Endogenous opioid mechanisms partially mediate P2X3/P2X2/3-related antinociception in rat models of inflammatory and chemogenic pain but not neuropathic pain (original) (raw)
Abstract
- P2X3/P2X2/3 receptors have emerged as important components of nociception. However, there is limited information regarding the neurochemical systems that are affected by antagonism of the P2X3/P2X2/3 receptor and that ultimately contribute to the ensuing antinociception. In order to determine if the endogenous opioid system is involved in this antinociception, naloxone was administered just prior to the injection of a selective P2X3/P2X2/3 receptor antagonist, A-317491, in rat models of neuropathic, chemogenic, and inflammatory pain.
- Naloxone (1–10 mg kg−1, i.p.), dose-dependently reduced the antinociceptive effects of A-317491 (1–300 _μ_mol kg−1, s.c.) in the CFA model of thermal hyperalgesia and the formalin model of chemogenic pain (2nd phase), but not in the L5–L6 spinal nerve ligation model of neuropathic allodynia. In comparison experiments, the same doses of naloxone blocked or attenuated the actions of morphine (2 or 8 mg kg−1, s.c.) in each of these behavioral models.
- Injection of a peripheral opioid antagonist, naloxone methiodide (10 mg kg−1, i.p.), did not affect A-317491-induced antinociception in the CFA and formalin assays, suggesting that the opioid component of this antinociception occurred within the CNS. Furthermore, this utilization of the central opioid system could be initiated by antagonism of spinal P2X3/P2X2/3 receptors since the antinociceptive actions of intrathecally delivered A-317491 (30 nmol) in the formalin model were reduced by both intrathecally (10–50 nmol) and systemically (10 mg kg−1, i.p.) administered naloxone.
- This utilization of the opioid system was not specific to A-317491 since suramin-, a nonselective P2X receptor antagonist, induced antinociception was also attenuated by naloxone.
- In in vitro studies, A-317491 (3–100 _μ_M) did not produce any agonist response at δ opioid receptors expressed in NG108-15 cells. A-317491 had been previously shown to be inactive at the κ and μ opioid receptors. Furthermore, naloxone, at concentrations up to 1 mM, did not compete for [3H] A-317491 binding in 1321N1 cells expressing human P2X3 receptors.
- Taken together, these results indicate that antagonism of spinal P2X3/P2X2/3 receptors results in an indirect activation of the opioid system to alleviate inflammatory hyperalgesia and chemogenic nociception.
Keywords: P2X3, ATP, neuropathic, allodynia, hyperalgesia, inflammation, formalin, opioid
Introduction
Adenosine 5′-triphosphate (ATP) is recognized as an important neurotransmitter in nociceptive transmission (Burnstock, 1996; Burnstock & Wood, 1996; Cook & McCleskey, 2002). ATP is a nonselective agonist for each of the seven ionotropic P2X purinoceptor subtypes currently identified (Ralevic & Burnstock, 1998; Bianchi et al., 1999; Jacobson et al., 2002). Additionally, several receptor subtypes in the metabotropic P2Y receptor family also respond to ATP (Abbracchio et al., 2003). While more than one of these P2 receptor subtypes are likely involved in the transmission or modulation of nociceptive signals (Collo et al., 1996; Vulchanova et al., 1996; 1997; Cockayne et al., 2000; Souslova et al., 2000; Okada et al., 2002; Yoshida et al., 2002; Tsuda et al., 2003; Wismer et al., 2003), the evidence supporting a role for P2X3/P2X2/3 receptors in nociception is perhaps the most compelling (North, 2004).
P2X3/P2X2/3 receptors are highly expressed in small diameter dorsal root ganglion neurons that relay nociceptive signals from the periphery to the spinal cord (Chen et al., 1995). Following an injury to the sciatic nerve, neuronal expression of the P2X3 receptor is elevated in both the dorsal root ganglion and the ipsilateral spinal cord (Novakovic et al., 1999). Behaviorally, P2X3 (−/−) gene-ablated mice show a significant attenuation in spontaneous pain behaviors following administration of ATP or formalin (Cockayne et al., 2000; Souslova et al., 2000), and animals treated with P2X3 anti-sense oligonucleotides exhibit reduced thermal hyperalgesia and mechanical allodynia (Barclay et al., 2002; Honore et al., 2002). Furthermore, it has been recently reported that systemic and site-specific administration of A-317491, the first non-nucleotide antagonist that has high affinity and selectivity for blocking P2X3 homomeric and P2X2/3 heteromeric channels, is antinociceptive in rat models of chronic inflammatory and neuropathic pain (Jarvis et al., 2002; McGaraughty et al., 2003; Wu et al., 2004).
Antinociception induced by administration of a P2X3/P2X2/3 receptor antagonist likely does not utilize a single pathway to affect the various forms of pathological nociception. Indeed, it has been demonstrated that A-317491 acts through a different combination of sites to reduce either neuropathic or inflammatory pain (McGaraughty et al., 2003). Furthermore, it has been suggested that ATP-induced mechanical allodynia is mediated by P2X2/3 receptors on capsaicin-insensitive neurons with slow-desensitizing ATP currents while thermal and nocifensive behaviors are mediated by P2X3 receptors on capsaicin-sensitive neurons with fast-desensitizing ATP responses (Tsuda et al., 2000). The utilization of different pathways to affect the various forms of pathological pain likely results in pathway-specific interactions with endogenous neuropharmacological systems. While it is known that both peripheral and spinal application of ATP can trigger the release of glutamate into the spinal dorsal horn (Gu & MacDermott, 1997; Tsuda et al., 1999; Wismer et al., 2003), there is very limited information regarding the neurochemical systems that are affected by antagonism of the P2X3/P2X2/3 receptor and that ultimately contribute to the ensuing antinociception. Endogenous systems other than glutamate, such as substance P, nitric oxide, GABA, and opioids may also be involved in this P2X3/P2X2/3-related antinociception (Bland-Ward & Humphrey, 1997; Fukuhara et al., 2000; Hugel & Schlichter, 2000; Ueda et al., 2000; Nakatsuka et al., 2001). Thus, even though a clear role for P2X3/P2X2/3 receptors has been demonstrated in many models of pathological nociception, the downstream mechanisms that are recruited following administration of a selective P2X3/P2X2/3 antagonist are not well understood.
