Anatomy of a proficient enzyme: The structure of orotidine 5′-monophosphate decarboxylase in the presence and absence of a potential transition state analog (original) (raw)
Abstract
Orotidine 5′-phosphate decarboxylase produces the largest rate enhancement that has been reported for any enzyme. The crystal structure of the recombinant Saccharomyces cerevisiae enzyme has been determined in the absence and presence of the proposed transition state analog 6-hydroxyuridine 5′-phosphate, at a resolution of 2.1 Å and 2.4 Å, respectively. Orotidine 5′-phosphate decarboxylase folds as a TIM-barrel with the ligand binding site near the open end of the barrel. The binding of 6-hydroxyuridine 5′-phosphate is accompanied by protein loop movements that envelop the ligand almost completely, forming numerous favorable interactions with the phosphoryl group, the ribofuranosyl group, and the pyrimidine ring. Lysine-93 appears to be anchored in such a way as to optimize electrostatic interactions with developing negative charge at C-6 of the pyrimidine ring, and to donate the proton that replaces the carboxylate group at C-6 of the product. In addition, H-bonds from the active site to O-2 and O-4 help to delocalize negative charge in the transition state. Interactions between the enzyme and the phosphoribosyl group anchor the pyrimidine within the active site, helping to explain the phosphoribosyl group's remarkably large contribution to catalysis despite its distance from the site of decarboxylation.
Orotidine 5′-phosphate decarboxylase (ODCase) (EC 4.1.1.23) is responsible for de novo synthesis of uridine 5′-phosphate, an essential precursor of RNA and DNA. In neutral solution, orotidine 5′-monophosphate (OMP) undergoes spontaneous decarboxylation to uridine 5′-phosphate with a half-time of 78 million years (1). At the ODCase active site, the same reaction proceeds with a half-time of 18 msec (2). Comparison of _k_cat/_K_m with _k_non indicates that ODCase surpasses other enzymes in its proficiency‖ as a catalyst, achieving a remarkable affinity for the altered substrate in the transition state (1). In addition to surmounting this formidable kinetic barrier, the ODCase reaction is of special interest in view of its lack of precedent in biological chemistry. The substrate is devoid of an effective repository for the negative charge that is generated at C-6 when CO2 is eliminated, yet the enzyme functions without metals or other cofactors. Enzymatic decarboxylation of OMP is also remarkable in the importance (for catalysis) of a seemingly irrelevant part of the substrate. By its presence, the 5′-phosphoryl group contributes a factor of ≈108-fold to _k_cat/_K_m, in spite of its considerable distance from the site of chemical transformation of the substrate (3). As a first step toward understanding these unusual properties, we have investigated the crystal structure of recombinant Saccharomyces cerevisiae ODCase alone and in complex with a postulated transition state analogue, 6-hydroxyuridine 5′-phosphate (BMP) (_K_i = 9 × 10−12 M (4)].
Materials and Methods
Recombinant yeast ODCase was expressed in Escherichia coli SS6130 (pBGM88) and was purified as described (5). Crystals of the native enzyme were grown at 4°C by the hanging drop vapor diffusion method using 0.1 M sodium/Hepes (pH 7.0), 10% isopropanol, and 20% (wt/vol) polyethylene glycol 4000 as the precipitant. These orthorhombic crystals belong to space group P212121 with unit cell dimensions of a = 90.1 Å, b = 116.2 Å, c = 117.0 Å. Crystals of ODCase complexed with BMP were grown at 22°C in 0.18 M sodium phosphate and 18% (wt/vol) polyethylene glycol 3350. Crystals of the inhibitory complex belong to space group P21 with unit cell parameters of a = 79.89 Å, b = 79.97 Å, c = 98.20 Å, and β = 108.59°. X-ray data for the apo and BMP-enzyme complex were collected at −180°C on a Rigaku (Tokyo) RU-200 rotating anode generator equipped with an MSC Raxis 4 area detector. Data were measured and scaled with denzo (6) and scalepack (6). Multiwavelength anomalous dispersion (MAD) data for one crystal of selenomethionyl native enzyme were collected on the Industrial Macromolecular Crystallography Association beamline at the Argonne National Laboratories by using a MAR 2 × 2 charge-coupled device detector for all MAD data sets collected. Data were measured and scaled by using hkl2000 (6) and scalepack (6). MAD phasing, x-ray crystallographic refinement, and molecular replacement were carried out in cns (7). Solvent flattening, electron density averaging, and histogram matching of the MAD phased electron density map were accomplished with dm (8, 9) to 2.3-Å resolution, and the resulting map was used to model all atoms of the apo yeast orotidine decarboxylase. A total of 20 ordered selenomethionine sites were identified with five sites belonging to each molecule by using cns (7). These five sites for each of the four molecules were used to locate the noncrystallographic symmetry. Water molecules were added to the x-ray refinement by using a 3_F_obs − 2_F_calc electron density map for peaks greater than 1.5σ with distance constraints. The resulting Ramachandran plot for both the apo and BMP complex showed all amino acids within energy favorable regions.
