An in vitro electrophysiological study on the effects of phenytoin, lamotrigine and gabapentin on striatal neurons (original) (raw)
Abstract
- We performed intracellular recordings from a rat corticostriatal slice preparation in order to compare the electrophysiological effects of the classical antiepileptic drug (AED) phenytoin (PHT) and the new AEDs lamotrigine (LTG) and gabapentin (GBP) on striatal neurons.
- PHT, LTG and GBP affected neither the resting membrane potential nor the input resistance/membrane conductance of the recorded cells. In contrast, these agents depressed in a dose-dependent and reversible manner the current-evoked repetitive firing discharge.
- These AEDs also reduced the amplitude of glutamatergic excitatory postsynaptic potentials (EPSPs) evoked by cortical stimulation. However, substantial pharmacological differences between these drugs were found. PHT was the most effective and potent agent in reducing sustained repetitive firing of action potentials, whereas LTG and GBP preferentially inhibited corticostriatal excitatory transmission. Concentrations of LTG and GBP effective in reducing EPSPs, in fact, produced only a slight inhibition of the firing activity of these cells.
- LTG, but not PHT and GBP, depressed cortically-evoked EPSPs increasing paired-pulse facilitation (PPF) of synaptic transmission, suggesting that a presynaptic site of action was implicated in the effect of this drug. Accordingly, PHT and GBP, but not LTG reduced the membrane depolarizations induced by exogenously-applied glutamate, suggesting that these drugs preferentially reduce postsynaptic sensitivity to glutamate released from corticostriatal terminals.
- These data indicate that in the striatum PHT, LTG and GBP decrease neuronal excitability by modulating multiple sites of action. The preferential modulation of excitatory synaptic transmission may represent the cellular substrate for the therapeutic effects of new AEDs whose use may be potentially extended to the therapy of neurodegenerative diseases involving the basal ganglia.
Keywords: Antiepileptic drugs, epilepsy, excitatory amino acids, intracellular recordings, striatum, synaptic transmission
Introduction
Despite substantial progress in the treatment of epileptic seizures made in the past two decades, epilepsy still remains a significant therapeutic challenge. Advances in the understanding of the pathogenesis of this disease have allowed the development of new pharmacological compounds with different mechanisms of action. These new antiepileptic drugs (AEDs), used in monotherapy or in association with traditional drugs, offer hope for patients with uncontrolled epilepsy. Different mechanisms of action have been proposed to explain the clinical effects of old and new AEDs: modulation of voltage-dependent sodium channels, modulation of voltage-dependent calcium channels, enhancement of GABA-mediated neuronal inhibition, reduction of glutamate-mediated excitatory transmission (Harden, 1994; Upton, 1994; Walker & Sander, 1994; Macdonald & Kelly, 1994; Calabresi et al., 1996b; Macdonald, 1996). The latter mechanism seems to be particularly important for the therapeutic effects of some new AEDs such as lamotrigine (LTG) and gabapentin (GBP). It has been shown, in fact, that LTG potently inhibited the release of excitatory amino acids evoked by the sodium channel activator veratridine while it was much less effective in inhibiting the release of acetylcholine or GABA (Leach et al., 1986). GBP prolonged the onset latency of epileptic seizures following intraperitoneal injection of the glutamate receptor agonist NMDA (Bartoszyk, 1982). Accordingly, the efficacy of GBP against tonic seizure in mice was antagonized by the administration of serine, an agonist at the glycine receptor on the NMDA receptor channel complex (Oles et al., 1990).
AEDs may attenuate or prevent seizures through effects on pathologically altered neurons of seizure foci or, alternatively, by reducing the spread of excitation from seizure foci to additional brain regions. It has been suggested that the basal ganglia play an important role in the initiation and propagation of seizure activity (Faeth et al., 1954; Engel et al., 1978; Amato et al., 1982; Gale, 1992). The main excitatory projection to the basal ganglia is represented by the corticostriatal glutamatergic pathway and activity of striatal neurons affects the excitability of the cortex by modulating the thalamocortical glutamatergic pathway (Divac et al., 1977; Calabresi et al., 1996a). An increased activity of GABAergic striatal neurons may result in an excessive inhibition of the substantia nigra pars reticulata and of the internal segment of the globus pallidus which exert an inhibitory action on the thalamus. This effect may finally result in an increased excitation of the cortex by the thalamus.
In the present study we have evaluated the electrophysiological actions of PHT, a classical AED, and of the new compounds LTG and GBP on striatal projecting spiny cells intracellularly recorded in a corticostriatal slice preparation in order to compare their effects on the intrinsic neuronal activity to those exerted on the excitatory corticostriatal synaptic transmission. An abnormal activity of the corticostriatal glutamatergic pathway has also been postulated to play a pathogenetic role in neurodegenerative diseases involving the basal ganglia such as Parkinson's disease and Huntington's disease (Calabresi et al., 1996a). Thus, the characterization of the electrophysiological effects produced by AEDs on the corticostriatal glutamatergic transmission might also provide possible insights for the treatment of these neurodegenerative disorders.
Methods
Preparation and maintenance of the slices
Adult male Wistar rats (150–250 g) were used for all the experiments. The preparation and maintenance of coronal slices have been described previously (Calabresi et al., 1990a, 1995a, 1997b, 1998). Briefly, corticostriatal coronal slices (200–300 μm) were prepared from tissue blocks of the brain with the use of a vibratome. A single slice was transferred to a recording chamber and submerged in a continuously flowing Krebs solution (35°C, 2–3 ml min−1) gassed with 95% O2- 5% CO2. The composition of the control solution was (in mM): NaCl, 126 KCl, 2.5 MgCl2, 1.2 NaH2PO4, 1.2 CaCl2, 2.4 Glucose, 11 NaHCO3 25.
