A burst of auxilin recruitment determines the onset of clathrin-coated vesicle uncoating (original) (raw)
Abstract
Clathrin-coated pits assemble on a membrane and pinch off as coated vesicles. The released vesicles then rapidly lose their clathrin coats in a process mediated by the ATPase Hsc70, recruited by auxilin, a J-domain-containing cofactor. How is the uncoating process regulated? We find that during coat assembly small and variable amounts of auxilin are recruited transiently but that a much larger burst of association occurs after the peak of dynamin signal, during the transition between membrane constriction and vesicle budding. We show that the auxilin burst depends on domains of the protein likely to interact with lipid head groups. We conclude that the timing of auxilin recruitment determines the onset of uncoating. We propose that, when a diffusion barrier is established at the constricting neck of a fully formed coated pit and immediately after vesicle budding, accumulation of a specific lipid can recruit sufficient auxilin molecules to trigger uncoating.
Keywords: hsc70, endocytosis, lipids
The cycle of clathrin-coated vesicle formation and disassembly requires coordinated interaction of a large number of proteins. Clathrin forms a lattice surrounding the invaginating membrane. Various adaptor proteins provide the links required for sorting cargo proteins into assembling coated pits. Additional proteins regulate coat formation and coat disassembly. Coat assembly is relatively steady, whereas disassembly is abrupt. The entire cycle typically lasts for 30–90 s (1).
Biochemical and genetic observations indicate that Hsc70 and its cofactor, auxilin, are directly involved in the uncoating process (2). Hsc70, an abundant and ubiquitous cytosolic protein, has an ATPase domain linked to a claw-like, substrate-binding domain (3). Through their J-domain, auxilins recruit Hsc70 molecules to their substrate, clathrin-coated vesicles (4–8). Mammalian cells express two auxilin variants, the brain-specific auxilin1 (Aux1) and the ubiquitous cyclin G-associated kinase (GAK), also called auxilin2. Auxilins have a region with high sequence similarity to the phosphatase and C2 domains of PTEN (9), a central segment with binding sites for dynamin (10), AP-2, and clathrin (8, 11, 12) and a C-terminal J-domain. In addition, GAK contains an N-terminal Ser/Thr kinase domain that phosphorylates in vitro the μ-chains of AP-1 and AP-2 clathrin adaptors (8, 13). Hsc70 promotes dissociation of clathrin coats in vitro by a mechanism that depends on ATP hydrolysis and on Hsc70 recruitment by substoichiometric amounts of auxilin (14). The C-terminal half of Aux1, lacking the PTEN-like domain, can also support uncoating in vitro (15). The position of this fragment within the clathrin lattice, in contact with three different clathrin legs, has been determined by cryoelectron microscopy (16).
Coated pits assemble continuously until the coated vesicles pinch off, and only then does the coat dissociate. Partially assembled lattices should be able to recruit both auxilins and the ATP-bound Hsc70 constitutively present in the cytosol, and therefore they should uncoat prematurely. Premature uncoating might be prevented, either by activating bound auxilin only after finishing coat growth or by restricting auxilin recruitment to completed coated vesicles. To work out which of these two possibilities determines the onset of uncoating, we used live-cell imaging to follow the dynamics of auxilin recruitment into assembling endocytic clathrin coats. We find that small and variable amounts of auxilin accumulate and dissociate during the growing phase, whereas much larger amounts arrive during the rapid transition between membrane invagination and budding of the coated vesicle. This late burst of auxilin requires its phosphatase-like domain and correlates strongly with the rupture of physical continuity between the plasma membrane and the invaginated vesicular membrane. We further demonstrate that Aux1 binds to specific phosphoinositides in vitro and that the PTEN-like region of auxilin is required for this binding. We propose that the onset of uncoating is determined by a precise timing of auxilin recruitment to the coat. This timing may be set by a rapid change in the concentration of a specific phosphoinositide.
Results
Auxilins Are Present in All Isolated Clathrin-Coated Vesicles but only in a Small Fraction of Clathrin-Coated Structures at the Cell Surface.
To work out what determines the onset of uncoating, we first studied by fluorescence microscopy the association of auxilins with clathrin-coated structures in fixed cells. Auxilins were present in only a fraction of clathrin-coated structures at the cell surface. Whereas all fluorescent spots containing EGFP-Aux1 and ≈70% of the EGFP-GAK spots colocalized with clathrin or AP-2, only a small fraction (10 ± 3%; n = 150) of the clathrin or AP-2 spots colocalized with auxilins (Fig. 1a). We found a similar colocalization of clathrin or AP-2 with endogenous Aux1 in astrocytes or with GAK in the epithelial kidney cell line BSC1 (Fig. 1b). In the latter case, clathrin and AP-2 were visualized by stable expression of EGFP-LCa or σ2-EGFP. Few of the spots of endogenous auxilins failed to coincide with either LCa or σ2. In contrast to the presence of auxilins in only a fraction of the structures containing clathrin or AP-2, we detected them in all isolated coated vesicles (Fig. 1c). This result suggests that auxilin associates with coated pits at one particular time of their assembly cycle, presumably at a late step of pit formation or immediately after coated-vesicle budding.
Fig. 1.