In an effort to further delineate the endogenous antinociceptive mechanisms that are activated following an antagonistic block of P2X3/P2X2/3 receptors, the opioid antagonist naloxone was administered prior to A-317491 in animal models of neuropathic, chemogenic, and inflammatory pain. Although A-317491 does not have any significant direct activity at the various opioid receptors (Jarvis et al., 2002), it has been demonstrated that the antinociceptive actions of some nonopioid analgesics can be at least partially mediated by an indirect utilization of the endogenous opioid system (Gouarderes et al., 1996; Schreiber et al., 2000; Tejwani & Rattan, 2000; Vanegas & Tortorici, 2002).
Methods
Animal preparation
All animal handling and experimental protocols were approved by Abbott's Institutional Animal Care and Use Committee (IACUC). Male Sprague–Dawley rats (260–350 g; Charles River, MA, U.S.A.) were used for all experiments and were housed in a temperature-controlled room with a 12/12-h day/night cycle. Food and water were available ad libitum.
Implantation of intrathecal catheters
In some cases, animals were implanted with chronic indwelling catheters to permit the direct delivery of study compounds onto the spinal cord. Under halothane inhalation anesthesia, PE-5 catheters (external PE-10, Marsil Enterprises, CA, U.S.A.) were inserted through the cisternal membrane at the base of the skull down to the lumbar enlargement (8.5 cm). Rats were not tested for at least 7 days after surgery. Animals demonstrating motor dysfunction or dehydration at any point following surgery were immediately euthanized.
Behavioral models of pathological nociception
Complete Freund's adjuvant-induced chronic thermal hyperalgesia
Chronic inflammatory hyperalgesia was induced by the injection of complete Freund's adjuvant (CFA, 50%, 150 _μ_l) into the plantar surface of the rat's right hindpaw 48 h prior to testing. On the day of testing, animals were acclimatized for 30 min to Plexiglas holding chambers (18 × 29 × 12.5 cm3) that rested on a temperature-regulated (30°C) glass surface. Thermal nociceptive thresholds were determined according to the method described by Hargreaves et al. (1988). Briefly, a radiant heat source (8 V, 50 W projector bulb) was focused through the glass surface onto the plantar surface of the hindpaw. Upon paw withdrawal, the heat stimulus was automatically deactivated and the rat's latency to withdraw was recorded to the nearest 0.1 s. Each animal's latency score was an average of two trials, which were separated by at least 5 min. Both the injured and uninjured hindpaws were similarly tested. Withdrawal latencies after injection of vehicle into a hindpaw did not differ from latencies observed in uninjected animals (unpublished observations).
Mean withdrawal latencies were compared within groups (inflamed vs noninflamed paws) and between drug- and vehicle-injected groups. ‘Reversal in hyperalgesia' scores for each animal were calculated by the following formula:
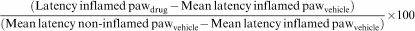
In cases of negative values, the scores were designated as 0 (no reversal in hyperalgesia).
L5–L6 ligation model of neuropathic allodynia
Under halothane inhalation anesthesia, rats received a unilateral tight ligation of the L5 and L6 spinal nerves (Kim & Chung, 1992). Experiments were conducted 1–3 weeks after surgery. Tactile allodynia was determined by measuring paw withdrawal to a series of graded von Frey hair (Stoelting, Wood Dale, IL, U.S.A.) stimulations using the up–down method described by Dixon (1980). Briefly, rats were placed on an elevated mesh bottom floor with a 1.27 × 1.27 cm2 grid to provide access to the hindpaw plantar surface. An inverted, clear plastic cage (29 × 18 × 12 cm3) was placed over each rat. Each von Frey filament was presented perpendicularly to the hindpaw ipsilateral to the injury, and held in this position for approximately 8 s with enough force to cause a slight buckle in the filament. Positive responses included sharp withdrawal or flinching behavior during stimulation. Each rat was tested in three sequential trials. Only those rats with a mean baseline threshold score of less than 4.5 × g on the ipsilateral hindpaw were used in this study. Allodynic responses were not observed in any rat following stimulation of the hindpaw contralateral to the injury (maximum force tested was 15 × g).
Formalin model of chemogenic nociception
Experimentally naïve animals were placed in individual mirrored (45°) Plexiglas cages (26 × 22 × 16 cm3) and allowed to acclimate to the testing environment for 15 min. Formalin (5% in 50 _μ_l) was then injected into the dorsal surface of the right hind paw using a 29.5 gauge needle. The number of nocifensive events (paw flinching, licking, guarding) for each animal was recorded during 1-min periods with each period being separated by 5 min. Flinching behaviors were recorded only during the second phase of the formalin assay.
Drug administration procedures
Naloxone antagonism
Three separate experiments were conducted to investigate the actions of naloxone to attenuate P2X3/P2X2/3 antagonist produced antinociception, and whether or not this effect was centrally or peripherally mediated. In one experiment, animals were pretreated with naloxone (1–10 mg kg−1, i.p.) or vehicle 5 min prior to the systemic injection of A-317491 (10–300 _μ_mol kg−1, s.c.) for each of the three behavioral assays. For comparison, the same doses of naloxone were used to antagonize the antinociceptive effects of systemically administered morphine (2–8 mg kg−1, s.c.) in the CFA, neuropathic, and formalin assays. Testing was started 30 min after administration of A-317941 or morphine. A similar administration paradigm was used to measure the effects of naloxone (10 mg kg−1, i.p.) on the nonselective P2X antagonist suramin (40 mg kg−1, s.c.) in the CFA model of thermal hyperalgesia. In another experiment, naloxone methiodide (10 mg kg−1, i.p.) or vehicle was administered 5 min ahead of A-317491 (100 _μ_mol kg−1, s.c.) in the CFA and formalin assays. Finally, naloxone was injected intrathecally (10–50 nmol, 5 min pretreatment), or systemically (10 mg kg−1, i.p., 30 min pretreatment) prior to the intrathecal injection of A-317491 (30 nmol) in the formalin assay. The volume of each intrathecal injection was 10 _μ_l followed by a 10 _μ_l sterile water flush. Testing commenced 5 min after the intrathecal injection of A-317491.