Recording technique
In all the experiments the intracellular recording electrodes were filled with 2 M KCl (30–60 MΩ). An Axoclamp 2A amplifier was used for recordings either in current-clamp or in voltage-clamp mode. In single-electrode voltage-clamp mode the switching frequency was 3 kHz. The headstage signal was continuously monitored on a separate oscilloscope. Traces were displayed on an oscilloscope and stored in a digital system. For synaptic stimulation, bipolar electrodes were used. These stimulating electrodes were located either in the cortical areas close to the recording electrode or in the white matter between the cortex and the striatum in order to activate corticostriatal fibres.
Morphological identification of the recorded cells
In some experiments biocytin (Sigma) was used in the intracellular electrode in order to stain the neurons. In these cases, biocytin at concentration of 2–4% was added to a 0.5 M KCl pipette solution. Slices containing neurons stained with biocytin were fixed in paraformaldehyde (in 0.1 M phosphate buffer at pH 7.4) overnight and processed according to published protocols (Horikawa & Armstrong, 1988). In several cases, sections were further processed to make permanent staining of biocytin loaded cells.
Data analysis and drug applications
Values given in the text and in the figures are mean±s.e.mean of changes in the respective cell populations. Student's _t_-test (for paired and unpaired observations) was used to compare the means. Drugs were applied by dissolving them to the desired final concentration in the saline and by switching the perfusion from control saline to drug-containing saline. Drug solutions entered the recording chamber within 40 s after a three ways tap had been turned on. Biocytin, tetrodotoxin (TTX), glutamate and phenytoin (PHT) were from Sigma; gabapentin (GBP) and lamotrigine (LTG) were respectively from Parke-Davis and Glaxo-Wellcome. GBP was solved in water while PHT and LTG were solved in DMSO. The maximal final concentration of the solvent was 1 : 300. This concentration of DMSO did not produce per se detectable electrophysiological changes.
Results
Properties of the recorded neurons
Conventional sharp-microelectrode intracellular recordings were obtained from 82 electrophysiologically identified ‘principal' spiny cells. The main characteristics of these cells have been described in detail previously both in vivo (Wilson & Groves, 1980; Calabresi et al., 1990b) and in vitro (Kita et al., 1984; Calabresi et al., 1990a, 1998; Jiang & North, 1991; Cepeda et al., 1994). These cells had high resting membrane potential (−84±5 mV), relatively low apparent input resistance (38±8 MΩ) when measured at the resting potentials from the amplitude of small (<10 mV) hyperpolarizing electrotonic potentials, action potentials of short duration (1.1±0.3 ms) and high amplitude (102±4 mV). They were silent at rest and showed membrane rectification and tonic firing activity during depolarizing current pulses. In 12 of the 82 recorded spiny neurons, the electrophysiological identification was confirmed by a morphological analysis obtained by using biocytin staining (data not shown).
Effects of PHT, LTG and GBP on intrinsic membrane properties of striatal spiny neurons
Bath application of PHT, LTG and GBP (5–10 min), at the concentrations employed in this study (10–300, 3–300, 10–1000 μM respectively), did not alter intrinsic membrane properties of the recorded cells such as resting membrane potential and current-voltage relationship measured in the subthreshold range. The current-voltage relationship was measured in voltage-clamp recordings before and during the application of the drugs and obtained by measuring the steady-state current during the application of voltage steps (1–3 s duration) both in positive and negative directions from the holding potential (_n_=12; data not shown).
Effects of PHT, LTG and GBP on the firing activity induced by depolarizing current steps
Striatal spiny neurons responded to a 700 ms depolarizing pulse with a sustained repetitive firing of action potentials which was blocked by the sodium channel blocker TTX (1 μM) (data not shown). In order to compare the effects of different doses of PHT, LTG and GBP we assumed as control value a number of 20–25 action potentials; this frequency of discharge was obtained by injecting 0.6–1 nA of current intensity through the intracellular recording electrode. Bath application (5–10 min) of PHT (10–300 μM), LTG (10–300 μM) and GBP (100–1000 μM), produced a dose-dependent decrease of the number of sodium-dependent action potentials elicited by the current step. This effect was reversible after 25–30 min wash out of the drugs. The minimal effect was obtained with 30 μM PHT, 30 μM LTG and 300 μM GBP, whilst maximal responses were achieved with 300 μM PHT, 300 μM LTG and 1 mM GBP respectively (Figure 1). The potency of these AEDs was expressed as the extrapolated EC50 value which was 42.8 μM for PHT, 48.9 μM for LTG, 320.4 μM for GBP (Table 1). It should be noted, however, that PHT and GBP were more effective than LTG in reducing action potential discharge. In fact, saturating doses of PHT (300 μM) and of GBP (1 mM) reduced the firing frequency to 20% of the control values. Conversely, saturating doses of LTG (300 μM) reduced the firing discharge to about 40% of the control. Even higher doses of LTG (1 mM, _n_=3) did not cause further reduction of this parameter (data not shown). Interestingly, all these three AEDs were more effective in suppressing the action potentials triggered during the last part of the depolarizing pulse suggesting that a certain activation of the voltage dependent sodium channels was required for the action of these compounds.
Figure 1.