Auxilins are present in clathrin-coated structures. Shown are confocal sections of selected cells (a and b) or of a clathrin-coated vesicle sample (c). Arrows point to examples of colocalization between auxilins and the indicated markers. (a) U373mg astrocytes stably expressing EGFP-Aux1 or EGFP-GAK were stained with antibodies specific for clathrin heavy chain or β1/β2-subunits of AP-1 and AP-2 complexes. EGFP-tagged Aux1 and GAK also gave a strong, diffuse signal, probably originating from excess auxilin at the plasma membrane and/or in the cytosol. (b) U373mg astrocytes (top two rows) and BSC1 cells (bottom two rows) stably expressing EGFP-LCA or σ2-EGFP were stained with antibodies specific for Aux1 or Aux1/GAK. (c) Calf brain clathrin-coated vesicles were stained with antibodies specific for clathrin heavy chain and Aux1/GAK. (Scale bars: 2 μm.)
Auxilin Is Transiently Recruited to the Plasma Membrane at Late Stages of Coated Vesicle Formation.
To characterize the temporal behavior of auxilins as they associate with clathrin coats, we used live-cell imaging to follow the recruitment of Aux1 and GAK to the plasma membrane of astrocytes (see Cells) (Figs. 6–8, which are published as supporting information on the PNAS web site). The fluorescence signals of Aux1 (green in Fig. 2a) or GAK (green in Fig. 2b) showed small, low-amplitude fluctuations during clathrin coat assembly and a major burst at the end of the growth phase (red) (Movie 1, which is published as supporting information on the PNAS web site). Every instance of disappearance of a clathrin-coated pit (e.g., corresponding to the uncoating of an endocytic vesicle) had an associated Aux1 or GAK burst; these bursts were detected in at least three consecutive time frames, and their intensities were higher than the average local background by one or more standard deviations. The probability of an incorrect assignment by our tracking algorithm is <10−4. The maximal peak signal of the EGFP-Aux1 bursts corresponds to ≈20–40 molecules, because their intensities were four to five times lower than the EGFP-LCa signals from coated vesicles observed in BSC1 cells just before uncoating (≈100–120 molecules; ref. 1). In the majority of cases (≈90% of 148 events) a single burst started immediately before the clathrin signal reached its maximum, hence just before completion of coat assembly (clathrin-coated pit stage) (Fig. 2c). In the remaining few cases there were two or more recruitment events just before the end of the growth phase (Fig. 2c). We observed no significant correlation between the sizes of the clathrin coats before uncoating (estimated by the maximum net signal of clathrin) and the number of auxilin molecules recruited in the final burst (Fig. 2d). We likewise found no correlation between the time required to build the coat (growth phase) and the time used to uncoat it (Fig. 2e, uncoating phase).
Fig. 2.

Aux1 or GAK are synchronously recruited at a late stage during coated vesicle formation. (a) Confocal time series acquired every ≈2 s from U373mg astrocytes stably expressing EGFP-Aux1 together with tomato-LCa (expressed transiently for 24 h); examples of clathrin-coated structures are shown in the kymograph view (Left); the fluorescence intensity plot (Right) shows that Aux1 is recruited transiently to the clathrin spot in small and variable amounts during the growth phase and in a significantly larger burst at the onset of uncoating. (b) As in a, but cells transiently coexpressing EGFP-GAK and tomato-LCa (16 h). (c) Bar plot showing that the most prominent Aux1 burst occurs during the uncoating phase. Similar results were obtained with all other cells expressing Aux1 or GAK (data not shown). (d) Scatter plot of the maximum fluorescence intensities of Aux1 and clathrin recruited to a given spot shows no correlation (r = 0.08) of the number of auxilin molecules recruited with the size of the coated vesicle. (e) Scatter plot shows no correlation (r = 0.2) between the durations of the uncoating and growth phases of a coated vesicle (n = 196) in cells expressing EGFP-Aux1.
We confirmed that the recruitment of auxilin to fully formed clathrin-coated structures happens during the transition from a coated structure still close to the plasma membrane (e.g., a coated pit ready to pinch off) to a structure that moves away from it (e.g., a budded coated vesicle). We did so by measuring the fluorescence intensity of EGFP-Aux1 while rapidly alternating between total internal reflection fluorescence (TIRF) and wide-field acquisition modes. In ≈55% of the EGFP-Aux1 objects the TIRF signal disappeared faster than the wide-field signal (Fig. 9_a_, which is published as supporting information on the PNAS web site), indicating that Aux1 was present in objects that had escaped the evanescent field (≈140 nm) by moving away from the ventral plasma membrane toward the cell interior. In the remaining cases the simultaneous disappearance of both fluorescence signals indicated that the Aux1 association and dissociation happened in objects that remained within the evanescent field, close to the plasma membrane, until complete uncoating (Fig. 9_b_). We observed a similar behavior for GAK (Fig. 9 c and d).
The PTEN Homology Region of Aux1 Is Required for the Burst of Recruitment to a Clathrin-Coated Pit.
To map the regions of Aux1 (Fig. 3a) required for coated pit/vesicle recruitment, we generated a series of truncation mutants of EGFP-Aux1 and studied their cellular localization and dynamics when coexpressed with labeled clathrin light chain (tomato-LCa) (Fig. 3b). The recruitment of Aux1 and GAK (data not shown) lacking the J-domain showed the same temporal pattern as full-length auxilin, indicating that this domain is not required for association with coats in cells. Removal of the complete PTEN homology domain strongly impaired the burst of Aux1 recruitment (Movie 2, which is published as supporting information on the PNAS web site); removal of just the phosphatase-like domain produced a similar perturbation (Movie 3, which is published as supporting information on the PNAS web site). It is unlikely that the targeting defects elicited by these truncated mutants are due to a major misfolding of Aux1, because the mutants are recruited to the clathrin microcages and other lattices formed in cells upon transient hypertonic shock with sucrose (17) (Fig. 10_a_, which is published as supporting information on the PNAS web site). The transient appearance of small amounts of auxilin during coat assembly is also unimpaired. We did not observe any detectable decrease in the endocytosis of transferrin (Fig. 7), suggesting that these variants are not dominant, presumably because the available endogenous WT auxilins are sufficient to sustain uncoating.