Results and Discussion
The structure of ODCase was solved by MAD using selenomethionine-labeled enzyme (Tables 1 and 2). The resulting 2.3-Å MAD phased electron density map was solvent-flattened and averaged to yield a readily interpretable electron density map (Fig. 1). Each ODCase subunit of this dimeric enzyme folds as an α/β barrel with eight central β sheets surrounded by nine α helices (Fig. 2), similar to the fold first described for triosephosphate isomerase (10). The model of the unliganded enzyme included all 267 monomer residues and was refined to 2.1-Å resolution. Residues 207–218, although considerably more mobile than the rest of the model, were nevertheless included in the refinement, yielding _R_factor and _R_free values of 21.8 and 25.5%, respectively. The ODCase-BMP complex was solved by molecular replacement to 2.4-Å resolution by using the dimer model from the ligand-free ODCase refined model. _R_factor and _R_free of the complex were 21.3 and 25.8%, respectively, and included all 267 residues with a well ordered loop for residues 207–218. Crystals of the ligand-free and the BMP complex contained two dimer molecules in the asymmetric unit. Neither structure showed the presence of metals or other cofactors that could participate in catalysis. The N and C termini are located on the same face of each monomer (Fig. 2) and distal to the active site and subunit–subunit interface (Fig. 3), explaining the widely exploited ability of this enzyme to yield functional protein fusions (11–13).
Table 1.
Data collection and phasing statistics
| Data set | Wavelength, Å | Resolution, Å | Overall I/σ | Unique reflections | Completeness, % | _R_sym, %* | _R_anom, %† | _f_′, _e_− | f", _e_− | Phasing acentric | Power centric |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Native | 1.0281 | 20–2.3 | 37.5 | 99,748 | 99.5 | 3.2 | |||||
| SML1 | 0.9793 | 20–2.3 | 41.6 | 99,690 | 99.4 | 3.8 | 2.1 | −7.9 | 3.0 | 1.57 | 1.53 |
| SML2 | 0.9795 | 20–2.3 | 39.4 | 99,710 | 99.4 | 4.1 | 1.9 | −8.3 | 3.8 | 1.49 | 1.47 |
| SML3 | 0.9350 | 20–2.3 | 33.4 | 98,546 | 98.9 | 5.3 | 1.7 | −2.6 | 0.55 | 0.89 | 0.89 |
Table 2.
Refinement statistics
| Data set | Resolution*, Å | Completeness†, % | _R_factor, %‡ | _R_free, %‡ | rms deviations | Model atoms | Average B Factor | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Bonds, Å | Angles, ° | Protein§ | Water | Ligand | Protein | Water | Ligand | |||||
| Apo | 2.1 (4.9%) | 97.1 (88.2) | 21.8 (23.3) | 25.5 (26.8) | 0.008 | 1.33 | 2059 (4) | 874 | 25.1 | 34.3 | ||
| BMP | 2.4 (6.1%) | 91.3 (78.4) | 21.3 (23.1) | 25.8 (27.3) | 0.010 | 1.50 | 2059 (4) | 501 | 32 (4) | 29.3 | 35.5 | 27.5 |
Figure 1.