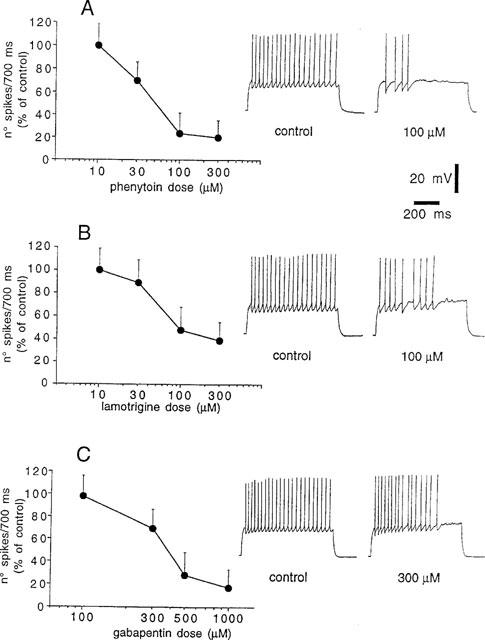
PHT, LTG and GBP inhibit current-evoked firing discharge of striatal spiny neurons. (A) The graph shows the dose-response curve obtained at various concentrations of PHT on the firing activity evoked by pulses of positive current (0.6–1 nA intensity, 700 ms duration). Each data point was obtained from at least four single experiments. The right part of the figure shows single voltage responses to a depolarizing pulse (0.8 nA) recorded from a striatal spiny neuron under control condition and during the application of 100 μM PHT. The resting membrane potential (RMP) of the cell was −87 mV and was constant throughout the experiment. (B) The graph shows the dose-response curve obtained at various concentrations of LTG on the firing activity evoked by pulses of positive current (0.6–1 nA intensity, 700 ms duration). Each data point was obtained from at least four single experiments. The right part of the figure shows single voltage responses to a depolarizing pulse (0.7 nA) recorded from a striatal spiny neuron under control condition and during the application of 100 μM LTG. The RMP of the cell was −90 mV and was constant throughout the experiment. (C) The graph shows the dose-response curve obtained at various concentrations of GBP on the firing activity evoked by pulses of positive current (0.6–1 nA intensity, 700 ms duration). Each data point was obtained from at least four single experiments. The right part of the figure shows single voltage responses to a depolarizing pulse (0.9 nA) recorded from a striatal spiny neuron under control condition and during the application of 300 μM GBP. The RMP of the cell was −85 mV and was constant throughout the experiment.
Table 1.
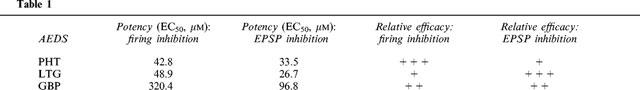
Effects of PHT, LTG and GBP on corticostriatal glutamatergic EPSPs
Spiny neurons respond to a single cortical stimulation by producing excitatory postsynaptic potentials (EPSPs) whose physiological and pharmacological characteristics have been previously described (Calabresi et al., 1990a, 1992a,1992b; Jiang & North, 1991). We studied the pharmacological action of these three AEDs on corticostriatal glutamatergic EPSPs. Bath application of PHT (3–300 μM), LTG (3–300 μM) and GBP (10–500 μM) significantly reduced the EPSP amplitude in a dose-dependent and reversible manner. The minimal effective concentration of PHT, LTG and GBP was 10, 10 and 100 μM respectively, while maximal inhibitory effects were obtained with 300 μM PHT, 300 μM LTG and 500 μM GBP (Figure 2). The EC50 value for this action was 33.5 μM for PHT, 26.7 μM for LTG, 96.8 μM for GBP (Table 1). It has to be stressed that LTG was also more effective than PHT and GBP in reducing the EPSP amplitude. In fact, saturating doses of PHT (300 μM) and GBP (500 μM) decreased the EPSP amplitude respectively to 70 and 55% of the control values. Conversely, saturating doses of LTG (300 μM) reduced the EPSP amplitude to about 45% of the control value.
Figure 2.
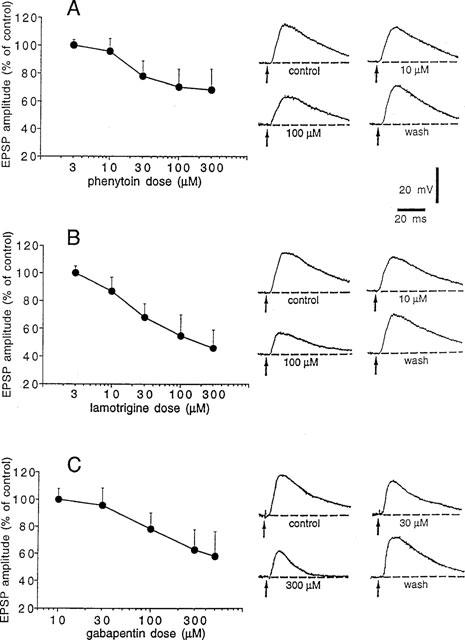
PHT, LTG and GBP inhibit excitatory postsynaptic potentials (EPSPs) evoked by cortical stimulation in striatal spiny neurons. (A) The graph shows the dose-response curve obtained at various concentrations of PHT on the amplitude of corticostriatal EPSPs. Each data point was obtained from at least four single experiments. The right part of the figure shows averages (four single sweeps) of EPSPs recorded from a striatal spiny neuron under control condition, during the application of two different concentrations of PHT and after 30 min of wash out. Each concentration was applied for 10 min. The RMP of the neuron was −89 mV and was constant throughout the experiment. (B) The graph shows the dose-response curve obtained at various concentrations of LTG on the amplitude of corticostriatal EPSPs. Each data point was obtained from at least four single experiments. The right part of the figure shows averages (four single sweeps) of EPSPs recorded from a striatal spiny neuron under control condition, during the application of two different concentrations of LTG and after 30 min of wash out. Each concentration was applied for 10 min. The RMP of the neuron was −89 mV and was constant throughout the experiment. (C) The graph shows the dose-response curve obtained at various concentrations of GBP on the amplitude of corticostriatal EPSPs. Each data point was obtained from at least four single experiments. The right part of the figure shows averages (four single sweeps) of EPSPs recorded from a striatal spiny neuron under control condition, during the application of two different concentrations of GBP and after 30 min of wash out. Each concentration was applied for 10 min. The RMP of the neuron was −88 mV and was constant throughout the experiment.