Fig. 3.

The PTEN homology region of Aux1 is required for the major recruitment to clathrin-coated structures. (a) Domain organization of WT and mutant forms of Aux1 used in our experiments. The domain organization of Aux1 and GAK are similar, with an additional Ser/Thr kinase located at the N terminus of GAK (data not shown). (b) Confocal time series acquired every 2 s from U373mg astrocytes stably expressing WT EGFP-Aux1 or the corresponding auxilin deletion mutants together with tomato-LCa (all expressed transiently for 24 h). Selected examples are presented as equally normalized kymographs (Left). The maximum amount of WT and truncated auxilin recruited to a given clathrin coat is shown. (c) Aux1 interacts with specific phosphoinositides in vitro. Shown is a representative chemiluminescence image obtained from the lipid–protein overlay assay using Aux1 purified from insect cells. The strip contained the following lipids: lysophosphatidic acid (LPA), lysophosphocholine (LPC), PI, PI (3)-phosphate (PI3P), PI (4)-phosphate (PI4P), PI (5)phosphate (PI5P), phosphatidylethanolamine (PE), phosphatidylcholine (PC), sphingosine-1-phosphate (S1P), PI (3, 4)-biphosphate (PI3,4P2), PI (3, 5)-biphosphate (PI3,5P2), PI (4, 5)-biphosphate (PI4,5P2), PI (3, 4, 5)-triphosphate (PI3,4,5P3), phosphatidic acid (PA), and phosphatidylserine (PS). The position of the spots (red circles) is shown. The bar plot shows the binding (average ± SE) of Aux1 determined in nine independent experiments (five using Aux1 purified from four bacterial preparations and four from two insect cell preparations).
How can the PTEN-like region influence recruitment? The homology with the phosphatase domain of PTEN and the presence of a C2 lipid-binding domain suggest that it might recognize lipid head groups. In a lipid overlay assay we found that recombinant Aux1 binds phosphatidylinositol (PI) (3)-phosphate, PI (4)phosphate, and PI (3, 4)-biphosphate more strongly than various other phosphoinositides or negatively charged lipids (Fig. 3c). In contrast, a truncated form of Aux1, lacking residues 1–546 (e.g., without the PTEN-like region), did not bind any of these lipids (Fig. 11_b_, which is published as supporting information on the PNAS web site).
Dynamin Function Is Required for the Major Recruitment of Auxilin.
The abrupt recruitment of EGFP-Aux1 at the transition from a fully formed coated pit to a coated vesicle suggests a relationship to dynamin activity. We examined the correlation of dynamin and auxilin bursts [using transiently expressed dynamin2 (Dyn2)–monomeric red fluorescent protein (mRFP) and stably expressed EGFP-Aux1] and found that all of the Aux1 correlated with a small but significant peak in the dynamin signal, generally ≈1–2 s before the auxilin maximum (Fig. 4a and Fig. 12_b_, which is published as supporting information on the PNAS web site). We then explored the relationship between the dynamics of these two proteins by three complementary approaches (1). We depleted endogenous Dyn2 in cells stably expressing EGFP-Aux1 by treatment with small interfering RNA oligonucleotides (18). We could not detect EGFP-Aux1 recruitment to the plasma membrane (Movie 4, which is published as supporting information on the PNAS web site) in any cell that showed the expected block in transferrin uptake (Fig. 13_a_, which is published as supporting information on the PNAS web site). These latter cells still displayed punctate clathrin and AP-2 patterns (Fig. 13 b and c) (2). We transiently overexpressed the dominant negative mutant Dyn2-K44A (19) fused to mRFP. Expression of this mutant impaired transferrin uptake and reduced EGFP-Aux1 recruitment to spots on the plasma membrane (Fig. 10_b_ and Movie 5, which is published as supporting information on the PNAS web site); control cells overexpressing WT Dyn2-mRFP showed no perturbations in clathrin-mediated endocytosis or Aux1 dynamics (Movie 6, which is published as supporting information on the PNAS web site) (3). We treated cells with dynasore, a small molecule that acutely, specifically, and reversibly inhibits the dynamin GTPase, thereby blocking transferrin uptake and locking coated pits at stages before budding (20). Dynasore abolished the final burst of auxilin recruitment (Fig. 4b); a substantially weaker auxilin signal (25–50% less than normal) remained associated with the locked pits.
Fig. 4.

Aux1 recruitment follows dynamin peak and requires normal dynamin function. (a) Selected frames from a time series acquired every 2 s of a small area of U373mg astrocytes expressing EGFP-Aux1 stably and Dyn2-mRFP transiently (24 h). Normalized fluorescence intensity as a function of time of the selected coated pit is shown. A bar plot shows the distribution of occurrences of Aux1 major bursts in relation to the time of appearance of Dyn2 peaks (n = 326). These data were obtained by first tracking the auxilin bursts, then extending the mask back in time for an additional five time frames (≈10 s). Finally, the relative timing corresponding to the appearance of the maximum fluorescence signals for Aux1 and Dyn2 was determined automatically. Accuracy of the method was verified manually on a subset of the observations (see additional examples in Fig. 12_b_). (b) Dynasore, a chemical inhibitor of dynamin, prevents the appearance of the auxilin burst. U373mg astrocytes stably expressing EGFP-Aux1 (green) and transiently (24 h) expressing tomato-LCa (red) were incubated with medium containing 0.5% DMSO or 80 μM dynasore/0.5% DMSO at 37°C for 5 min. After this treatment, confocal time series were recorded every 2 s (− Dynasore) or 6 s (+ Dynasore), and examples are displayed as kymograph views (Top and Middle) and as plots of normalized fluorescence intensities as a function of time (Bottom).