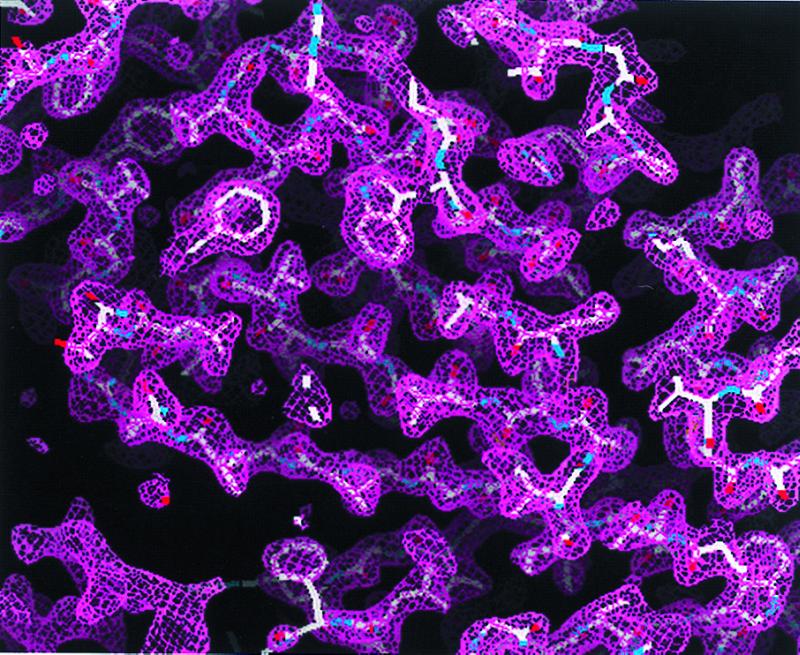
Experimental MAD phased electron density map at 2.3-Å resolution contoured at 1 σ. The electron density map was readily interpretable for modeling of yeast ODCase.
Figure 2.
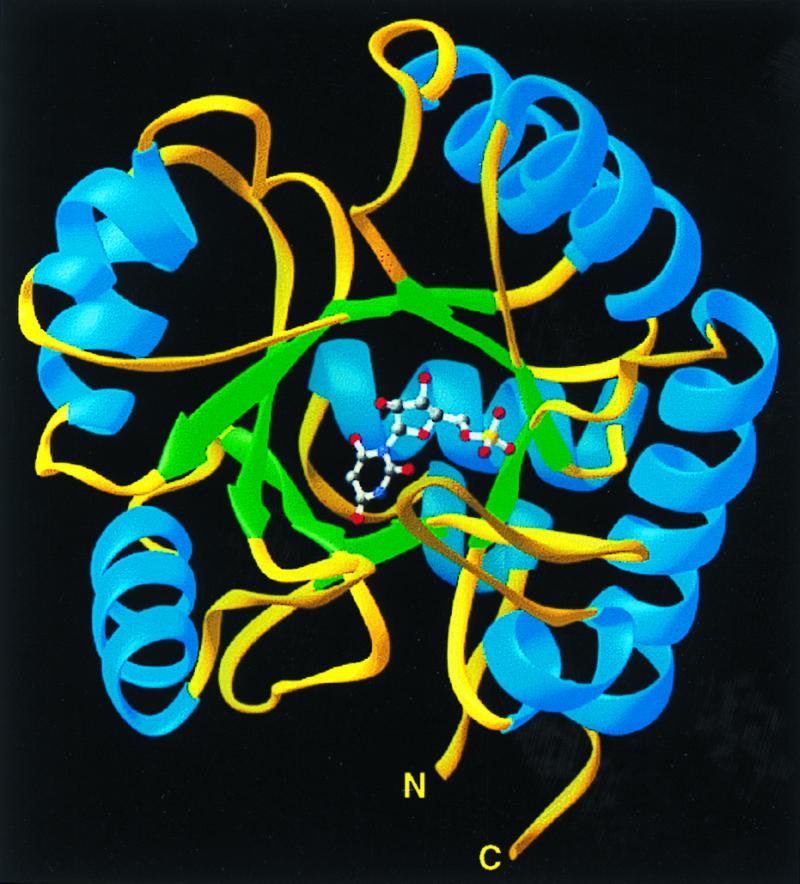
Ribbons (25) depiction of the ODCase monomer with bound inhibitor BMP positioned at the open end of the α/β barrel.