Preparation of compounds
For systemic injections, A-317491 (synthesized at Abbott Laboratories, IL, U.S.A.), morphine, naloxone, and naloxone methiodide (all from Sigma-Aldrich, MO, U.S.A.) were dissolved in saline prior to administration. A solution of dimethylsulfoxide (10%), 2-hydroxypropyl-_β_-cyclodextrin (34%), and saline (at physiological pH) was used as the vehicle for the intrathecal delivery of A-317491, naloxone, and morphine. Suramin (Sigma-Aldrich) was dissolved in water for systemic administration.
Statistics
Statistical significance on group means was measured by an ANOVA followed by a Fisher's PLSD post hoc analysis (P<0.05). Data are presented as mean±s.e.m.
In vitro assays
The pharmacological selectivity of A-317491 (10 _μ_M) had been previously evaluated by use of standardized assay protocols (Cerep, Celle l'Evescault, France) against each opioid receptor subtype and several other cell-surface receptors, ion channels, transport sites, and enzymes (Jarvis et al., 2002). Although A-317491 was inactive at the κ and μ receptors, 10 _μ_M did produce a weak effect at the δ receptor (Jarvis et al., 2002). Therefore, a more detailed analysis of A-317491 was conducted to examine its actions at the δ opioid receptor. To this end, cytosolic Ca2+ concentrations were measured as described previously (Jarvis et al., 2002), to determine the effects of A-317491 on 1 _μ_M [D-Pen(2), D-Pen(5)]-enkephalin (DPDPE)-induced activity in endogenously expressed δ receptors found on NG108-15 cells.
In order to determine if naloxone could prevent A-317491 binding to P2X3 receptors, a binding assay was performed according to Jarvis et al. (2004) with 1321N1 cells expressing human P2X3 receptors. Briefly, 3 nM of the [3H] A-317491 radioligand and 20 _μ_g membrane protein were added in each assay tube. A 10 mM stock of naloxone was prepared in dH2O, and further diluted in dH2O to obtain (10 ×) concentrations. A total of 25 ml of the (10 ×) solutions was added per assay tube. Final assay volume was 250 ml. Incubation time was 30 min at 4°C (on ice). Assay was terminated by rapid filtration over a Whatman GF/B glass fiber filter mat. For comparison, the effects of ATP on [3H] A-317491 binding were also examined. A 10 mM stock solution of ATP was prepared in dH2O, and diluted with additional dH2O to obtain (10 ×) solutions.
Results
Naloxone antagonism of A-317491-induced antinociception
In CFA animals, significant hyperalgesia (P<0.01) was observed on the inflamed but not the noninflamed hindpaw. Withdrawal latencies to thermal stimulation in vehicle-treated rats in the A-317491 experiments were 4.75±0.15 s (inflamed paw) and 9.97±0.28 s (noninflamed paw), and in the morphine experiments the latencies were 3.43±0.36 s (inflamed paw) and 11.14±0.45 s (noninflamed paw). Administration of 30 _μ_mol kg−1 (s.c.) of A-317491 was antihyperalgesic (Figure 1a), increasing (P<0.01) withdrawal latencies in the inflamed hindpaw. Pretreatment with 10 mg kg−1 of naloxone significantly (P<0.01) attenuated the A-317491-induced antinociception (Figure 1a). Naloxone (5–10 mg kg−1) also attenuated (P<0.01) the antihyperalgesic effects of morphine (2 mg kg−1, s.c., Figure 1b). Withdrawal latencies were unaffected by the injection of naloxone alone (10 mg kg−1, i.p., Figure 1a and b). Pretreatment with the high dose of naloxone (10 mg kg−1, i.p.), also reduced the dose-related efficacy of A-317491 (10–300 _μ_mol kg−1, s.c.) in the CFA assay (Figure 2).
Figure 1.

Naloxone (1–10 mg kg−1, i.p.) attenuated, in a dose-related manner, the antihyperalgesic actions of (a) A-317491 (30 _μ_mol kg−1, s.c.) and (b) morphine (2 mg kg−1, i.v.) in the CFA model of thermal hyperalgesia. Withdrawal latencies of the noninflamed hindpaws were unaffected by administration of morphine (11.04±0.78 s) or A-317491 (10.21±0.3 s) compared to the vehicle-treated groups (11.14±0.45 and 9.97±0.28 s, respectively); _n_=6–7 per group, *P<0.05, **P<0.01 vs vehicle group; ++P<0.01 vs A-317491 or morphine group, respectively.
Figure 2.

At 10 mg kg−1, i.p. naloxone shifted the A-317491 (10–300 _μ_mol kg−1, s.c.) dose–response curve to the right, thereby decreasing the potency of A-317491 to reduce CFA-induced thermal hyperalgesia on the inflamed hindpaw. Compared to the vehicle control group (10.69±0.31 s), the noninflamed hindpaw was unaffected by administration of any dose of A-317491 (range of 10.75±0.6 to 10.76±0.8 s) or by naloxone-A-317491 (range of 10.3±0.14 to 11.21±0.72 s); _n_=6 per group, **P<0.01 vs vehicle group.
In the second phase of the formalin assay, 70.05±4.06 (A-317491 experiment) and 68.0±3.87 (morphine experiment) instances of nocifensive behaviors were observed from vehicle-treated animals during the recording period. These events were not significantly affected by the administration of naloxone (10 mg kg−1, i.p.), but were reduced (P<0.01) by the injection of A-317491 (100 _μ_mol kg−1, s.c.) or morphine (2 mg kg−1, s.c., Figure 3a and b). The antinociceptive actions of A-317491 and morphine were significantly attenuated by administration of 5 mg kg−1 (P<0.05) and 10 mg kg−1 (P<0.01) of naloxone.
Figure 3.

Naloxone (1–10 mg kg−1, i.p.) reduced the antinociceptive actions of (a) A-317491 (100 _μ_mol kg−1, s.c.) and (b) morphine (2 mg kg−1, s.c.) in the formalin model of chemogenic nociception; _n_=4–8 per group, *P<0.05, **P<0.01 vs vehicle group; +P<0.05, ++P<0.01 vs A-317491 or morphine group, respectively.