Auxilin and dynamin have been reported to interact directly in vitro through a region of auxilin that lies between the PTEN homology domain and the clathrin-binding domain (10). Could this interaction account for the correlations described in the preceding paragraph? At least two lines of evidence suggest otherwise. First, the actual dynamics are different. Dynamin is recruited steadily to coated pits, with an incremental burst at the time of pinching (1), whereas the auxilin burst begins essentially at baseline. Second, truncated Aux1 that lacks the PTEN-like region but retains the dynamin-binding segment exhibits no late burst in intensity, whereas its early, low-level, transient recruitment appears to be normal. We therefore believe that a direct interaction between the two proteins cannot account for the major auxilin burst.
Discussion
Our principal finding is that auxilin recruitment to a coated pit occurs primarily in a burst just as growth is complete and as budding occurs. We conclude that the timing of this recruitment is the process that triggers rapid uncoating shortly after assembly has finished. There is also a low level of transient auxilin incorporation during the growth phase of a coated pit. One function for these auxilins (and the Hsc70 that they presumably bring along) might be to release a few clathrin triskelions from the growing edge of the pit, perhaps to correct errors appearing during assembly. Another might be to promote local disassembly, possibly required to accommodate a change in curvature of the underlying membrane in response to incorporation of cargo of larger size. Auxilin could also have a function distinct from its role as an Hsc70 cochaperone.
We can imagine two general ways in which the major burst of auxilin recruitment could be timed. In one class of mechanism, interactions with clathrin, APs, dynamin, or other proteins would recruit auxilin, and the regulation of those interactions, e.g., by phosphorylation or other modification, would in turn determine the timing of auxilin association. A model based exclusively on the modulation of protein–protein interactions would, however, require coordinated modification of all 20–40 auxilin molecules during the major burst or of an equivalent number of partner proteins distributed throughout the coat. It is difficult to imagine how local modifications can be coordinated across so large a structure. A second kind of mechanism, based on topological constraints, provides a simpler explanation to account for the major burst of auxilin recruitment before uncoating. A specific lipid species, generated by an enzymatic modification within the coated membrane and perhaps recognized by the PTEN domain, might act as a recruiting signal for auxilin. For example, it is known that synaptojanin, a phosphoinositide phosphatase that binds proteins linked to endocytosis, is needed for the normal completion of the coated vesicle cycle (21–28). This enzyme dephosphorylates PI (4, 5)-biphosphate phosphoinositides to create PI (4)phosphate (29), one of the lipids shown here to bind auxilin, which could be the recruiting signal for auxilin. During all stages of coat formation before vesicle budding, the underlying membrane remains connected to the plasma membrane, and any such lipid will rapidly diffuse into the surrounding membrane. It will therefore fail to reach the concentration threshold needed to capture enough auxilin for productive uncoating. Only when a diffusion barrier is established at the connecting neck or after the vesicle has been topologically isolated from the plasma membrane (i.e., after fission) can the concentration of the postulated lipid increase sufficiently to support the coordinated recruitment of a large number of auxilin molecules (Fig. 5). The internal location of the Aux1 C-terminal portion seen by cryoelectron microscopy reconstruction would allow the PTEN region to extend inward and contact the membrane (16).
Fig. 5.
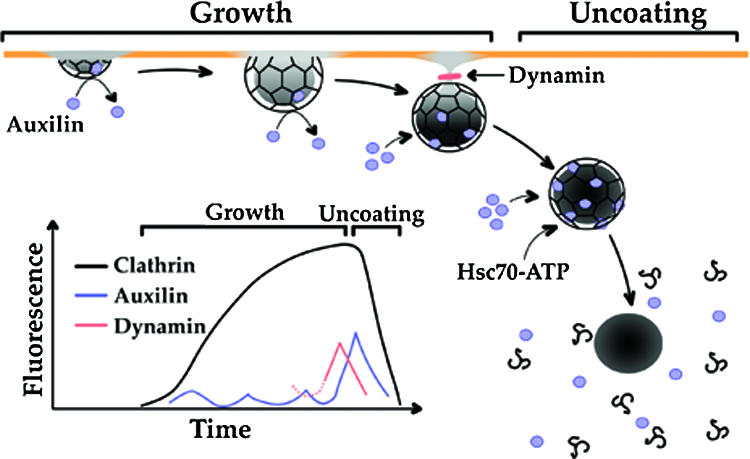
Model for the recruitment of auxilin. The principal stages leading to the auxilin burst and its relation to the onset of clathrin coat disassembly are shown. The plasma membrane is represented by the orange line; the membrane containing the lipid signal used to recruit auxilin is shown in gray. During the growth phase, cargo, clathrin, adaptors, and other coat components continuously accumulate. Continued growth results in deep membrane invaginations, until membrane constriction and fission coordinated by a final burst of dynamin occur. After budding, the coated vesicle moves away from the membrane, and uncoating rapidly ensues. Small and variable amounts of auxilin associate to and dissociate from growing pits, while a large burst of auxilin recruitment starts during membrane constriction and ends with the onset of uncoating. At this stage, sufficient amounts of ATP-bound Hsc70 are captured by auxilin to drive coat disassembly. In this model, a lipid species (indicated by the gray shadowing of the membrane), generated within the coated membrane and perhaps recognized by the PTEN-like domain of auxilin, acts as a recruiting signal for auxilin. Before vesicle budding, the underlying membrane remains connected to the plasma membrane, and any such lipid will rapidly diffuse into the surrounding membrane. It will therefore fail to reach the concentration threshold needed to capture enough auxilin to ensue coat disassembly.