Figure 3.

Ribbon drawing of dimeric ODCase showing the symmetrically opposed active sites occupied by BMP and the positions of the N and C termini of each subunit.
In each monomer, BMP is bound in a cavity near the “open” end of the α/β barrel (Figs. 2 and 4). Comparison of the models calculated for ODCase in the absence and presence of BMP indicates that ligand binding evokes significant loop movements (Fig. 5). As a result, the ligand is sequestered from solvent, forming 10 H-bonds with active site residues, in which the electronegative atoms are separated by 2.9 Å or less (Fig. 6).
Figure 4.

Stereo view of the ODCase active site with ligand BMP. The electron density is colored magenta and contoured at 1.5 σ from a composite simulated-annealing omit map [cns 0.5 (7)] at 2.4-Å resolution. The α carbons and water molecules are labeled in green.
Figure 5.
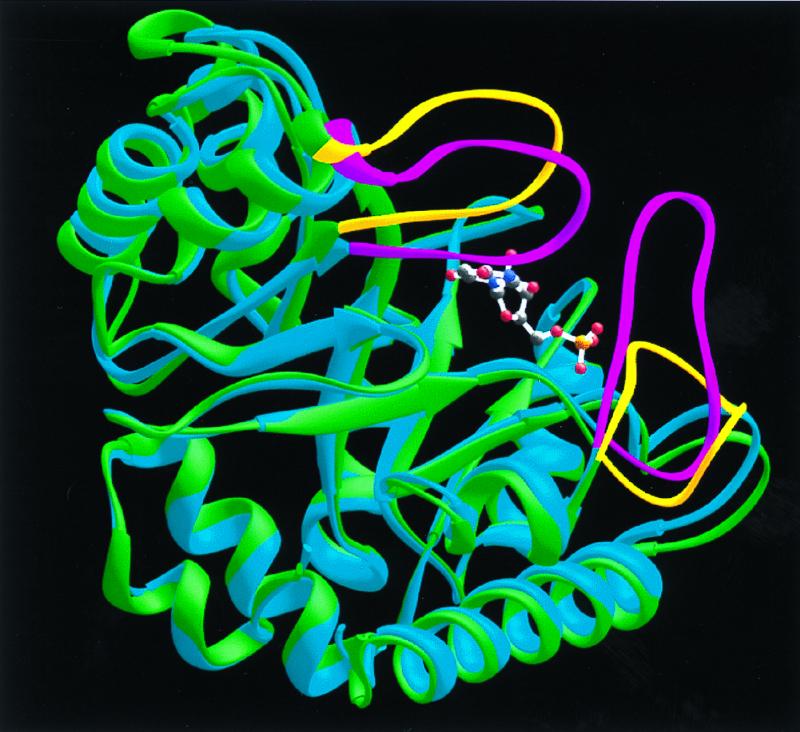
Superposition of native (green) and BMP-liganded (blue) monomers to illustrate the protein loop movements that accompany BMP binding. The loops of the native enzyme are shown in yellow. Upon ligand binding, these protein loops (shown in magenta for the liganded monomer) close around the ligand and sequester it from solvent.
Figure 6.