Effects of PHT, LTG and GBP on paired-pulse facilitation
In order to study whether the depression of corticostriatal synaptic transmission by these compounds was dependent on pre- or postsynaptic sites of action, we measured synaptic responses to a pair of stimuli before, during and after the application of two different concentrations of PHT, LTG and GBP (30 and 100 μM, 30 and 100 μM, 100 and 500 μM respectively). In these experiments interstimulus interval was of 60 ms. Paired-pulse modification of neurotransmission has been studied extensively and is attributed to a presynaptic change in release probability (Manabe et al., 1993; Schulz et al., 1994). An increase in the ratio of the second pulse response to the first pulse response (EPSP2/EPSP1) indicates a decrease in the release probability. The decrease in transmitter release probability is consistent with the observations that manipu-lations depressing transmitter release usually increase the magnitude of this ratio also at corticostriatal synapses (Calabresi et al., 1997a). PHT and GBP depressed corticostriatal EPSPs without significantly affecting paired-pulse facilitation (PPF). This finding suggests that the inhibitory effect of these AEDs did not exclusively involve a presynaptic site of action. Conversely, the depression of the EPSP by 30 μM LTG, which was similar to those caused by 100 μM PHT and 500 μM GBP, was coupled to a clear increase of PPF as expected by a prevalent presynaptic site of action. Interestingly, at higher concentrations (100 μM), LTG reduced corticostriatal synaptic transmission without changing EPSP2/EPSP1 ratio. The latter observation indicates that, at this higher dose, LTG also affects a postsynaptic site of action (Figure 3).
Figure 3.
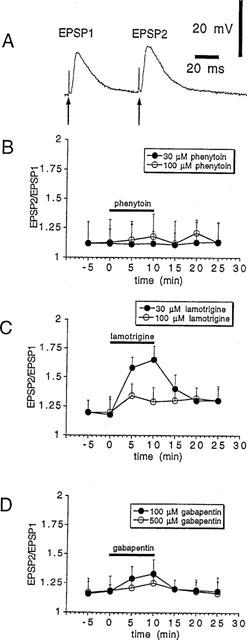
LTG but not PHT and GBP increases paired-pulse facilitation. (A) The trace shows synaptic responses to a pair of stimuli recorded with interstimulus interval of 60 ms under control condition. (B) The graph shows the ratio of the second pulse response to the first pulse response (EPSP2/EPSP1) before, during, and after the application of two different concentrations of PHT (black bar). (C) The graph shows the ratio of EPSP2/EPSP1 before, during, and after the application of two different concentrations of LTG (black bar). (D) The graph shows the ratio of EPSP2/EPSP1 before, during, and after the application of two different concentrations of GBP (black bar).
Effects of PHT, LTG and GBP on membrane depolarizations induced by exogenous glutamate
The experiments dealing with the paired-pulse facilitation raised the possibility that the depression of the EPSP amplitude induced by PHT, GBP but not by low doses of LTG could be caused by a reduction of the sensitivity of the postsynaptic cell to glutamate. In order to explore this possibility, we have measured the amplitude of the membrane depolarizations induced by brief bath application of exogenous glutamate (0.3–1 mM for 10–20 s) before, during and after the administration of these three compounds. As shown in Figure 4, two different doses of both PTH and GBP, which caused per se a depression of cortically-evoked EPSPs uncoupled to change in PPF (30–100 μM and 100–300 μM respectively), decreased in a dose-dependent manner the glutamate-induced membrane depolarizations. We also tested the postsynaptic sensitivity to glutamate before and during the application of lamotrigine. Neither 30 μM nor 100 μM LTG significantly affected the postsynaptic sensitivity to glutamate (_P_>0.05).
Figure 4.
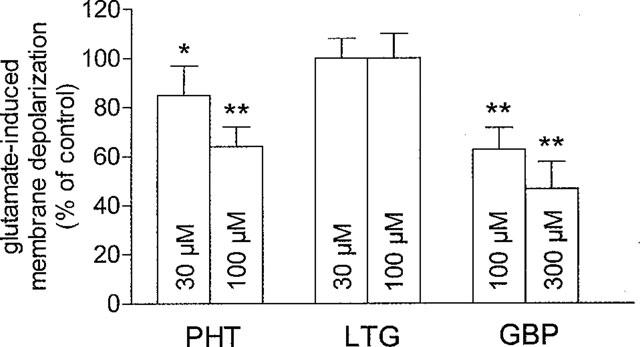
PHT and GBP but not LTG decrease postsynaptic sensitivity to exogenous glutamate. The graph shows that PHT and GBP exert a dose-dependent inhibition of membrane depolarizations induced by brief (10–20 s) applications of exogenous glutamate (0.3–1 mM) while LTG fails to affect these responses. Each data point was obtained from at least four single experiments (*=P<0.05; **=_P_>0.01). These experiments were performed in the presence of 1 μM TTX in order to avoid possible contaminations of the postsynaptic responses by a TTX-sensitive release of excitatory transmitter.