In vehicle-treated neuropathic rats, the threshold for withdrawal from von Frey hair stimulation to the ipsilateral hindpaw was 2.46±0.2 × g (A-317491 experiment) and 2.43±0.47 × g (morphine experiment). Mechanical sensitivity was not affected by the injection of 10 mg kg−1of naloxone (Figure 4a). Administration of A-317491 (100 _μ_mol kg−1, s.c.) raised the withdrawal threshold (P<0.05) to 5.94±0.66 × g, but naloxone pretreatment (10 mg kg−1, i.p.) did not attenuate the antiallodynic action of A-317491 in the L5–L6 nerve ligation model of neuropathic pain (Figure 4a). In contrast, the potent action of morphine (8 mg kg−1, s.c.) was completely blocked by preadministration of 10 mg kg−1of naloxone (Figure 4b).
Figure 4.

In (a), naloxone (10 mg kg−1, i.p.) did not alter the antiallodynic action of A-317491 (100 _μ_mol kg−1, s.c.) in the L5–L6 ligation model of neuropathic pain. However, in (b) the same dose of naloxone (10 mg kg−1, i.p.) blocked the action of 8 mg kg−1 (s.c.) of morphine. Mean withdrawal threshold of the contralateral ‘uninjured' hindpaw was 13.76±0.34 g prior to administration of drugs. _n_=4–6 per group, **P<0.01 vs vehicle group; ++P<0.01 vs morphine group.
Contributions from central or peripheral opioids to A-317491-induced antinociception
Systemic administration of naloxone methiodide (10 mg kg−1, i.p.), which does not readily cross the blood–brain barrier, did not affect the antinociceptive actions of A-317491 (100 _μ_mol kg−1, s.c.) in either the CFA or formalin assays (Figure 5a). Doses equal to or lower than 10 mg kg−1 of naloxone methiodide have been shown to attenuate peripheral-opioid antinociception (Reichert et al., 2001; Furst et al., 2005). However, intrathecal (30 and 50 nmol, P<0.05 and 0.01, respectively) and systemic (10 mg kg−1, i.p., P<0.01) injections of naloxone reduced the antinociceptive effects of intrathecally delivered A-317491 (30 nmol) on formalin-induced nocifensive behaviors (Figure 5b and c).
Figure 5.

In (a), systemic administration of naloxone methiodide (10 mg kg−1, i.p.), which does not readily cross the blood–brain barrier, did not affect the antinociceptive actions of A-317491 (100 _μ_mol kg−1, s.c.) in the CFA and formalin models, _n_=5–6 per group. The effects of intrathecal A-317491 (30 nmol) were reduced by either (b) intrathecal (10–50 nmol) or (c) systemic (10 mg kg−1, i.p.) delivery of naloxone in the formalin assay; *P<0.05, **P<0.01 vs vehicle group; +P<0.05, ++P<0.01 vs respective A-317491 group.
Naloxone antagonism of suramin-induced antinociception
In the CFA assay, the nonselective P2X antagonist, suramin (40 mg kg−1, s.c.) significantly raised the withdrawal latencies of the injured paw from 5.28±0.31 s (vehicle-treated rats) to 8.05±0.63 s (Figure 6). Pretreatment with 10 mg kg−1 (i.p.) of naloxone significantly (P<0.01) attenuated the antihyperalgesic action of suramin.
Figure 6.

Naloxone (10 mg kg−1, i.p.) reduced the antinociceptive activity of suramin (40 mg kg−1, s.c.), a nonselective P2X receptor antagonist, in the CFA model of thermal hyperalgesia. Withdrawal latencies of the noninflamed hindpaws were unaffected by administration of suramin (10.2±0.42 s) compared to the vehicle-treated group (10.74±0.43 s); _n_=6 per group, **P<0.01 vs vehicle group; ++P<0.01 vs suramin group.
In vitro assays
A-317491 (10 _μ_M) was previously demonstrated to be a highly selective compound with no significant activity at the μ and κ opioid receptors, but weak activity was observed at the δ receptor (Jarvis et al., 2002). Subsequently, by measuring antagonism of DPDPE-evoked (1 _μ_M) cytosolic Ca2+ concentrations in NG108-15 cells, the IC50 of A-317491 (3–100 _μ_M) was determined to be 20 _μ_M at the δ receptor (Figure 7b). Application of A-317491 at the doses tested did not evoke any agonist activity at the δ receptor (Figure 7a). Additionally, it was determined that naloxone (up to 1 mM) did not compete for specific binding with A-317491 (3 nM) to P2X3 receptors (Figure 8). For comparison, ATP inhibited A-317491 binding with an IC50 of 22.7 nM (Figure 8).
Figure 7.

(a) Representative changes in intracellular calcium concentrations in NG108-15 cells by A-317491 and DPDPE. Following pipetting equilibration, application of 30 _μ_M A-317491 (X) alone did not produce a significant increase in intracellular calcium concentrations. Similar application of 1 _μ_M DPDPE (Y), in the absence of A-317491, resulted in a rapid increase in intracellular calcium concentrations (large peak). Application of 30 _μ_M A-317491 significantly attenuated the amplitude of the DPDPE-evoked calcium response. (b) A-317491 produced a concentration-dependent block of 1 _μ_M DPDPE-evoked increase in intracellular calcium concentrations (IC50=20 _μ_M).
Figure 8.

Representative concentration–effect curves for the ability of ATP and naloxone to compete for 3 nM [3H] A-317491 binding to 1321N1 cells expressing human P2X3 receptors. Naloxone, at concentrations up to 1 mM, did not compete for [3H] A-317491 binding. In contrast, ATP inhibited 3 nM [3H] A-317491 binding in a concentration-dependent fashion with an IC50 of 22.7 nM.