Contacts formed between ODCase and the phosphate group (green dashed lines), the ribose (magenta dashed lines), and the pyrimidine ring (the cyan dashed lines) of the postulated transition state analog inhibitor BMP (shown in red). Active site residues contributed by loop 95–103 of the opposite subunit (represented by the green ribbon) are designated by an asterisk. The numbers adjacent to each dashed line were measured between electronegative atoms and represent the average value derived from all four subunits in the asymmetric unit. Contacts between amino acid residues are indicated by thin, black dashed lines. These distances between electronegative atoms are Gly-214(CO) → Lys-156(NH), 2.7 Å; Asn-120(CO) → Lys-93(NH), 2.8 Å; Asn-120(NH2) → Asp-91(COO−), 2.7 Å; Asp-91(COO−) → Lys-93(NH3+), 2.5 Å; Asp-91(COO−) → Lys-59(NH3+), 2.7 Å; Ser-35(OH) → Lys-59(NH3+), 3.1 Å; and Ser-35(OH) → Asp-37(COO−), 2.9 Å.
H-bonds between BMP and ODCase can be divided into four groups based on their position and function in the free and inhibited enzymes. One group of H-bonds between BMP and enzyme involves a scaffold of residues whose positions are little affected by ligand binding, changing their Cα positions by 0.6 Å, on average. These scaffold H-bonds are formed from Gly-234(NH) to the phosphoryl group, from Asp-37 to the 3′-OH group, from Lys-93 to O-6, and from Arg-235 to the phosphoryl group (Fig. 6). Arg-235 was included in this group based on function because its Cα rms deviation movement is 0.96 Å. This change results from rotation of the guanidinium side chain, of sequence Gly-Arg-Gly, from solvent toward the inhibitor phosphoryl group to establish three 2.7–2.9 Å H-bonds. In a second group, residues from the opposite subunit extend across the dimer interface into the binding pocket, forming H-bonds from Asp-96* and Thr-100* to the 2′-OH group of BMP. These contacts are made possible by a moderate repositioning of Loop 95*-103* (Cα rms deviation of 1.3–3.1 Å) as BMP is bound. Third, Loop 151–165 shifts position, moving toward the bound ligand (Cα rms deviation of 1.3–4.1 Å) so that the peptide amide of Ser-154 contacts O-4 of BMP. The remaining residues of Loop 151–165 provide a “hydrophobic umbrella” covering O-4 and C-5 of the pyrimidine ring. The fourth set of interactions involves a mobile domain, Loop 207–217, which is disordered in the free enzyme but well organized in the inhibitor–enzyme complex. Loop 207–217 provides H-bonds from Tyr-217 to the phosphoryl group and from Gln-215 to O-2 of the pyrimidine ring. As a consequence of their combined movements, Loop 151–165 and Loop 207–217 contact each other, enclosing BMP and sealing off the active site from bulk solvent (Fig. 5).
Contacts between the enzyme and the ribosyl moiety of BMP involve both the 2′- and 3′-OH groups (Fig. 6). The 2′-OH group participates in two strong H-bonds with the side-chains of Asp-96* (2.4 Å) and Thr-100* (2.8 Å) of the opposite subunit whereas the 3′-OH forms a 2.8-Å H-bond with the carboxylate group of Asp-37. Together, these three H-bonds tether the BMP ribosyl moiety to the enzyme, constraining the position of the pyrimidine ring within the active site. The interaction between the ligand's ribosyl OH groups and the side chains of Asp-96* and Thr-100* of the second subunit is consistent with the proposal that the active species of yeast ODCase is a dimer similar to that shown for the bifunctional human uridine 5′-phosphate synthase and its ODCase domain (14).
TIM-barrel enzymes that act on phosphorylated substrates typically feature a flexible loop proximal to the protein C terminus that becomes ordered upon ligand binding, establishing contacts with the substrate's phosphoryl group (15). Loop 207–218 becomes ordered upon BMP binding, resulting in an H-bond (2.7 Å) between the OH group of Tyr-217 and a phosphoryl oxygen. This conformational change makes possible four additional H-bonds (Fig. 6) from the phosphoryl group to Gly-234(NH) (2.9 Å) and Arg-235 (2.9, 3.1, and 2.8 Å). The extreme importance of the phosphoryl group for ligand recognition is also reflected in the behavior of ODCase substrates and inhibitors. In the yeast enzyme, we find that the presence of the BMP phosphoryl group results in a binding affinity (_K_i = 9 × 10−12 M) ≈6 orders of magnitude greater than that of the nucleoside (9.3 × 10−6 M) or base (6 × 10−6 M). Furthermore, comparison of _k_cat/_K_m for OMP (3 × 107 s−1⋅M−1) with _k_cat/_K_m for orotidine (1.3 s−1⋅M−1) indicates an even greater contribution by the phosphoryl group to transition state affinity. Similar observations have been reported for the human enzyme (3), which, unlike the yeast enzyme, is a bifunctional protein.