Discussion
The main finding of the present study is that PHT, LTG and GBP decrease the excitability of striatal spiny neurons by modulating multiple electrophysiological properties of these cells. Although previous data concerning the mechanisms of action of these AEDs have been obtained by using different experimental preparations (McLean & Macdonald, 1983; Leach et al., 1986; Yaari et al., 1986; Cheung et al., 1992; Rock et al., 1993; Kuo & Bean, 1994; Taylor, 1994; Calabresi et al., 1996b; Stefani et al., 1997), our study represents the first analysis in which multiple electrophysiological effects have been investigated in the same preparation in order to determine the sensitivity of each electrophysiological parameter to these compounds.
Intrinsic resting properties of striatal spiny neurons such as membrane potential, input resistance and membrane conductance were not significantly modified even by high concentrations of PHT, LTG and GBP. In contrast, we found that all these three AEDs depressed both the current-evoked repetitive firing discharge and the amplitude of EPSPs evoked by cortical stimulation. However, substantial pharmacological differences in the potency (EC50) and in the efficacy of these three agents were observed (Table 1). The EC50 measured for the inhibitory action on firing activity and synaptic transmission were close for PHT while these values were significantly different for LTG and GBP. Moreover, both LTG and GBP were more effective in reducing corticostriatal EPSPs than the current-evoked firing discharge. These data may suggest that, although these two new AEDs reduce the neuronal excitability by targeting multiple sites of action, they preferentially decrease excitatory synaptic transmission in the striatum. Moreover, PHT was the most potent and effective agent in reducing sustained repetitive firing, whereas LTG showed the major potency and efficacy in reducing corticostriatal EPSPs. GBP was more potent in reducing EPSP amplitude than in decreasing firing frequency, although high concentrations of this drug were also very effective in reducing the latter parameter.
The suppression of voltage-dependent sodium currents, detected as an inhibition of the firing activity, is a common action of many neuroprotective and anticonvulsivant drugs. The interaction with sodium channels has been demonstrated for phenytoin (Yaari et al., 1986), carbamazepine (McLean & Macdonald, 1986), oxcarbazepine (Calabresi et al., 1995b), gabapentin (Wamil & McLean, 1994), lamotrigine (Cheung et al., 1992), felbamate (Pisani et al., 1995), riluzole (Benoit & Escande, 1991; Siniscalchi et al., 1997). In the present study we have shown that PHT, LTG and GBP interfere with TTX-sensitive action potentials also in striatal neurons. All these three drugs reduced repetitive firing activity in a use-dependent manner as indicated by the evidence that the action potentials occurring in the late phase of the current-induced depolarization were preferentially inhibited. These data may indicate that in the striatum, such as in other brain regions, a sustained activation of the voltage-dependent sodium channels is required for the action of these AEDs presumably by stabilizing sodium channels in the inactivated state.
Synaptic excitatory transmission is emerging as a promising site of intervention for new AEDs and a reduction of glutamatergic transmission has been claimed as part of the mechanism of action of some new anticonvulsants (Walker & Sander, 1994; Macdonald & Kelly, 1994; Calabresi et al., 1995b, 1996b; Pisani et al., 1995). PHT, LTG and GBP reduced cortically-evoked EPSPs with apparently different relative order of potency and efficacy (Table 1). These differential pharmacological profiles, probably, originate from different mechanisms of action. The experiments dealing with the paired-pulse facilitation, in fact, suggest that LTG affects corticostriatal glutamatergic transmission with a prominent presynaptic site of action whereas PHT and GBP preferentially reduce postsynaptic sensitivity to glutamate. Changes in paired-pulse facilitation of synaptic transmission are commonly considered a good index of a presynaptic mechanism of action (Manabe et al., 1993; Schulz et al., 1994; Calabresi et al., 1997a). Accordingly, LTG reduced corticostriatal EPSPs with a clear increase in PPF and, even at saturating doses, failed to affect the membrane depolarizations induced by exogenous glutamate. Conversely, PHT and GBP decreased corticostriatal EPSPs without affecting the PPF; these AEDs also reduced the membrane depolarizations induced by exogenously-applied glutamate, suggesting a postsynaptic site of action. Interestingly, the doses required to achieve this latter effect were similar to those required to reduce corticostriatal synaptic transmission, indicating that a reduction of postsynaptic sensitivity to glutamate may play a major role in the inhibitory action exerted by PHT and GBP on glutamatergic transmission.
Therapeutic plasma levels of PHT and LTG are usually reported to be around 10–20 μM. The concentration of PHT in the cerebrospinal fluid is equal to the unbound fraction in the plasma (McNamara, 1996). The therapeutic GBP plasma levels are generally in the range of 10–50 μM. Limited data indicate that concentrations of GBP in cerebrospinal fluid are approximately 5–35% of those in plasma while concentrations in brain tissue are approximately 80% of those in plasma (McLean, 1994).
It is interesting to note that the resting membrane potential of striatal spiny neurons recorded in vitro is rather negative (around −85 mV in the presence of 2.5 external potassium concentration). If one assumes that the AEDs are exerting at least part of their effects via an interaction with the inactivated state of the Na+ channel population, this could explain why relatively high concentrations of AEDs are required to exert their effects. Nevertheless, it should be considered that striatal spiny neurons intracellularly recorded in vivo show a characteristic pattern of spontaneous activity consisting of long periods of silence (‘down' state) separated by brief episodes that cause firing discharge (‘up' state). These depolarizing episodes were attributed to maintained, coordinated synaptic excitation from cerebral cortex, whereas the ‘down' state was attributable to the activation of a strong inwardly rectifying potassium conductance (Calabresi et al., 1990b; Wilson & Kawaguchi, 1996). Thus, it is likely that in vivo the concentrations of AEDs required to achieve firing and synaptic inhibition during the ‘up' state are lower than those necessary to inhibit striatal spiny neurons in vitro.