Four findings support this proposal (1). Auxilin association with the clathrin lattice is relatively weak (16), and a strong association will probably require an additional set of contacts (2). All known perturbations that block budding but still retain clathrin coats at the plasma membrane also prevent late auxilin recruitment and thus prevent uncoating. The perturbations include removal of dynamin by small interfering RNA treatment and inhibition of dynamin function either by expression of Dyn2K44A mutant or by treatment of the cells with dynasore. Cholesterol depletion, by brief incubation of the cells with cyclodextrins, also retains clathrin arrays at the plasma membrane, blocks budding, impairs auxilin recruitment (Movie 7, which is published as supporting information on the PNAS web site), and prevents coordinated uncoating. Thus, discontinuity between the plasma membrane and a fully formed vesicle appears essential for auxilin recruitment before uncoating (3). Removal either of the entire PTEN homology domain of auxilin or of just the phosphatase-like domain completely prevents the final burst of auxilin, even though binding sites for dynamin, AP-2, clathrin, and Hsc70 are still present on the truncated auxilins. The C2 domain of PTEN has been reported to confer nonspecific recognition of lipid head groups (30), and it seems likely that the auxilin C2 domain functions similarly. The phosphatase-like domain of auxilin lacks the residues in PTEN essential for catalytic activity toward phosphoinositides, but the extent of sequence identity is sufficiently high that recognition of phosphoinositides is possible (4). Purified full-length Aux1 (but not a truncated form lacking its PTEN-like region) displays preferential binding to phosphoinositides in a lipid overlay assay. Treatment with wortmannin or LY-294002 (Fig. 12_a_) did not alter coat dynamics in a noticeable way; this lack of an effect could be due to incomplete PI 3-kinase inhibition (31) or to the presence of a pool of PI (3)-phosphate lipids with a relatively slow turnover. Future studies will be needed to identify the lipid species recognized by auxilin and to test its role in triggering uncoating.
Materials and Methods
Plasmids, Oligos, Transfections, and Protein Purification.
Full-length bovine Aux1 (910 residues) was amplified by PCR from a full-length cDNA clone and inserted into pEGFP-C1 (CLONTECH), resulting in pEGFP-Aux1. The C-terminal J-domain of Aux1 was removed by using an internal BamHI site to create a truncation after M812 (pEGFP-Aux1ΔJ). The N-terminal PTEN homology region or the phosphatase-like domain was removed by QuikChange mutagenesis (Stratagene) by incorporating a HindIII site after Q419 or a BglII site after L216. The resulting HindIII/SacII or BglII/SacII fragment was transferred into pEGFP-C1, resulting in pEGFP-ΔPTEN Aux1 or pEGFP-ΔPDAux1. Full-length rat Dyn2 WT or K44A (874 residues) EGFP fusions were a generous gift of Sandy Schmidt (The Scripps Research Institute, La Jolla, CA). mRFP was amplified from pRSETb and substituted for EGFP in WT and K44A versions of Dyn2-EGFP. Full-length LCa and σ2 fused to EGFP (pEGFP-LCa and pσ2-EGFP, respectively) were previously described (1). The sequence corresponding to the tandem repeat tomato ORF (476 residues) was amplified by PCR and substituted for EGFP in pEGFP-LCa using Age1/BsrGI sites (pTom-LCa). All constructs were verified by restriction digest and sequence analysis. All transfections were carried out by using FuGENE 6 (Roche Diagnostics, Indianapolis), and cells were evaluated 16–48 h later or selected with geneticin to obtain stable cell lines. The sequence of the double-stranded small interfering RNA oligo (Dharmacon, Lafayette, CO) targeting human Dyn2 was the following: sense strand, 5′-GGAGAUUGAAGCAGAGACCTT-3′; antisense strand, 5′-GGUCUCUGCUUCAAUCUCCTG-3′. Small interfering RNA oligo targeting CD4 was used as a negative silencer control. Transfections of oligos were performed by using Oligofectamine (Invitrogen) following the manufacturer's recommendations. Cells were tested 96 h after transfection.
For bacterial expression of auxilin we transferred Aux1 from pEGFP-Aux1 into pGEX-4T-1 using BglII and SalI restriction enzymes, resulting in full-length Aux1 fused to GST (pGEX-4T-1-Aux1), which was then used to transform BL21-pLys cells. Expression was achieved after overnight growth at 37°C in LB medium supplemented with chloramphenicol/kanamycin, followed by inoculation of 10 ml into 1 liter of fresh LB medium and further allowed to grow for 2–3 h at room temperature (OD560 ≈ 0.5–0.7) before adding isopropyl-13-d-thiogalactoside (0.1 mM final). Cells were harvest by centrifugation after a further incubation for 16 h, washed with cold PBS, resuspended, and lysed by sonication at 4°C (four pulses of 1 min, 30 s of cooling) in the presence of complete protease inhibitors (Roche Molecular Biochemicals) and 1 mM PMSF. The lysate was clarified by centrifugation (34,000 × g, 20 min, 4°C), and the GST-Aux1 protein was isolated by using glutathione affinity columns. The yield is consistently very low (0.07–0.15 mg/liter of culture), and the preparation consists of full-length Aux1 and several fragments.