Catalysis presents two problems for any enzyme. First, the binding affinity of the altered substrate in the transition state should be as large as possible, to maximize “efficiency” (the second order rate constant _k_cat/_K_m) relative to _k_non. Second, the first order rate constant _k_cat should be as large as possible relative to _k_non, to maximize substrate turnover. This second objective can be accomplished only to the extent that the enzyme's binding affinity for the altered substrate in the transition state exceeds its affinity for the substrate in the ground state. ODCase achieves both of these objectives more effectively than any other enzyme whose properties have been reported, by a reaction without precedent among other decarboxylases, and it is of interest to ask how this is accomplished.
To enhance the rate of OMP decarboxylation, this catalyst must presumably stabilize a carbanion (or carbene), generated at C-6 upon elimination of CO2, more effectively than does solvent water. This reaction shows a 13C kinetic isotope effect on _k_cat/_K_m of sufficient magnitude to indicate that cleavage of the scissile C—C bond is almost fully rate determining (16). The potency of BMP as an inhibitor may be explained (4) by its resemblance to a carbanionic intermediate proposed by Beak and Siegel (17) and would be expected to contact the same enzyme residues that participate in OMP decarboxylation. Thus, interactions between active site residues and the pyrimidine ring of BMP revealed by the structure of the inhibited enzyme are of central mechanistic interest. In BMP (Fig. 7), —O− replaces the —COO− at C-6 of substrate OMP. The structure of the BMP-ODCase complex suggests that BMP binds as the 2-syn isomer, with substituent ribose rotated away from the 6-oxygen atom, as in the major conformation of orotidine in solution (18).
Figure 7.

Decarboxylation of orotidylate (OMP) via a nitrogen ylide, according to a mechanism proposed by Beak and Siegel (17). Because both the 2-syn and 2-anti isomers of BMP contain negative charges at positions corresponding to the 2- and 6-positons of the carbanionic intermediate, tight binding of either isomer of this potential transition state analogue would be compatible with the Beak–Siegel mechanism. The position of Lys-93 in the transition state, inferred from its position in the BMP complex, is shown on the right.
Previously, it was shown that replacement of Lys-93 of the native yeast enzyme with cysteine yielded a mutant enzyme with significantly reduced activity (19). We have also found that replacement of Lys-93 by alanine markedly reduces the activity of the recombinant yeast enzyme. Mechanisms previously proposed for elimination of CO2 from OMP involve the generation of a carbanion at C-6, which could in principle be stabilized by donation of an enzyme proton at C-2 (17) or C-4 (20), by the protonated ɛ-amino group of Lys-93. In the x-ray structure, however, C-2 and C-4 are seen to be associated not with Lys-93 but instead with the amide group of Gln-215 and the peptide NH group of Ser-154, respectively. These NH groups are too weakly acidic for complete proton transfer but do furnish H-bonds that appear to stabilize the BMP complex and presumably have a stabilizing character on the carbanion that is generated during decarboxylation.