It has recently been proposed the use of old and new AEDs as possible neuroprotective agents in the therapy of acute (ischaemia) and neurodegenerative (Parkinson's disease and Huntington's disease) disorders involving the basal ganglia (Zipp et al., 1993; Meldrum, 1994; Lynch et al., 1995; Koroshetz & Moskowitz, 1996; Olson et al., 1997; Shinotoh et al., 1997; Starr et al., 1997). Thus, the differential pharmacological profiles reported for PHT, LTG and GBP might help not only in understanding the antiepileptic mechanisms of action of old and new AEDs, but also in the design of new clinical protocols for the treatment of acute and chronic diseases involving the striatum.
Acknowledgments
We wish to thank M. Tolu for the technical assistance. This study was supported by BIOMED Project to P.C. (No. BMH4-97-2215) and by HURST-CNR Biotechnology Program L-95/95 to G.B.
References
- AMATO G., CRESCIMANNO G., SORBERA F., LA GRUTTA V. Relationship between the striatal system and amygdaloid paroxysmal activity. Exp. Neurol. 1982;77:492–504. doi: 10.1016/0014-4886(82)90223-0. [DOI] [PubMed] [Google Scholar]
- BARTOSZYK G.D. Gabapentin and convulsions provoked by excitatory amino acids. Naunyn Schmiedebergs Arch. Pharmacol. 1982;324:R24. [Google Scholar]
- BENOIT E., ESCANDE D. Riluzole specifically blocks inactivated Na channels in myelinated nerve fibre. Pflügers Arch. 1991;419:603–609. doi: 10.1007/BF00370302. [DOI] [PubMed] [Google Scholar]
- CALABRESI P., CENTONZE D., PISANI A., BERNARDI G. Endogenous adenosine mediates the presynaptic inhibition induced by aglycemia at corticostriatal synapses. J. Neurosci. 1997a;17:4509–4516. doi: 10.1523/JNEUROSCI.17-12-04509.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CALABRESI P., CENTONZE D., PISANI A., SANCESARIO G., GUBELLINI P., MARFIA G.A., BERNARDI G. Striatal spiny neurons and cholinergic interneurons express differential ionotropic glutamatergic responses and vulnerability: implications for ischemia and Huntington's disease. Ann. Neurol. 1998;43:586–597. doi: 10.1002/ana.410430506. [DOI] [PubMed] [Google Scholar]
- CALABRESI P., DE MURTAS M., PISANI A., STEFANI A., SANCESARIO G., MERCURI N.B., BERNARDI G. Vulnerability of medium spiny striatal neurons to glutamate: role of Na+/K+ ATPase. Eur. J. Neurosci. 1995a;7:1674–1683. doi: 10.1111/j.1460-9568.1995.tb00689.x. [DOI] [PubMed] [Google Scholar]
- CALABRESI P., DE MURTAS M., STEFANI A., PISANI A., SANCESARIO G., MERCURI N.B., BERNARDI G. Action of GP47779, the active metabolite of oxcarbazepine, on corticostriatal system. I. Modulation of corticostriatal synaptic transmission. Epilepsia. 1995b;36:990–996. doi: 10.1111/j.1528-1157.1995.tb00957.x. [DOI] [PubMed] [Google Scholar]
- CALABRESI P., MAGARINOS ASCONE C., CENTONZE D., PISANI A., SANCESARIO G., D'ANGELO V., BERNARDI G. Opposite membrane potential changes induced by glucose deprivation in striatal spiny neurons and in large aspiny interneurons. J. Neurosci. 1997b;17:1940–1949. doi: 10.1523/JNEUROSCI.17-06-01940.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CALABRESI P., MAJ R., PISANI A., MERCURI N.B., BERNARDI G. Long-term synaptic depression in the striatum: physiological and pharmacological characterization. J. Neurosci. 1992a;12:4224–4233. doi: 10.1523/JNEUROSCI.12-11-04224.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CALABRESI P., MERCURI N.B., BERNARDI G. Synaptic and intrinsic control of membrane excitability of neostriatal neurons. II. An in vitro analysis. J. Neurophysiol. 1990a;63:663–675. doi: 10.1152/jn.1990.63.4.663. [DOI] [PubMed] [Google Scholar]
- CALABRESI P., MERCURI N.B., BERNARDI G. Synaptic and intrinsic control of membrane excitability of neostriatal neurons. I. An in vivo analysis. J. Neurophysiol. 1990b;63:651–662. doi: 10.1152/jn.1990.63.4.651. [DOI] [PubMed] [Google Scholar]
- CALABRESI P., PISANI A., MERCURI N.B., BERNARDI G. Long-term potentiation in the striatum is unmasked by removing the voltage-dependent blockade of NMDA receptor channel. Eur. J. Neurosci. 1992b;4:929–935. doi: 10.1111/j.1460-9568.1992.tb00119.x. [DOI] [PubMed] [Google Scholar]
- CALABRESI P., PISANI A., MERCURI N.B., BERNARDI G. The corticostriatal projection: from synaptic plasticity to dysfunctions of the basal ganglia. Trends Neurosci. 1996a;19:19–24. doi: 10.1016/0166-2236(96)81862-5. [DOI] [PubMed] [Google Scholar]