Discussion
P2X3/P2X2/3 receptors have emerged as key players in ATP-induced nociception. These receptors are expressed in relevant sensory afferents (Chen et al., 1995) and are upregulated following a neuropathic or inflammatory injury (Novakovic et al., 1999; Xu & Huang, 2002). Furthermore, gene-ablation or treatment with P2X3 antisense oligonucleotides results in antinociception (Cockayne et al., 2000; Souslova et al., 2000; Barclay et al., 2002; Honore et al., 2002). Administration of A-317491, the first potent and selective P2X3/P2X2/3 receptor antagonist, has revealed clear roles for both spinal and peripheral P2X3/P2X2/3 receptors in alleviating pathological nociception in a broad spectrum of animal pain models (Jarvis et al., 2002; McGaraughty et al., 2003). The present study demonstrates that the endogenous opioid system plays an important role in some forms of antinociception produced by injection of a P2X3/P2X2/3 receptor antagonist. Administration of naloxone, an opioid antagonist, attenuated A-317491-induced antinociception in the CFA and formalin models of pathological nociception. Thus, injection of a potent and selective P2X3/P2X2/3 receptor antagonist appears to utilize the endogenous opioid system to reduce both thermal hyperalgesia and chemogenic nociception.
This opioid-related effect of A-317491 was most likely indirect and due to an activation of ‘downstream' opioid mechanisms since A-317491 has little to no direct activity at opioid receptors. It has been previously shown that A-317491 is inactive at μ and κ opioid receptors (Jarvis et al., 2002). In the current study, at concentrations up to 100 _μ_M, A-317491 did not produce an agonist response at the δ opioid receptor. Although A-317491 was found to have weak antagonist (IC50=20 _μ_M) activity at the δ opioid receptor, it is unlikely that free plasma concentrations of A-317491 ever reached sufficient levels to affect the δ receptor. Systemic bioavailability of A-317491 is approximately 80%, but the compound is highly (>99%) protein bound (Jarvis et al., 2002). Thus, the estimated free plasma concentration following systemic injection of 100 _μ_mol kg−1 of A-317491 is approximately 300 nM, which is approximately 10-fold less than that required to block δ receptors in vitro. Furthermore, the antinociceptive effect of suramin, a structurally dissimilar nonselective P2X receptor antagonist, was also reduced by naloxone.
The indirect utilization of the endogenous opioid system to attenuate nociception has been demonstrated following administration of several pharmacological agents including nonsteroidal anti-inflammatory drugs (NSAIDS), antidepressants, _α_2-adrenoreceptor agonists, and neuropeptide FF agonists (Gouarderes et al., 1996; Schreiber et al., 2000; Tejwani & Rattan, 2000; Vanegas & Tortorici, 2002). Thus, pharmacologically diverse compounds can indirectly tap into the advantageous actions of endogenous opioids. Along with the pharmacological diversity, there is likely a large diversity in mechanisms and regions that are used to trigger the opioid activity. Although the current experiments do not explain how antagonism of the P2X3/P2X2/3 receptors indirectly initiates opioid activity to decrease nociception, the mechanism may include enhancing the release of opioids from primary afferents or second-order spinal neurons (Zadina et al., 1999; Przewlocki & Przewlocka, 2001), or limiting the effects of antiopioid transmitters like CCK (Wiesenfeld-Hallin et al., 2002). Nevertheless, whatever mechanism triggered the activation of the opioid system, it did not result in tolerance to A-317491, since the antihyperalgesic effectiveness of A-317491 does not diminish with repeated dosing (Jarvis et al., 2002).
Inflammatory hyperalgesia can be reduced by endogenous opioids acting at both central and peripheral sites (Przewlocki & Przewlocka, 2001). However, the contributions from endogenous opioids following antagonism of P2X3/P2X2/3 receptors appear to be both initiated and mediated by central sites. Administration of 10 mg kg−1 of naloxone methiodide, which acts as a peripheral opioid antagonist when given systemically, did not attenuate the antinociceptive actions of systemic A-317491 in either the CFA or formalin models of pathological nociception suggesting that the opioid contribution to the antinociception was mediated by central mechanisms. Doses lower than or equal to 10 mg kg−1 of naloxone methiodide have been demonstrated to attenuate peripherally mediated opioid antinociception (Reichert et al., 2001; Furst et al., 2005). The opioid utilization could also be triggered by activity at central P2X3/P2X2/3 receptors, since the antinociceptive actions of intrathecally delivered A-317491 were attenuated by systemic administration of naloxone. A central interaction between these mechanisms was confirmed by the effectiveness of intrathecally administered naloxone to reverse the actions of intrathecal A-317941 in the formalin assay.
The most likely site of A-317491 action to induce the opioid effects is the superficial laminae of the spinal dorsal horn (Cook et al., 1997; Vulchanova et al., 1997). However, a recent paper has suggested that when administered systemically, A-317491 is a peripherally acting P2X3/P2X2/3 antagonist (Wu et al., 2004). The authors base this assertion upon a low plasma-to-brain ratio (0.00825) for A-317491 following subcutaneous injections. Despite this very low ratio, the authors also measured 242.9±3.2 ng g−1 tissue of A-317491 (430 nM) in the brain following a 10 mg kg−1 dosing (about 16 _μ_mol kg−1). Considering that the _K_i value for A-317491 at the rat P2X3 receptor is 22±8 nM (Jarvis et al., 2002), the levels of A-317491 entering the central nervous system following systemic administration, particularly at doses higher than 16 _μ_mol kg−1, should be sufficient to produce spinal-mediated antinociception and trigger opioid utilization.
In contrast to the antihyperalgesic effects in the CFA and formalin models, the antiallodynic actions of A-317491 in the L5–L6 ligation model of neuropathic pain were not attenuated by naloxone. This suggests that antagonism of P2X3/P2X2/3 receptors can reduce at least one form of pathological nociception without triggering endogenous opioid mechanisms. Interestingly, a similar profile exists for the indirect interaction between neuropeptide FF and endogenous opioids (Altier et al., 2000). Naloxone is able to block the antinociceptive effects of neuropeptide FF in models of inflammation but not neuropathy (Altier et al., 2000). This lack of opioid utilization by neuropeptide FF and A-317491 in models of neuropathic pain may be related to the decreased efficacy of spinal opioids following nerve injury (Ossipov et al., 1995), particularly when measuring tactile allodynia (Bian et al., 1995; Lee et al., 1995). Thus, P2X3/P2X2/3-mediated antinociception is not totally reliant on contributions from endogenous opioids. It is likely that both opioid-independent and opioid-dependent mechanisms were involved in reducing CFA and formalin-related nociception, since naloxone, at least at the doses tested, did not completely block the actions of A-317491.