If bound substrate resembles BMP in conformation (Figs. 6 and 7), then formation of a carbanionic intermediate would generate negative charge at C-6 of the pyrimidine ring in the transition state, near the position observed for the positively charged ɛ-amino group of Lys-93. This suggests a different and highly important role in catalysis for Lys-93 in stabilizing the carbanionic transition state and in furnishing the proton that appears at C-6 of product uridine 5′-phosphate. To maximize _k_cat, it is of special importance that Lys-93 be prevented from interacting favorably with the 6-carboxylate group of substrate OMP, and model building suggests that the —COO− group of OMP cannot replace the O− group of BMP without severe steric crowding. The interaction of Lys-93 with the O− group of BMP is relatively favorable, foreshadowing the very great affinity that the enzyme evidently develops for the carbanion generated at C-6 in the transition state.‡‡
In the present structure, the importance of Lys-93 is further suggested by the existence of a network of contacts that involve Ser-35, Lys-59, Asp-91, Asn-120, and Asp-96* of the second subunit. In controlling the position of the substrate with respect to Lys-93, and generating the strong differential binding affinity on which catalysis depends, it is necessary that the position of the side-chain of Lys-93 be tightly constrained. Aspartate residues 91 and 96* appear to serve that purpose. Equally important is the exact position of the pyrimidine ring, imposed by the very numerous H-bonding interactions of the ribosyl and phosphoryl groups. The significance of these interactions can be judged by the contribution of the phosphoribosyl group to _k_cat/_K_m for OMP, ≈5 × 108-fold for the human enzyme (3). This appears to be one of the largest substituent effects that has been reported for any enzyme reaction, for a part of the substrate that does not undergo chemical transformation. Through this use of what Jencks (22) has termed the “anchor principle,” distant parts of a substrate can serve an important purpose in the catalytic process, maximizing _k_cat by maximizing the difference in binding affinity between the substrate in the ground state and the altered substrate in the transition state.
Finally, comparison of the structures of ODCase in the absence and presence of the proposed transition state analog BMP reveals conformational transitions that accompany ligand binding. Initially open to rapid substrate access (_k_cat/_K_m = 3 × 107 s−1⋅M−1), yeast ODCase exploits enzyme–ligand contacts, particularly contacts involving the phosphoryl group, to organize active site residues that participate in catalysis. The movement of Loop 207–218 that accompanies ligand binding brings the amide nitrogen of Gln-215 to a position 2.6 Å from O-2. Similarly, the movement of Loop 151–165 toward the ligand places the peptide amide of Ser-154 within 2.9 Å of the O-4 group of BMP. In this enzyme, as in others (23, 24), catalysis appears to depend on a conformation change that permits a remarkable number of binding contacts, removing the substrate from water in which it is highly unreactive and allowing maximal interaction between the enzyme and the altered substrate in the transition state. The importance of each of the contacts revealed by the BMP-ODCase structure is being tested by mutagenesis to provide additional insight into the elegant mechanism by which this enzyme brings about such a remarkable enhancement in the rate of this difficult reaction.
Acknowledgments
We thank R. Nolte, S. Williams, J. Chrzas, and the Industrial Macromolecular Crystallography Association beamline staff at Argonne National Labs for data collection. We also thank D. Porter for critical comments and suggestions. This work was supported, in part, by National Institutes of Health Grant GM-18325 (to R.W.) and National Institutes of Health Training Grant GM-08570 (to B.G.M.).
Abbreviations
ODCase
orotidine 5′-phosphate decarboxylase
OMP
orotidine 5′-monophosphate
BMP
6-hydroxyuridine 5′-phosphate
MAD
multiwavelength anomalous dispersion
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.rcsb.org [PDB ID codes 1DQW (native yeast ODCase) and 1DQX (enzyme complexed with BMP)].
‖
ODCase is extreme in its proficiency, defined as the second order rate constant for its reaction with the substrate (_k_cat/_K_m) divided by the rate constant for the nonenzymatic reaction (_k_non). Most enzymes have second-order rate constants that approach the diffusion limit within a factor of 1,000, as is necessary if they are to be useful at reasonable intracellular concentrations. However, the rate constant for the reactions that they are called on to catalyze vary over a range of more than 14 orders of magnitude.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.030409797.
‡‡
In the highly ordered environment of an enzyme's active site, electrostatic and H-bonding interactions can compete favorably with H-bonding interactions furnished by solvent water (for a review, see ref. 21).
