X chromosome evidence for ancient human histories (original) (raw)
Abstract
Diverse African and non-African samples of the X-linked PDHA1 (pyruvate dehydrogenase E1 α subunit) locus revealed a fixed DNA sequence difference between the two sample groups. The age of onset of population subdivision appears to be about 200 thousand years ago. This predates the earliest modern human fossils, suggesting the transformation to modern humans occurred in a subdivided population. The base of the PDHA1 gene tree is relatively ancient, with an estimated age of 1.86 million years, a late Pliocene time associated with early species of Homo. PDHA1 revealed very low variation among non-Africans, but in other respects the data are consistent with reports from other X-linked and autosomal haplotype data sets. Like these other genes, but in conflict with microsatellite and mitochondrial data, PDHA1 does not show evidence of human population expansion.
The estimated ages of the earliest modern human fossils from South Africa and the Levant are roughly 100–130 thousand years (1, 2), and a number of African transitional forms date from 100–200 thousand years before the present (Kyr BP) (3). The same time frame also emerges from genetic data as a period of splitting between African and non-African populations (4–9). The broad, approximate coincidence of the two types of data could have arisen from a history in which the morphological transition to modern forms occurred over a wide geographic range. The extent to which this is true and the details of such a process are issues that lie at the heart of some debates on the origins of modern humans.
According to African origins models, modern humans are posited to have originated in a subpopulation of archaic hominids in sub-Saharan Africa, with the location of this subpopulation undetermined (9–11). However, transitional fossils that display near modern characteristics have been found in widely disparate regions within Africa, including South Africa, East Africa, as well as North Africa, leaving the possibility that the ancestral population covered a wide geographic area (12). In contrast, a strict multiregional view supposes that modern humans arose in multiple, different regions of the Old World (13). Between these models there lie many intermediate scenarios of the geographic spread and structure of the ancestral population at the time of the transformation to modern humans. There are three common questions to these debates (9, 14, 15). How geographically spread were ancestors of modern humans? Were they divided into subpopulations? If they were divided, how much geneflow occurred between subpopulations?
One effective approach to address these questions is to generate comparative haplotype data sets for multiple regions of the genome. Multilocus data sets permit detailed historical estimates for each genomic region studied, and the comparison among genomic regions can be used to distinguish those forces that have acted on individual genomic regions (i.e., natural selection) from evolutionary forces that have affected the entire genome (e.g., population size, populations size changes, population subdivision, and gene flow) (16).
We have generated a comparative DNA sequence data set from a diverse Old World sample for a portion of the X chromosome gene for the pyruvate dehydrogenase E1 α subunit, or PDHA1. We then compared our results to those reported for other X chromosome and autosomal haplotype studies.
METHODS
DNA sequences of a 4,200-base region of the PDHA1 gene were collected from 35 male individuals, including 6 French, 7 Chinese, 5 Vietnamese, 1 Mongolian, 4 Southern Bantu-speakers, 6 Senegalese, 3 African Pygmies, and 3 Khoisan speakers; as well as from 2 male common chimpanzees (Pan troglodytes). The sequenced region corresponds to nucleotide bases 11,158 through 15,351 of the PDHA1 published sequence (17) and includes exons 7–10 and their intervening introns. Initial PCR amplification used genomic DNA and primers corresponding to positions 10,528–10,552 and 15,579–15,602 of the published sequence. One chimpanzee sample came originally from the Southwest Foundation (San Antonio, TX) (individual number 4X0377), and the second came from Yerkes Primate Center (Atlanta, GA) (individual number C0389). The PCR fragments were gel purified and used as templates for subsequent PCR amplification to generate DNA sequencing templates. PCR products in the range of 700–900 bases were sequenced directly and simultaneously on both strands by using a LI-COR 4200 automated sequencer (Lincoln, NE). We were also able to obtain DNA sequence (1,600 bases) from the 5′ end of this region of PDHA1 from an orangutan (Pongo pygmaeus, Yerkes Primate Center, Sumatran male named Gelar). The DNA sequences have been submitted to GenBank (accession numbers AF125053–AF125089), and a file of the aligned sequences is available upon request to J.H.
DNA sequences were aligned by eye and basic population genetic analyses were conducted by using the sites computer program (18). Maximum likelihood analyses (19–21) were conducted with the genetree program distributed by R. C. Griffiths (University of Oxford, Oxford). The maximum likelihood method is based on the standard coalescent model (22) and assumes an infinite sites mutational model in an equilibrium Wright–Fisher population. Mutations are assumed to occur along branches by a Poisson process of rate θ/2, where θ is the population mutation parameter. The estimates of θ and other parameters are conditional upon the full information in the sequence data set as represented by the gene tree.
RESULTS AND DISCUSSION
Within the human sample, there are 25 polymorphic sites (Fig. 1) and 5 insertions/deletions. There were three differences between the two chimpanzee sequences and 35 fixed differences between chimpanzees and humans. A striking haplotype distribution is evident in Fig. 1, with no haplotypes shared between the African and the non-African populations and one fixed difference (site 544) between these two groups. The probability of obtaining a sample with a gene tree structure that could support a fixed difference can be calculated under a null hypothesis of panmixia. In general, this type of pattern is quite unlikely even with small samples, and in this case the probability is exceedingly remote (P = 9.4 × 10−11) (23). Thus, even though a larger sample could be expected to reveal some shared haplotypes, the presence of a fixed difference among a moderate number of DNA sequences is a strong indication of historical population subdivision. Other genetic studies, including DNA sequence studies (4, 11, 24) and many studies of allelic variation (25), have also revealed significant population structure between Africans and non-Africans, but this is the first report of a fixed DNA sequence difference.