For insect cell expression of Aux1 we transferred Aux1 from pEGFPAux1 into the pFastBac HT vector (Invitrogen) using BglII and PstI. This plasmid was used to transform DH10Bac cells to produce, upon recombination, the bacmids coding for full-length N-terminal His6-tagged Aux1. Sf9 insect cells were transfected with the resulting bacmids, and the amplified virus was used to transduce High Five insect cells (1–2 × 106 cells per ml; volume, 500 ml infected with a 1:600 dilution of viral stock). Cells were harvested 56–72 h after infection, and Aux1 was purified by using TALON metal affinity resin (CLONTECH) as described (32). This procedure provides a moderate yield of mostly full-length His6-tagged Aux1 (0.5–0.7 mg/liter of culture) and with no detectable contaminants observed by Coomassie blue staining.
Cells.
Sf9 and High Five insect cells were grown in ExCell 420 medium at 28°C in spinner flasks. BSC1, HeLa, HEK-293, and U373mg astrocytes were grown in DMEM supplemented with 10% FBS, 2 mM l-glutamine, penicillin (50 units/ml), streptomycin (50 mg/ml), and nonessential amino acids (0.1 mM). The human U373mg astrocytes offer the proper cellular context to study the brain-specific endogenous Aux1. U373mg cell lines, stably expressing EGFP fusion chimeras of Aux1, EGFP-clathrin light chain (LCa), or σ2-EGFP were generated by selection with 0.5–0.7 mg/ml geneticin. The rates of the growth and uncoating phases of clathrin-coated structures observed by using LCa and AP-2 fused to fluorescent proteins is very similar (Fig. 8 a_–_c). The relative expression level of EGFP-Aux1 in the crude microsomal fraction containing clathrin-coated vesicles was 4–5 times higher than the expression level of endogenous Aux1; there was no significant replacement of endogenous Aux1 associated with this fraction. Simultaneous expression of EGFP-Aux1 with tomato-LCa or with mRFP-dynamin was performed by transient expression of the red fluorescent proteins in cells stably expressing EGFP-Aux1 or by transient coexpression of the green and red fluorescent proteins. Cells were seeded on glass coverslips and imaged 16–72 h later at ≈50–70% confluency. EGFP-Aux1 was recruited to diffraction-limited spots at the ventral (Fig. 6 a and b and Movie 8_a_, which is published as supporting information on the PNAS web site) and dorsal (Movie 8_b_) surfaces of the cell with a residence time of 8 ± 1.5 s (n = 150–270 particles per cell analyzed from six cells; Fig. 6_c_). We obtained comparable results upon transient expression of EGFP-Aux1 in HeLa, HEK-293, and BSC1 cells (Movie 8_c_) and of EGFP-GAK in astrocytes (Fig. 6 d_–_f and Movie 9, which is published as supporting information on the PNAS web site). Expression of EGFP-tagged Aux1 or GAK did not affect the uptake of transferrin (Fig. 7) or the duration of coat formation and uncoating (Fig. 8 d and e). The red fluorescent protein tomato fused to clathrin LCa does not disturb the dynamic behavior of endocytic-coated structures or the endocytosis of transferrin (Fig. 8_f_).
Immunofluorescence.
Cells were fixed in 3% paraformaldehyde in PBS for 10 min at room temperature, washed with PBS, blocked with 50 mM NH4Cl in PBS for 15 min at room temperature, and finally incubated with 2% BSA in PBS for 30 min at room temperature. All antibodies were diluted in PBS supplemented with 2% BSA and 0.1% saponin. Aux1 was labeled with the mouse monoclonal antibody 100/4 that recognizes only Aux1 (33). Aux1 and GAK were simultaneously labeled with a rabbit polyclonal antibody. Clathrin heavy chain, β-adaptins, and dynamin were labeled with the mouse monoclonal antibodies X22 (34), 10A (35), and Hudy-1 (Upstate Biotechnology), respectively. Alexa Fluor 488-, Alexa Fluor 594-, or Alexa Fluor 647-labeled goat anti-mouse or anti-rabbit were used as secondary antibodies (Molecular Probes).
Lipid–Protein Overlay Assay.
PIP Strips hydrophobic membranes, spotted with 15 different biologically active lipids (Echelon Biosciences, Salt Lake City, UT), were used to determine lipid–Aux1 interactions following the manufacturer's instructions. Bound Aux1 was detected by using either a mouse monoclonal antibody specific for His6 (His-1, Sigma) or a rabbit polyclonal antibody specific for Aux1/GAK followed by incubation with appropriate secondary antibodies conjugated to horseradish peroxidase and ECL (Amersham Pharmacia). The intensity of each spot was corrected by the signal of the membrane immediately adjacent to the spot by using image j (36).
Live-Cell Imaging.
Cells grown on 25-mm-diameter coverslips (no. 1.5, Electron Microscopy Sciences, Hatfield, PA) were washed with sterile PBS and placed into an open perfusion chamber, and prewarmed medium was added. The chamber was inserted into a sample holder (20/20 Technology, Wilmington, NC) placed on top of the microscope stage enclosed by an environmental chamber (37°C). The cells were maintained in a humidified environment of 95% air and 5% CO2. Most experiments were done by using MEM without phenol red supplemented with 20 mM Hepes (HMEM) and 0.5 g/dl BSA. Experiments carried in the presence of 80 μM dynasore or 10 mM methyl-β-cyclodextrin were done with PBS supplemented with 0.1 mM CaCl2, 1 mM MgCl2, glucose (4.5 g/liter), and 1% Nuserum. To test the effect of inhibition of PI 3-kinases in EGFP-Aux1 recruitment and dynamics we incubated cells with 0.5–10 μM wortmannin or 30 μM LY-294002 in HMEM for up to 30 min. HMEM containing the inhibitors was added fresh during the recording of the time lapse. Transferrin uptake followed using Alexa Fluor 594- or Alexa Fluor 647-labeled human transferrin (Molecular Probes) added to the media.
