Tieman, D.M., Lund, S.T., Jones, J.B., Stall, R.E., and Klee, H.J. (2000). Response to Xanthomonas campestris pv. vesicatoria in tomato involves regulation of ethylene receptor gene expression. Plant Physiol. 123**,** 81–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conklin, P.L., Williams, E.H., and Last, R.L. (1996). Environmental stress sensitivity of an ascorbic acid–deficient Arabidopsis mutant. Proc. Natl. Acad. Sci. USA 93**,** 9970–9974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross, A.R., and Jones, O.T.G. (1986). The effect of the inhibitor diphenylene iodonium on the superoxide-generating system of neutrophils. Biochem. J. 237**,** 111–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich, R.A., Delaney, T.P., Uknes, S.J., Ward, E.R., Ryals, J.A., and Dangl, J.L. (1994). Arabidopsis mutants simulating disease resistance response. Cell 77**,** 565–577. [DOI] [PubMed] [Google Scholar]
- Drew, M.C., He, C.-J., and Morgan, P.W. (2000). Programmed cell death and aerenchyma formation in roots. Trends Plant Sci. 5**,** 123–127. [DOI] [PubMed] [Google Scholar]
- Elstner, E.F., Osswald, W., and Youngman, R.J. (1985). Basic mechanisms of pigment bleaching and loss of structural resistance in spruce (Picea abies) needles: Advances in phytomedical diagnostics. Experientia 41**,** 591–597. [Google Scholar]
- Greenberg, J.T., and Ausubel, F.M. (1993). Arabidopsis mutants compromised for the control of cellular damage during pathogenesis and aging. Plant J. 4**,** 327–341. [DOI] [PubMed] [Google Scholar]
- Greenberg, J.T., Guo, A., Klessig, D.F., and Ausubel, F.M. (1994). Programmed cell death in plants: A pathogen-triggered response activated coordinately with multiple defense functions. Cell 77**,** 551–563. [DOI] [PubMed] [Google Scholar]
- Guzman, P., and Ecker, J.R. (1990). Exploiting the triple response of Arabidopsis to identify ethylene-related mutants. Plant Cell 2**,** 513–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath, R.L., and Taylor, G.E., Jr. (1997). Physiological processes and plant responses to ozone exposure. Ecol. Stud. 127**,** 317–368. [Google Scholar]
- Jabs, T., Dietrich, R.A., and Dangl, J.L. (1996). Initiation of runaway cell death in an Arabidopsis mutant by extracellular superoxide. Science 273**,** 1853–1856. [DOI] [PubMed] [Google Scholar]
- Kangasjärvi, J., Talvinen, J., Utriainen, M., and Karjalainen, R. (1994). Plant defense systems induced by ozone. Plant Cell Environ. 17**,** 783–794. [Google Scholar]
- Kley, D., Kleinman, M., Sandermann, H., Jr., and Krupa, S. (1999). Photochemical oxidants: State of the science. Environ. Pollut. 100**,** 19–24. [DOI] [PubMed] [Google Scholar]
- Kliebenstein, D.J., Dietrich, R.A., Martin, A.C., Last, R.L., and Dangl, J.L. (1999). LSD1 regulates salicylic acid induction of copper zinc superoxide dismutase in Arabidopsis thaliana. Mol. Plant-Microbe Interact. 12**,** 1022–1026. [DOI] [PubMed] [Google Scholar]
- Koch, J.R., Scherzer, A.J., Eshita, S.M., and Davis, K.R. (1998). Ozone sensitivity in hybrid poplar is correlated with a lack of defense-gene activation. Plant Physiol. 118**,** 1243–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb, C., and Dixon, R.A. (1997). The oxidative burst in plant disease resistance. Annu. Rev. Plant Physiol. Plant Mol. Biol. 48**,** 251–275. [DOI] [PubMed] [Google Scholar]
- Landry, L.G., Chapple, C.C.S., and Last, R.L. (1995). Arabidopsis mutants lacking phenolic sunscreens exhibit enhanced ultraviolet-B injury and oxidative damage. Plant Physiol. 109**,** 1159–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langebartels, C., Ernst, D., Kangasjärvi, J., and Sandermann, H., Jr. (2000). Ozone effects on plant defense. Methods Enzymol. 319**,** 520–535. [DOI] [PubMed] [Google Scholar]
- Mehlhorn, H., and Wellburn, A.R. (1987). Stress ethylene formation determines plant sensitivity to ozone. Nature 327**,** 417–418. [Google Scholar]
- Murashige, T., and Skoog, F. (1962). A revised medium for rapid growth and bioassays with tobacco tissue culture. Physiol. Plant. 15**,** 473–497. [Google Scholar]
- Norman-Setterblad, C., Vidal, S., and Palva, E.T. (2000). Interacting signal pathways control defense gene expression in Arabidopsis in response to cell wall–degrading enzymes from Erwinia carotovora. Mol. Plant-Microbe Interact. 13**,** 430–438. [DOI] [PubMed] [Google Scholar]
- O'Donnell, P.J., Calvert, C., Atzorn, R., Wasternack, C., Leyser, H.M.O., and Bowles, D.J. (1996). Ethylene as a signal mediating the wound response of tomato plants. Science 274**,** 1914–1917. [DOI] [PubMed] [Google Scholar]
- Örvar, B.L., McPherson, J., and Ellis, B.E. (1997). Pre-activating wounding response in tobacco prior to high-level ozone exposure prevents necrotic injury. Plant J. 11**,** 203–212. [DOI] [PubMed] [Google Scholar]