References
- 1.Radzicka A, Wolfenden R. Science. 1995;267:90–93. doi: 10.1126/science.7809611. [DOI] [PubMed] [Google Scholar]
- 2.Bell J B, Jones M E. J Biol Chem. 1991;266:12662–12667. [PubMed] [Google Scholar]
- 3.Miller B G, Traut T W, Wolfenden R. Bioorg Chem. 1998;26:283–288. [Google Scholar]
- 4.Levine H L, Brody R, Westheimer F H. Biochemistry. 1980;19:4990–4999. doi: 10.1021/bi00563a010. [DOI] [PubMed] [Google Scholar]
- 5.Miller B G, Smiley J A, Short S A, Wolfenden R. J Biol Chem. 1999;274:23841–23843. doi: 10.1074/jbc.274.34.23841. [DOI] [PubMed] [Google Scholar]
- 6.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 7.Brünger A T, Adams P D, Clore G M, DeLano W L, Gros R, Grosse-Kunstleve W, Jiang J-S, Kuszewski J, Nilges M, Pannu N S, et al. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 8.Cowtan K. Protein Crystallogr. 1994;31:34–38. [Google Scholar]
- 9.Collaborative Computing Project 4. Acta Crystallogr D. 1994;50:760–776. [Google Scholar]
- 10.Banner D W, Bloomer A C, Petsko G A, Phillips D C, Pogson C I, Wilson I A, Corran P H, Furth A J, Milman J D, Offord R E, et al. Nature (London) 1975;255:609–614. doi: 10.1038/255609a0. [DOI] [PubMed] [Google Scholar]
- 11.Alani E, Kleckner N. Genetics. 1987;117:5–12. doi: 10.1093/genetics/117.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cullin C, Minvielle-Sebastia L. Yeast. 1994;10:105–112. doi: 10.1002/yea.320100110. [DOI] [PubMed] [Google Scholar]
- 13.Schneider B L, Seufert W, Steiner B, Yang Q H, Futcher A B. Yeast. 1995;11:1265–1274. doi: 10.1002/yea.320111306. [DOI] [PubMed] [Google Scholar]
- 14.Yablonski M J, Pasek D A, Han B-D, Jones M E, Traut T W. J Biol Chem. 1996;271:10704–10708. doi: 10.1074/jbc.271.18.10704. [DOI] [PubMed] [Google Scholar]
- 15.Farber G F, Petsko G A. Trends Biochem Sci. 1990;15:228–234. doi: 10.1016/0968-0004(90)90035-a. [DOI] [PubMed] [Google Scholar]
- 16.Smiley J A, Paneth P, O'Leary M H, Bell J B, Jones M E. Biochemistry. 1991;30:6216–6223. doi: 10.1021/bi00239a020. [DOI] [PubMed] [Google Scholar]
- 17.Beak P, Siegel B. J Am Chem Soc. 1976;98:3601–3606. doi: 10.1021/ja00428a035. [DOI] [PubMed] [Google Scholar]
- 18.Hruska F. J Am Chem Soc. 1971;93:1795–1797. doi: 10.1021/ja00736a046. [DOI] [PubMed] [Google Scholar]
- 19.Smiley J A, Jones M E. Biochemistry. 1992;31:12162–12168. doi: 10.1021/bi00163a027. [DOI] [PubMed] [Google Scholar]
- 20.Lee J K, Houk K N. Science. 1997;276:942–945. doi: 10.1126/science.276.5314.942. [DOI] [PubMed] [Google Scholar]
- 21.Warshel A. J Biol Chem. 1998;273:27035–27038. doi: 10.1074/jbc.273.42.27035. [DOI] [PubMed] [Google Scholar]
- 22.Jencks W P. Adv Enzymol Relat Areas Mol Biol. 1975;43:219–410. doi: 10.1002/9780470122884.ch4. [DOI] [PubMed] [Google Scholar]
- 23.Wolfenden R. Bioorg Med Chem. 1999;7:647–652. doi: 10.1016/s0968-0896(98)00247-8. [DOI] [PubMed] [Google Scholar]
- 24.Wolfenden R. Mol Cell Biochem. 1974;3:207–211. doi: 10.1007/BF01686645. [DOI] [PubMed] [Google Scholar]
- 25.Carson M. Methods Enzymol. 1997;277:493–505. [PubMed] [Google Scholar]