Image Acquisition.
All spinning-disk confocal-based imaging experiments were conducted by using the microscope setup previously described (1) captured with slidebook 4 software (Intelligent Imaging Innovations, Denver). A second microscope was equipped with a TIRF slider (Zeiss), illuminated with a 40-mW solid-state laser (Crystal Laser, Reno, NV) for TIRF, and selected with a computer-controlled acoustooptical tunable filter and with a 175-W Xe lamp Lambda DG-4 (Sutter Instruments, Novato, CA) for wide-field illumination; the images were acquired with a charge-coupled device camera (Cascade, Photometrics, Tucson) set with no electronic amplification using a ×100 objective lens (n.a. 1.45, Zeiss). The TIRF laser beam was focused at an off-axis position in the back focal plane of lens by using a TIRF slider (Zeiss), with an angle of incidence above the critical angle necessary to achieve total internal reflection. The spherical aberration was corrected with a computer-driven SACS device (Intelligent Imaging Innovations). Exposure times were 100–300 ms.
Image Processing and Analysis.
An image analysis application (iab) was developed with matlab 7 (Mathworks, Natick, MA) to identify and track fluorescent objects by using the same criteria previously described (1) and also to carry out all data analysis. Objects tracks were defined as follows: background smoothing followed by a Gaussian, Laplacian, intensity threshold filtering and three-dimensional connectivity of resulting masks. Object tracks with the following characteristics were eliminated: (i) objects with masks diameters larger than three to five pixels (378–500 nm, depending on the camera used); (ii) objects tracks shorter than three consecutive time frames; (iii) objects present in the first or last frame of the time series; (iv) objects whose centroid moved more than two pixels between consecutive time frames; (v) tracks corresponding to merge or dissociation of nearby objects; (vi) tracks of coated pits appearing before the disappearance of another at the same site. All particle tracks were validated manually. Statistical analysis was performed by using matlab 7 or sigmaplot 7.0 (SYSTAT, Point Richmond, CA).
Supplementary Material
Supporting Information
Acknowledgments
We are grateful to Drs. E. Ungewickell (Hannover Medical School, Hannover, Germany), S. Sever (Massachusetts General Hospital, Boston), and R. Tsien (University of California, San Diego) for providing the full-length cDNAs for mRFP, tomato, Aux1, GAK, and antibodies specific for auxilins. Part of the imaging equipment was acquired with the generous support from the Perkin Fund. This work was supported by National Institutes of Health Grant R01 GM075252-01.
Abbreviations
Aux1
auxilin 1
GAK
cyclin G-associated kinase
mRFP
monomeric red fluorescent protein
PI
phosphatidylinositol
TIRF
total internal reflection fluorescence
Dyn2
dynamin2
Footnotes
Conflict of interest statement: No conflicts declared.
References
- 1.Ehrlich M., Boll W., Van Oijen A., Hariharan R., Chandran K., Nibert M. L., Kirchhausen T. Cell. 2004;118:591–605. doi: 10.1016/j.cell.2004.08.017. [DOI] [PubMed] [Google Scholar]
- 2.Lemmon S. K. Curr. Biol. 2001;11:R49–R52. doi: 10.1016/s0960-9822(01)00010-0. [DOI] [PubMed] [Google Scholar]
- 3.Jiang J., Prasad K., Lafer E. M., Sousa R. Mol. Cell. 2005;20:513–524. doi: 10.1016/j.molcel.2005.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greener T., Zhao X., Nojima H., Eisenberg E., Greene L. E. J. Biol. Chem. 2000;275:1365–1370. doi: 10.1074/jbc.275.2.1365. [DOI] [PubMed] [Google Scholar]
- 5.Jiang J. W., Taylor A. B., Prasad K., Ishikawa-Brush Y., Hart P. J., Lafer E. M., Sousa R. Biochemistry. 2003;42:5748–5753. doi: 10.1021/bi034270g. [DOI] [PubMed] [Google Scholar]
- 6.Jiang R. F., Greener T., Barouch W., Greene L., Eisenberg E. J. Biol. Chem. 1997;272:6141–6145. doi: 10.1074/jbc.272.10.6141. [DOI] [PubMed] [Google Scholar]
- 7.Ungewickell E., Ungewickell H., Holstein S. E., Lindner R., Prasad K., Barouch W., Martin B., Greene L. E., Eisenberg E. Nature. 1995;378:632–635. doi: 10.1038/378632a0. [DOI] [PubMed] [Google Scholar]
- 8.Umeda A., Meyerholz A., Ungewickell E. Eur. J. Cell Biol. 2000;79:336–342. doi: 10.1078/S0171-9335(04)70037-0. [DOI] [PubMed] [Google Scholar]
- 9.Lee J. O., Yang H., Georgescu M. M., Di Cristofano A., Maehama T., Shi Y., Dixon J. E., Pandolfi P., Pavletich N. P. Cell. 1999;99:323–334. doi: 10.1016/s0092-8674(00)81663-3. [DOI] [PubMed] [Google Scholar]