- CALABRESI P., SINISCALCHI A., PISANI A., STEFANI A., MERCURI N.B., BERNARDI G. A field potential analysis on the effects of lamotrigine, GP 47779, and felbamate in neocortical slices. Neurology. 1996b;47:557–562. doi: 10.1212/wnl.47.2.557. [DOI] [PubMed] [Google Scholar]
- CEPEDA C., WALSH J.P., PEACOCK W., BUCKWALD N.A., LEVINE M.S. Neurophysiological, pharmacological and morphological properties of human caudate neurons recorded in vitro. Neuroscience. 1994;59:89–103. doi: 10.1016/0306-4522(94)90101-5. [DOI] [PubMed] [Google Scholar]
- CHEUNG H., KAMP D., HARRIS E. An in vitro investigation of the action of lamotrigine on neuronal-activated sodium channels. Epilepsy Res. 1992;13:107–112. doi: 10.1016/0920-1211(92)90065-2. [DOI] [PubMed] [Google Scholar]
- DIVAC I., FONNUM F., STORM-MATHISON J. High affinity uptake of glutamate in terminals of corticostriatal axons. Nature. 1977;266:377–378. doi: 10.1038/266377a0. [DOI] [PubMed] [Google Scholar]
- ENGEL J., WOLFSON L., BROWN L. Anatomical correlates of electrical and behavioral events related to amygdala kindling. Ann. Neurol. 1978;3:538–544. doi: 10.1002/ana.410030615. [DOI] [PubMed] [Google Scholar]
- FAETH W.H., WALKER A.E., ANDY O.J. The propagation of cortical and sub-cortical epileptic discharge. Epilepsia. 1954;3:37–48. doi: 10.1111/j.1528-1157.1954.tb03152.x. [DOI] [PubMed] [Google Scholar]
- GALE K. Subcortical structures and pathways involved in convulsive seizure generation. J. Clin. Neurophysiol. 1992;9:264–277. doi: 10.1097/00004691-199204010-00007. [DOI] [PubMed] [Google Scholar]
- HARDEN C.L. New antiepileptic drugs. Neurology. 1994;44:787–795. doi: 10.1212/wnl.44.5.787. [DOI] [PubMed] [Google Scholar]
- HORIKAWA H., ARMSTRONG W.E. A versatile means of intracellular labelling: injection of biocytin and its detection with avidin conjugates. J. Neurosci. Methods. 1988;25:1–11. doi: 10.1016/0165-0270(88)90114-8. [DOI] [PubMed] [Google Scholar]
- JIANG Z.-C., NORTH R.A. Membrane properties and synaptic responses of rat striatal neurones in vitro. J. Physiol. 1991;443:533–553. doi: 10.1113/jphysiol.1991.sp018850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KITA T., KITA H., KITAI S.T. Passive electrical membrane properties of rat neostriatal neurons in an in vitro slice preparation. Brain Res. 1984;300:129–139. doi: 10.1016/0006-8993(84)91347-7. [DOI] [PubMed] [Google Scholar]
- KOROSHETZ W.J., MOSKOWITZ M.A. Emerging treatments for stroke in humans. Trends Pharmacol. Sci. 1996;17:227–233. doi: 10.1016/0165-6147(96)10020-1. [DOI] [PubMed] [Google Scholar]
- KUO C.-C., BEAN B.P. Na+ channels must deactivate to recover from inactivation. Neuron. 1994;12:819–829. doi: 10.1016/0896-6273(94)90335-2. [DOI] [PubMed] [Google Scholar]
- LEACH M.J., MARDEN C.M., MILLER A.A. Pharmacological studies on lamotrigine, a novel potential antiepileptic drug: II Neurochemical studies on the mechanism of action. Epilepsia. 1986;27:490–497. doi: 10.1111/j.1528-1157.1986.tb03573.x. [DOI] [PubMed] [Google Scholar]
- LYNCH III J.J., YU S.P., CANZONIERO L.M.T., SENSI S.L., CHOI D.W. Sodium channel blockers reduce oxygen-glucose deprivation-induced cortical neuronal injury when combined with glutamate receptor antagonists. J. Pharmacol. Exp. Ther. 1995;273:554–560. [PubMed] [Google Scholar]
- MACDONALD R.L., KELLY K.New antiepileptic drug mechanisms of action An appraisal of some new anticonvulsivants – a clinical perspective 1994New York: Wiley; 35–50.ed. Trimble, M. pp [Google Scholar]
- MACDONALD R.L.Cellular effects of antiepileptic drugs Epilepsy: a comprehensive textbook 1996Philadelphia: Lippincott-Raven Publishers; 1383–1391.eds. Engel, J., & Pedley, T.A. pp [Google Scholar]
- MCNAMARA J.O.Drugs effective in the therapy of the epilepsies Goodman & Gilman's; The pharmacological basis of therapeutics 1996New York: McGraw-Hill; 461–486.eds. Hardman, J.G. & Limbird, L.E. pp [Google Scholar]
- MANABE T., WYLLIE D.J.I., NICOLL R.A. Modulation of synaptic transmission and long-term potentiation: effects on paired pulse facilitation and EPSC variance in the CA1 region of the hippocampus. J. Neurophysiol. 1993;70:1451–1459. doi: 10.1152/jn.1993.70.4.1451. [DOI] [PubMed] [Google Scholar]
- MCLEAN M.J. Clinical pharmacokinetics of gabapentin. Neurology. 1994;44 suppl 5:S17–S22. [PubMed] [Google Scholar]