In conclusion, antagonism of P2X3/P2X2/3 receptors results in an indirect utilization of the opioid system to alleviate pathological nociception in animal models of inflammatory hyperalgesia and chemogenic pain, but not in a model of neuropathic allodynia. Moreover, our results suggest that this antinociception is triggered by direct antagonism of spinal P2X3/P2X2/3 receptors and is mediated by the spinal opioid system.
Abbreviations
ATP
adenosine 5′-triphosphate
CFA
complete Freund's adjuvant
References
- ABBRACCHIO M.P., BOEYNAEMS J.M., BARNARD E.A., BOYER J.L., KENNEDY C., MIRAS-PORTUGAL M.T., KING B.F., GACHET C., JACOBSON K.A., WEISMAN G.A., BURNSTOCK G. Characterization of the UDP-glucose receptor (re-named here the P2Y(14) receptor) adds diversity to the P2Y receptor family. Trends Pharmacol. Sci. 2003;24:52–55. doi: 10.1016/S0165-6147(02)00038-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ALTIER N., DRAY A., MENARD D., HENRY J.L. Neuropeptide FF attenuates allodynia in models of chronic inflammation and neuropathy following intrathecal or intracerebroventricular administration. Eur. J. Pharmacol. 2000;407:245–255. doi: 10.1016/s0014-2999(00)00668-3. [DOI] [PubMed] [Google Scholar]
- BARCLAY J., PATEL S., DORN G., WOTHERSPOON G., MOFFATT S., EUNSON L., ABDEL'AL S., NATT F., HALL J., WINTER J., BEVAN S., WISHART W., FOX A., GANJU P. Functional downregulation of P2X3 receptor subunit in rat sensory neurons reveals a significant role in chronic neuropathic and inflammatory pain. J. Neurosci. 2002;22:8139–8147. doi: 10.1523/JNEUROSCI.22-18-08139.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BIAN D., NICHOLS M.L., OSSIPOV M.H., LAI J., PORRECA F. Characterization of the antiallodynic efficacy of morphine in a model of neuropathic pain in rats. Neuroreport. 1995;6:1981–1984. doi: 10.1097/00001756-199510010-00007. [DOI] [PubMed] [Google Scholar]
- BIANCHI B.R., LYNCH K.J., TOUMA E., NIFORATOS W., BURGARD E.C., ALEXANDER K.M., PARK H.S., YU H., METZGER R., KOWALUK E., JARVIS M.F., VAN BIESEN T. Pharmacological characterization of recombinant human and rat P2X receptor subtypes. Eur. J. Pharmacol. 1999;376:127–138. doi: 10.1016/s0014-2999(99)00350-7. [DOI] [PubMed] [Google Scholar]
- BLAND-WARD P.A., HUMPHREY P.P. Acute nociception mediated by hindpaw P2X receptor activation in the rat. Br. J. Pharmacol. 1997;122:365–371. doi: 10.1038/sj.bjp.0701371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BURNSTOCK G. A unifying purinergic hypothesis for the initiation of pain. Lancet. 1996;347:1604–1605. doi: 10.1016/s0140-6736(96)91082-x. [DOI] [PubMed] [Google Scholar]
- BURNSTOCK G., WOOD J.N. Purinergic receptors: their role in nociception and primary afferent neurotransmission. Curr. Opin. Neurobiol. 1996;6:526–532. doi: 10.1016/s0959-4388(96)80060-2. [DOI] [PubMed] [Google Scholar]
- CHEN C.C., AKOPIAN A.N., SIVILOTTI L., COLQUHOUN D., BURNSTOCK G., WOOD J.N. A P2X purinoceptor expressed by a subset of sensory neurons. Nature. 1995;377:428–431. doi: 10.1038/377428a0. [DOI] [PubMed] [Google Scholar]
- COCKAYNE D.A., HAMILTON S.G., ZHU Q.M., DUNN P.M., ZHONG Y., NOVAKOVIC S., MALMBERG A.B., CAIN G., BERSON A., KASSOTAKIS L., HEDLEY L., LACHNIT W.G., BURNSTOCK G., MCMAHON S.B., FORD A.P. Urinary bladder hyporeflexia and reduced pain-related behaviour in P2X3-deficient mice. Nature. 2000;407:1011–1015. doi: 10.1038/35039519. [DOI] [PubMed] [Google Scholar]
- COLLO G., NORTH R.A., KAWASHIMA E., MERLO-PICH E., NEIDHART S., SURPRENANT A., BUELL G. Cloning of P2X5 and P2X6 receptors and the distribution and properties of an extended family of ATP-gated ion channels. J. Neurosci. 1996;16:2495–2507. doi: 10.1523/JNEUROSCI.16-08-02495.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COOK S.P., MCCLESKEY E.W. Cell damage excites nociceptors through release of cytosolic ATP. Pain. 2002;95:41–47. doi: 10.1016/s0304-3959(01)00372-4. [DOI] [PubMed] [Google Scholar]
- COOK S.P., VULCHANOVA L., HARGREAVES K.M., ELDE R., MCCLESKEY E.W. Distinct ATP receptors on pain-sensing and stretch-sensing neurons. Nature. 1997;387:505–508. doi: 10.1038/387505a0. [DOI] [PubMed] [Google Scholar]
- DIXON W.J. Efficient analysis of experimental observations. Annu. Rev. Pharmacol. Toxicol. 1980;20:441–462. doi: 10.1146/annurev.pa.20.040180.002301. [DOI] [PubMed] [Google Scholar]
- FUKUHARA N., IMAI Y., SAKAKIBARA A., MORITA K., KITAYAMA S., TANNE K., DOHI T. Regulation of the development of allodynia by intrathecally administered P2 purinoceptor agonists and antagonists in mice. Neurosci. Lett. 2000;292:25–28. doi: 10.1016/s0304-3940(00)01427-0. [DOI] [PubMed] [Google Scholar]
- FURST S., RIBA P., FRIEDMANN T., TIMAR J., AL-KHRASANI M., OBARA I., MAKUCH W., SPETEA M., SCHUTZ J., PRZEWLOCKI R., PRZEWLOCKA B., SCHMIDHAMMER H. Peripheral versus central antinociceptive actions of 6-amino acid-substituted derivatives of 14-O-methyloxymorphone in acute and inflammatory pain in the rat. J. Pharmacol. Exp. Ther. 2005;312:609–618. doi: 10.1124/jpet.104.075176. [DOI] [PubMed] [Google Scholar]