- 10.Newmyer S. L., Christensen A., Sever S. Dev. Cell. 2003;4:929–940. doi: 10.1016/s1534-5807(03)00157-6. [DOI] [PubMed] [Google Scholar]
- 11.Scheele U., Alves J., Frank R., Duwel M., Kalthoff C., Ungewickell E. J. Biol. Chem. 2003;278:25357–25368. doi: 10.1074/jbc.M303738200. [DOI] [PubMed] [Google Scholar]
- 12.Scheele U., Kalthoff C., Ungewickell E. J. Biol. Chem. 2001;276:36131–36138. doi: 10.1074/jbc.M106511200. [DOI] [PubMed] [Google Scholar]
- 13.Korolchuk V. I., Banting G. Traffic. 2002;3:428–439. doi: 10.1034/j.1600-0854.2002.30606.x. [DOI] [PubMed] [Google Scholar]
- 14.Barouch W., Prasad K., Greene L., Eisenberg E. Biochemistry. 1997;36:4303–4308. doi: 10.1021/bi962727z. [DOI] [PubMed] [Google Scholar]
- 15.Holstein S. E., Ungewickell H., Ungewickell E. J. Cell Biol. 1996;135:925–937. doi: 10.1083/jcb.135.4.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fotin A., Cheng Y., Grigorieff N., Walz T., Harrison S. C., Kirchhausen T. Nature. 2004;432:649–653. doi: 10.1038/nature03078. [DOI] [PubMed] [Google Scholar]
- 17.Heuser J. E., Anderson R. G. J. Cell Biol. 1989;108:389–400. doi: 10.1083/jcb.108.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Veiga E., Cossart P. Nat. Cell Biol. 2005;7:894–900. doi: 10.1038/ncb1292. [DOI] [PubMed] [Google Scholar]
- 19.Damke H., Baba T., Warnock D. E., Schmid S. L. J. Cell Biol. 1994;127:915–934. doi: 10.1083/jcb.127.4.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Macia E., Ehrlich M., Massol R., Boucrot E., Brunner C., Kirchhausen T. Dev. Cell. 2006;10:839–850. doi: 10.1016/j.devcel.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 21.Micheva K. D., Kay B. K., McPherson P. S. J. Biol. Chem. 1997;272:27239–27245. doi: 10.1074/jbc.272.43.27239. [DOI] [PubMed] [Google Scholar]
- 22.McPherson P. S., Garcia E. P., Slepnev V. I., David C., Zhang X., Grabs D., Sossin W. S., Bauerfeind R., Nemoto Y., De Camilli P. Nature. 1996;379:353–357. doi: 10.1038/379353a0. [DOI] [PubMed] [Google Scholar]
- 23.Schuske K. R., Richmond J. E., Matthies D. S., Davis W. S., Runz S., Rube D. A., van der Bliek A. M., Jorgensen E. M. Neuron. 2003;40:749–762. doi: 10.1016/s0896-6273(03)00667-6. [DOI] [PubMed] [Google Scholar]
- 24.Harris T. W., Hartwieg E., Horvitz H. R., Jorgensen E. M. J. Cell Biol. 2000;150:589–600. doi: 10.1083/jcb.150.3.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gad H., Ringstad N., Low P., Kjaerulff O., Gustafsson J., Wenk M., Di Paolo G., Nemoto Y., Crun J., Ellisman M. H., et al. Neuron. 2000;27:301–312. doi: 10.1016/s0896-6273(00)00038-6. [DOI] [PubMed] [Google Scholar]
- 26.Verstreken P., Koh T. W., Schulze K. L., Zhai R. G., Hiesinger P. R., Zhou Y., Mehta S. Q., Cao Y., Roos J., Bellen H. J. Neuron. 2003;40:733–748. doi: 10.1016/s0896-6273(03)00644-5. [DOI] [PubMed] [Google Scholar]
- 27.Rusk N., Le P. U., Mariggio S., Guay G., Lurisci C., Nabi I. R., Corda D., Symons M. Curr. Biol. 2003;13:659–663. doi: 10.1016/s0960-9822(03)00241-0. [DOI] [PubMed] [Google Scholar]
- 28.Dickman D. K., Lu Z., Meinertzhagen I. A., Schwarz T. L. Curr. Biol. 2006;16:591–598. doi: 10.1016/j.cub.2006.02.058. [DOI] [PubMed] [Google Scholar]
- 29.Chung J. K., Sekiya F., Kang H. S., Lee C., Han J. S., Kim S. R., Bae Y. S., Morris A. J., Rhee S. G. J. Biol. Chem. 1997;272:15980–15985. doi: 10.1074/jbc.272.25.15980. [DOI] [PubMed] [Google Scholar]
- 30.Rizo J., Sudhof T. C. J. Biol. Chem. 1998;273:15879–15882. doi: 10.1074/jbc.273.26.15879. [DOI] [PubMed] [Google Scholar]
- 31.Domin J., Pages F., Volinia S., Rittenhouse S. E., Zvelebil M. J., Stein R. C., Waterfield M. D. Biochem. J. 1997;326:139–147. doi: 10.1042/bj3260139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sever S., Skoch J., Bacskai B. J., Newmyer S. L. Methods Enzymol. 2005;404:570–585. doi: 10.1016/S0076-6879(05)04050-4. [DOI] [PubMed] [Google Scholar]
- 33.Ahle S., Ungewickell E. J. Cell Biol. 1990;111:19–29. doi: 10.1083/jcb.111.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brodsky F. M. J. Cell Biol. 1985;101:2047–2054. doi: 10.1083/jcb.101.6.2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clairmont K. B., Boll W., Ericsson M., Kirchhausen T. Cell. Mol. Life Sci. 1997;53:611–619. doi: 10.1007/s000180050080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abramoff M. D., Magelhaes P. J., Ram S. J. Biophotonics Int. 2004;11:36–42. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information