- MCLEAN M.J., MACDONALD R.L. Carbamazepine and 10,11-epoxycarbamazepine produce use- and voltage-dependent limitation of rapidly firing action potentials of mouse central neurons in cell culture. J. Pharmacol. Exp. Ther. 1986;238:727–737. [PubMed] [Google Scholar]
- MCLEAN M.J., MACDONALD R.L. Multiple actions of phenytoin on mouse spinal cord neurons in cell culture. J. Pharmacol. Exp. Ther. 1983;227:779–789. [PubMed] [Google Scholar]
- MELDRUM B.S. The role of glutamate in epilepsy and other CNS disorders. Neurology. 1994;44 suppl 8:S14–S23. [PubMed] [Google Scholar]
- OLES R.J., SINGH L., HUGHES J., WOODRUFF G.N. The anticonvulsant action of gabapentin involves the glycine/NMDA receptor. Soc. Neurosci. 1990;16:783. [Google Scholar]
- OLSON W.L., GRUENTHAL M., MUELLER M.E., OLSON W.H. Gabapentin for parkinsonism: a double-blind, placebo-controlled, crossover trial. Am. J. Med. 1997;102:60–66. doi: 10.1016/s0002-9343(96)00381-6. [DOI] [PubMed] [Google Scholar]
- PISANI A., STEFANI A., SINISCALCHI A., MERCURI N.B., BERNARDI G., CALABRESI P. Electrophysiological actions of felbamate on rat striatal neurones. Br. J. Pharmacol. 1995;116:2053–2061. doi: 10.1111/j.1476-5381.1995.tb16411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROCK D.M., KELLY K.M., MACDONALD R.L. Gabapentin actions on ligand- and voltage-gated responses in cultured rodent neurons. Epilepsy Res. 1993;16:89–98. doi: 10.1016/0920-1211(93)90023-z. [DOI] [PubMed] [Google Scholar]
- SCHULZ P.E., COOK E.P., JOHNSTON D. Changes in paired-pulse facilitation suggest presynaptic involvement in long-term potentiation. J. Neurosci. 1994;14:5325–5337. doi: 10.1523/JNEUROSCI.14-09-05325.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHINOTOH H., VINGERHOETS F.J., LEE C.S., UITTI R.J., SCHULZER M., CALNE D.B., TSU J. Lamotrigine trial in idiopathic parkinsonism: a double-blind, placebo-controlled, crossover study. Neurology. 1997;48:1282–1285. doi: 10.1212/wnl.48.5.1282. [DOI] [PubMed] [Google Scholar]
- SINISCALCHI A., BONCI A., MERCURI N.B., BERNARDI G. Effects of riluzole on rat cortical neurones: an in vitro electrophysiological study. Br. J. Pharmacol. 1997;120:225–230. doi: 10.1038/sj.bjp.0700905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STARR M.S., STARR B.J., KAUR J. Stimulation of basal and L-DOPA-induced motor activity by glutamate antagonists in animal models of Parkinson's disease. Neurosci. Biobehav. Rev. 1997;21:437–446. doi: 10.1016/s0149-7634(96)00039-5. [DOI] [PubMed] [Google Scholar]
- STEFANI A., SPADONI F., BERNARDI G. Differential inhibition by riluzole, lamotrigine and phenytoin of sodium and calcium currents in cortical neurons: implication for neuroprotective strategies. Exp. Neurol. 1997;147:115–122. doi: 10.1006/exnr.1997.6554. [DOI] [PubMed] [Google Scholar]
- TAYLOR C.P. Emerging perspective on the mechanism of action of gabapentin. Neurology. 1994;44 suppl 5:S10–S16. [PubMed] [Google Scholar]
- UPTON N. Mechanisms of action of new antiepileptic drugs: rational design and serendipitous finding. Trends Pharmacol. Sci. 1994;15:456–463. doi: 10.1016/0165-6147(94)90059-0. [DOI] [PubMed] [Google Scholar]
- WALKER M.C., SANDER J.W. Developments in antiepileptic drug therapy. Curr. Opin. Neurol. 1994;7:131–139. doi: 10.1097/00019052-199404000-00009. [DOI] [PubMed] [Google Scholar]
- WAMIL A.W., MCLEAN M.J. Limitation by gabapentin of high-frequency action potential firing by mouse central neurons in cell culture. Epilepsy Res. 1994;17:1–11. doi: 10.1016/0920-1211(94)90074-4. [DOI] [PubMed] [Google Scholar]
- WILSON C.J., GROVES P.M. Fine structure and synaptic connections of the common spiny neuron of the rat neostriatum: a study employing intracellular inject of horse-radish peroxidase. J. Comp. Neurol. 1980;194:599–615. doi: 10.1002/cne.901940308. [DOI] [PubMed] [Google Scholar]
- WILSON C.J., KAWAGUCHI Y. The origins of two-state spontaneous membrane potential fluctuations of neostriatal spiny neurons. J. Neurosci. 1996;16:2651–2661. doi: 10.1523/JNEUROSCI.16-07-02397.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAARI Y., SELTZER M., PINCUS J. Phenytoin: mechanism of its anticonvulsivant action. Ann. Neurol. 1986;20:171–184. doi: 10.1002/ana.410200202. [DOI] [PubMed] [Google Scholar]
- ZIPP F., BAAS H., FISHER P.A. Lamotrigine-antiparkinsonian activity by blockade of glutamate release. J. Neural Transm. Park. Dis. Dement Sect. 1993;5:67–75. doi: 10.1007/BF02260916. [DOI] [PubMed] [Google Scholar]