- GOUARDERES C., JHAMANDAS K., SUTAK M., ZAJAC J.M. Role of opioid receptors in the spinal antinociceptive effects of neuropeptide FF analogues. Br. J. Pharmacol. 1996;117:493–501. doi: 10.1111/j.1476-5381.1996.tb15217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GU J.G., MACDERMOTT A.B. Activation of ATP P2X receptors elicits glutamate release from sensory neuron synapses. Nature. 1997;389:749–753. doi: 10.1038/39639. [DOI] [PubMed] [Google Scholar]
- HARGREAVES K., DUBNER R., BROWN F., FLORES C., JORIS J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- HONORE P., KAGE K., MIKUSA J., WATT A.T., JOHNSTON J.F., WYATT J.R., FALTYNEK C.R., JARVIS M.F., LYNCH K. Analgesic profile of intrathecal P2X3 antisense oligonucleotide treatment in chronic inflammatory and neuropathic pain states in rats. Pain. 2002;99:11–19. doi: 10.1016/s0304-3959(02)00032-5. [DOI] [PubMed] [Google Scholar]
- HUGEL S., SCHLICHTER R. Presynaptic P2X receptors facilitate inhibitory GABAergic transmission between cultured rat spinal cord dorsal horn neurons. J. Neurosci. 2000;20:2121–2130. doi: 10.1523/JNEUROSCI.20-06-02121.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JACOBSON K.A., JARVIS M.F., WILLIAMS M. Perspective: purine and pyrimidine (P2) receptors as drug targets. J. Med. Chem. 2002;45:4057–4093. doi: 10.1021/jm020046y. [DOI] [PubMed] [Google Scholar]
- JARVIS M.F., BIANCHI B., UCHIC J.T., CARTMELL J., LEE C.H., WILLIAMS M., FALTYNEK C. [3H]A-317491, a novel high-affinity non-nucleotide antagonist that specifically labels human P2X2/3 and P2X3 receptors. J. Pharmacol. Exp. Ther. 2004;310:407–416. doi: 10.1124/jpet.103.064907. [DOI] [PubMed] [Google Scholar]
- JARVIS M.F., BURGARD E.C., MCGARAUGHTY S., HONORE P., LYNCH K., BRENNAN T.J., SUBIETA A., VAN BIESEN T., CARTMELL J., BIANCHI B., NIFORATOS W., KAGE K., YU H., MIKUSA J., WISMER C.T., ZHU C.Z., CHU K., LEE C.H., STEWART A.O., POLAKOWSKI J., COX B.F., KOWALUK E., WILLIAMS M., SULLIVAN J., FALTYNEK C. A-317491, a novel potent and selective non-nucleotide antagonist of P2X3 and P2X2/3 receptors, reduces chronic inflammatory and neuropathic pain in the rat. Proc. Natl. Acad. Sci. U.S.A. 2002;99:17179–17184. doi: 10.1073/pnas.252537299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KIM S.H., CHUNG J.M. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50:355–363. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- LEE Y.W., CHAPLAN S.R., YAKSH T.L. Systemic and supraspinal, but not spinal, opiates suppress allodynia in a rat neuropathic pain model. Neurosci. Lett. 1995;199:111–114. doi: 10.1016/0304-3940(95)12034-2. [DOI] [PubMed] [Google Scholar]
- MCGARAUGHTY S., WISMER C.T., ZHU C.Z., MIKUSA J., HONORE P., CHU K.L., LEE C.H., FALTYNEK C.R., JARVIS M.F. Effects of A-317491, a novel and selective P2X3/P2X2/3 receptor antagonist, on neuropathic, inflammatory and chemogenic nociception following intrathecal and intraplantar administration. Br. J. Pharmacol. 2003;140:1381–1388. doi: 10.1038/sj.bjp.0705574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NAKATSUKA T., MENA N., LING J., GU J.G. Depletion of substance P from rat primary sensory neurons by ATP, an implication of P2X receptor-mediated release of substance P. Neuroscience. 2001;107:293–300. doi: 10.1016/s0306-4522(01)00342-6. [DOI] [PubMed] [Google Scholar]
- NORTH R.A. P2X3 receptors and peripheral pain mechanisms. J. Physiol. 2004;554:301–308. doi: 10.1113/jphysiol.2003.048587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NOVAKOVIC S.D., KASSOTAKIS L.C., OGLESBY I.B., SMITH J.A., EGLEN R.M., FORD A.P., HUNTER J.C. Immunocytochemical localization of P2X3 purinoceptors in sensory neurons in naive rats and following neuropathic injury. Pain. 1999;80:273–282. doi: 10.1016/s0304-3959(98)00225-5. [DOI] [PubMed] [Google Scholar]
- OKADA M., NAKAGA W.A.T., MINAMI M., SATOH M. Analgesic effects of intrathecal administration of P2Y nucleotide receptor agonists UTP and UDP in normal and neuropathic pain model rats. J. Pharmacol. Exp. Ther. 2002;303:66–73. doi: 10.1124/jpet.102.036079. [DOI] [PubMed] [Google Scholar]
- OSSIPOV M.H., LOPEZ Y., NICHOLS M.L., BIAN D., PORRECA F. The loss of antinociceptive efficacy of spinal morphine in rats with nerve ligation injury is prevented by reducing spinal afferent drive. Neurosci. Lett. 1995;199:87–90. doi: 10.1016/0304-3940(95)12022-v. [DOI] [PubMed] [Google Scholar]
- PRZEWLOCKI R., PRZEWLOCKA B. Opioids in chronic pain. Eur. J. Pharmacol. 2001;429:79–91. doi: 10.1016/s0014-2999(01)01308-5. [DOI] [PubMed] [Google Scholar]
- RALEVIC V., BURNSTOCK G. Receptors for purines and pyrimidines. Pharmacol. Rev. 1998;50:413–492. [PubMed] [Google Scholar]