- Orzáez, D., and Granell, A. (1997). DNA fragmentation is regulated by ethylene during carpel senescence in Pisum sativum. Plant J. 11**,** 137–144. [Google Scholar]
- Pellinen, R., Palva, T., and Kangasjärvi, J. (1999). Subcellular localization of ozone-induced hydrogen peroxide production in birch (Betula pendula) leaf cells. Plant J. 20**,** 349–356. [DOI] [PubMed] [Google Scholar]
- Penninckx, I.A.M.A., Thomma, B.P.H.J., Buchala, A., Métraux, J.-P., and Broekaert, W.F. (1998). Concomitant activation of jasmonate and ethylene response pathways is required for induction of a plant defensin gene in Arabidopsis. Plant Cell 10**,** 2103–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieterse, C.M.J., van Wees, S.C.M., van Pelt, J.A., Knoester, M., Laan, R., Gerrits, H., Weisbeek, P.J., and van Loon, L.C. (1998). A novel signaling pathway controlling induced systemic resistance in Arabidopsis. Plant Cell 10**,** 1571–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao, M.V., and Davis, K.R. (1999). Ozone-induced cell death occurs via two distinct mechanisms in Arabidopsis: The role of salicylic acid. Plant J. 17**,** 603–614. [DOI] [PubMed] [Google Scholar]
- Rate, D.N., Cuenca, J.V., Bowman, G.R., Guttman, D.S., and Greenberg, J.T. (1999). The gain-of-function Arabidopsis acd6 mutant reveals novel regulation and function of the salicylic acid signaling pathway in controlling cell death, defenses, and cell growth. Plant Cell 11**,** 1695–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reymond, P., and Farmer, E.E. (1998). Jasmonate and salicylate as global signals for defense gene expression. Curr. Opin. Plant Biol. 1**,** 404–411. [DOI] [PubMed] [Google Scholar]
- Runeckles, V.C., and Vaartnou, M. (1997). EPR evidence for superoxide anion formation in leaves during exposure to low levels of ozone. Plant Cell Environ. 20**,** 306–314. [Google Scholar]
- Sandermann, H., Jr., Ernst, D., Heller, W., and Langebartels, C. (1998). Ozone: An abiotic elicitor of plant defence reactions. Trends Plant Sci. 3**,** 47–50. [Google Scholar]
- Schraudner, M., Moeder, W., Wiese, C., van Camp, W., Inzé, D., Langebartels, C., and Sandermann, H., Jr. (1998). Ozone-induced oxidative burst in the ozone biomonitor plant, tobacco Bel W3. Plant J. 16**,** 235–245. [DOI] [PubMed] [Google Scholar]
- Sharma, Y.K., and Davis, K.R. (1997). The effects of ozone on antioxidant responses in plants. Free Radical Biol. Med. 23**,** 480–488. [DOI] [PubMed] [Google Scholar]
- Sharma, Y.K., León, J., Raskin, I., and Davis, K.R. (1996). Ozone-induced responses in Arabidopsis thaliana: The role of salicylic acid in the accumulation of defense-related transcripts and induced resistance. Proc. Natl. Acad. Sci. USA 93**,** 5099–5104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staswick, P.E., and Howell, S.H. (1992). Methyl jasmonate inhibition of root growth and induction of a leaf protein are decreased in an Arabidopsis thaliana mutant. Proc. Natl. Acad. Sci. USA 89**,** 6837–6840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tingey, D.T., Standley, C., and Field, R.W. (1976). Stress ethylene evolution: A measure of ozone effects on plants. Atmos. Environ. 10**,** 969–974. [DOI] [PubMed] [Google Scholar]
- Tuomainen, J., Betz, C., Kangasjärvi, J., Ernst, D., Yin, Z.H., Langebartels, C., and Sandermann, H., Jr. (1997). Ozone induction of ethylene emission in tomato plants: Regulation by differential transcript accumulation for the biosynthetic enzymes. Plant J. 12**,** 1151–1162. [Google Scholar]
- Vahala, J., Schlagnhaufer, C.D., and Pell, E.J. (1998). Induction of an ACC synthase cDNA by ozone in light-grown Arabidopsis thaliana leaves. Physiol. Plant. 103**,** 45–50. [Google Scholar]
- Van Camp, W., Van Montagu, M., and Inzé, D. (1998). H2O2 and NO: Redox signals in disease resistance. Trends Plant Sci. 3**,** 330–334. [Google Scholar]
- Wellburn, F.A.M., and Wellburn, A.R. (1996). Variable patterns of antioxidant protection but similar ethene emission differences in several ozone-sensitive and ozone-tolerant plant selections. Plant Cell Environ. 19**,** 754–760. [Google Scholar]
- Young, T.E., Gallie, D.R., and DeMason, D.A. (1997). Ethylene-mediated programmed cell death during maize endosperm development of wild-type and shrunken2 genotypes. Plant Physiol. 115**,** 737–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, I.-C., Parker, J., and Bent, A.F. (1998). Gene-for-gene disease resistance without the hypersensitive response in Arabidopsis dnd1 mutant. Proc. Natl. Acad. Sci. USA 95**,** 7819–7824. [DOI] [PMC free article] [PubMed] [Google Scholar]