- REICHERT J.A., DAUGHTERS R.S., RIVARD R., SIMONE D.A. Peripheral and preemptive opioid antinociception in a mouse visceral pain model. Pain. 2001;89:221–227. doi: 10.1016/s0304-3959(00)00365-1. [DOI] [PubMed] [Google Scholar]
- SCHREIBER S., BACKER M.M., HERMAN I., SHAMIR D., BONIEL T., PICK C.G. The antinociceptive effect of trazodone in mice is mediated through both mu-opioid and serotonergic mechanisms. Behav. Brain Res. 2000;114:51–56. doi: 10.1016/s0166-4328(00)00185-6. [DOI] [PubMed] [Google Scholar]
- SOUSLOVA V., CESARE P., DING Y., AKOPIAN A.N., STANFA L., SUZUKI R., CARPENTER K., DICKENSON A.H., BOYCE S., HILL R., NEBENUIS-OOSTHUIZEN D., SMITH A.J., KIDD E.J., WOOD J.N. Warm-coding deficits and aberrant inflammatory pain in mice lacking P2X3 receptors. Nature. 2000;407:1015–1017. doi: 10.1038/35039526. [DOI] [PubMed] [Google Scholar]
- TEJWANI G.A., RATTAN A.K. Antagonism of antinociception produced by intrathecal clonidine by ketorolac in the rat: the role of the opioid system. Anesth. Analg. 2000;90:1152–1156. doi: 10.1097/00000539-200005000-00028. [DOI] [PubMed] [Google Scholar]
- TSUDA M., KOIZUMI S., KITA A., SHIGEMOTO Y., UENO S., INOUE K. Mechanical allodynia caused by intraplantar injection of P2X receptor agonist in rats: involvement of heteromeric P2X2/3 receptor signaling in capsaicin-insensitive primary afferent neurons. J. Neurosci. 2000;20:RC90. doi: 10.1523/JNEUROSCI.20-15-j0007.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TSUDA M., SHIGEMOTO-MOGAMI Y., KOIZUMI S., MIZOKOSHI A., KOHSAKA S., SALTER M.W., INOUE K. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature. 2003;424:778–783. doi: 10.1038/nature01786. [DOI] [PubMed] [Google Scholar]
- TSUDA M., UENO S., INOUE K. In vivo pathway of thermal hyperalgesia by intrathecal administration of alpha,beta-methylene ATP in mouse spinal cord: involvement of the glutamate-NMDA receptor system. Br. J. Pharmacol. 1999;127:449–456. doi: 10.1038/sj.bjp.0702582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UEDA H., MATSUNAGA S., INOUE M., YAMAMOTO Y., HAZATO T. Complete inhibition of purinoceptor agonist-induced nociception by spinorphin, but not by morphine. Peptides. 2000;21:1215–1221. doi: 10.1016/s0196-9781(00)00262-x. [DOI] [PubMed] [Google Scholar]
- VANEGAS H., TORTORICI V. Opioidergic effects of nonopioid analgesics on the central nervous system. Cell Mol. Neurobiol. 2002;22:655–661. doi: 10.1023/A:1021896622089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VULCHANOVA L., ARVIDSSON U., RIEDL M., WANG J., BUELL G., SURPRENANT A., NORTH R.A., ELDE R. Differential distribution of two ATP-gated channels (P2X receptors) determined by immunocytochemistry. Proc. Natl. Acad. Sci. U.S.A. 1996;93:8063–8067. doi: 10.1073/pnas.93.15.8063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VULCHANOVA L., RIED M.S., SHUSTER S.J., BUELL G., SURPRENANT A., NORTH R.A., ELDE R. Immunohistochemical study of the P2X2 and P2X3 receptor subunits in rat and monkey sensory neurons and their central terminals. Neuropharmacology. 1997;36:1229–1242. doi: 10.1016/s0028-3908(97)00126-3. [DOI] [PubMed] [Google Scholar]
- WIESENFELD-HALLIN Z., XU X.J., HOKFELT T. The role of spinal cholecystokinin in chronic pain states. Pharmacol. Toxicol. 2002;91:398–403. doi: 10.1034/j.1600-0773.2002.910619.x. [DOI] [PubMed] [Google Scholar]
- WISMER C.T., FALTYNEK C.R., JARVIS M.F., MCGARAUGHTY S. Distinct neurochemical mechanisms are activated following administration of different P2X receptor agonists into the hindpaw of a rat. Brain. Res. 2003;965:187–193. doi: 10.1016/s0006-8993(02)04193-8. [DOI] [PubMed] [Google Scholar]
- WU G., WHITESIDE G.T., LEE G., NOLAN S., NIOSI M., PEARSON M.S., ILYIN V.I. A-317491, a selective P2X(3)/P2X(2/3) receptor antagonist, reverses inflammatory mechanical hyperalgesia through action at peripheral receptors in rats. Eur. J. Pharmacol. 2004;504:45–53. doi: 10.1016/j.ejphar.2004.09.056. [DOI] [PubMed] [Google Scholar]
- XU G.Y., HUANG L.Y. Peripheral inflammation sensitizes P2X receptor-mediated responses in rat dorsal root ganglion neurons. J. Neurosci. 2002;22:93–102. doi: 10.1523/JNEUROSCI.22-01-00093.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YOSHIDA K., NAKAGAWA T., KANEKO S., AKAIKE A., SATOH M. Adenosine 5′-triphosphate inhibits slow depolarization induced by repetitive dorsal root stimulation via P2Y purinoceptors in substantia gelatinosa neurons of the adult rat spinal cord slices with the dorsal root attached. Neurosci. Lett. 2002;320:121–124. doi: 10.1016/s0304-3940(02)00018-6. [DOI] [PubMed] [Google Scholar]
- ZADINA J.E., MARTIN-SCHILD S., GERALL A.A., KASTIN A.J., HACKLER L., GE L.J., ZHANG X. Endomorphins: novel endogenous mu-opiate receptor agonists in regions of high mu-opiate receptor density. Ann. NY Acad. Sci. 1999;897:136–144. doi: 10.1111/j.1749-6632.1999.tb07885.x. [DOI] [PubMed] [Google Scholar]