Figure 1.

Polymorphic base positions within humans. Each of the 25 polymorphic positions is listed by its position within the alignment. The base values of the chimpanzee sequences are shown for these positions, and the human haplotypes are shown with respect to the chimpanzee sequences. - indicates the haplotype base was the same as the chimpanzee; ∗ indicates a gap in a sequence relative to the chimpanzee, where a polymorphism fell within an insertion/deletion. The polymorphism at position 2,489 is associated with an amino acid variant (ATG, for methionine, and CTG for leucine). The polymorphism at position 544 is a synonymous variant (GCA and GCG for alanine). All other variants occurred in introns. The haplotype letter designations are the same as those shown in Fig. 2. The B1 haplotype is not included in Fig. 2 because position 3,306 was removed from the genetree analyses of the complete Old World sample (see text). The populations are as follows: B, South African Bantu speakers; S, Senegalese; K, Khoisan from the Angola/Namibia border; P, Pygmy from the Central African Republic; C, China; V, Vietnam; F, France; M, Mongolia. The C, V, and F samples are random subsets of DNAs used in a larger study (45).
Because of the haplotype distribution and strong pattern of population differentiation, we considered three sets of samples: the complete Old World sample; all African samples; and all non-African samples. Table 1 shows parameter estimates for these three groupings. The African samples revealed much more variation than the non-African samples. The estimates of the population mutation rate, 3_Nu_, differ by about a factor of 10, as do the estimates of effective population size, N. Within Africa and among the non-African samples, historical gene flow is suggested by the presence of haplotypes that are shared among populations (Fig. 1). The migration rate parameter estimate (Table 1) is highest among the African populations where all pairs of populations share one or more haplotypes, and haplotype C (that haplotype most similar to the non-African haplotypes) occurs in three of the four sampled populations. All of the African samples are sub-Saharan, and the apparent lack of population structure among them is similar to what has been found in other studies (4–6, 26).
Table 1.
Estimates of population genetic parameters
| Population | n | S | θw | θml | D | N | TMRCA | Nm |
|---|---|---|---|---|---|---|---|---|
| African | 16 | 23 | 6.93 | 5.78 | 0.66 | 23,833 | 3.88 | 4.81 |
| (2.98) | (2.19) | (0.88) | ||||||
| Non-African | 19 | 2 | 0.57 | 0.62 | −0.48 | 2,561 | 2.84 | 1.96 |
| (0.45) | (0.49) | (1.46) | ||||||
| Old World | 35 | 25 | 6.07 | 4.41 | 0.78 | 18,184 | 4.75 | – |
| (2.24) | (1.34) | (1.09) |
The data revealed very little evidence of historical recombination, or of homoplasy in general. With the exception of two low frequency polymorphisms (positions 1,232 and 3,306 in Fig. 1, hereafter excluded from gene tree analyses), the data fit a single most parsimonious tree. The two sites causing homoplasy probably arose either via repeated mutation or gene conversion, and not by crossing-over. Crossing-over can generate a large amount of apparent homoplasy, but the effect is a shift of apparent gene tree histories for large blocks of linked sequence (27). Fig. 2 shows the gene tree estimate rooted with the two chimpanzee sequences. Comparison of the position of haplotypes in Fig. 2 with the geographic locations listed in Fig. 1 reveals that African samples occur on both sides of the deepest node in the tree, whereas non-African samples are restricted to just one side. Parsimoniously then, Africa is the most likely location of the ancestral sequence.
Figure 2.

The gene tree estimate for PDHA1, with estimated ages of polymorphic mutations. Each mutation is identified by its position in the sequence and by its branch location (as determined by maximum parsimony and maximum likelihood). Mutations that generated new haplotypes are indicated at branch points. For branches with multiple mutations, the order of mutations in time is arbitrary. A line is drawn under the non-African haplotypes to the left, and under the African haplotypes to the right.
To estimate the time to the most recent common ancestral PDHA1 sequence, we assumed a chimpanzee/human speciation time of 5 million years before present (Myr BP) (changes in this value have a linear effect on all subsequent time estimates). Because some of the human/chimpanzee DNA sequence divergence is caused by variation within the common ancestral species (28), we used net divergence as an estimate divergence. This measure is equal to the average pairwise divergence between chimpanzee and human sequences, less the average of the two within species measures of pairwise variation, and it can be used to estimate how much divergence has accrued since the time of common ancestry (29). The net divergence was 40.42, and the average number of mutations between sequences across the base of the human gene tree (Fig. 2) was 15.05. By equating the former with 5 Myr BP, the estimated time of the base of the human PDHA1 gene tree is 1.86 Myr BP, a late Pliocene time associated with early species of Homo (30). This time estimate is also close to one generated from the TMRCA (the estimate time to the most recent common ancestral sequence) that was obtained from the maximum likelihood simulations. From Table 1, the TMRCA is 4.75⋅N generations. Substituting the estimated value of N, and again assuming 20 years per generation, the TMRCA based estimate of the base of the tree is 1.73 Myr BP. The TMRCA method assumes a well mixed population of constant size but is in good agreement with the value based only on the molecular clock. These ancient dates are unlikely to be an artifact caused by a recent increase in the mutation rate in the human branch of the gene tree. When the partial orangutan sequence is included as a root, it was possible to parsimoniously allocate 14 of the 35 fixed differences between humans and chimpanzees to either the human or the chimpanzee branch. Only four of these fixed differences appear to have arisen on the branch leading to modern humans.
By equating the net divergence between humans and chimpanzees (40.42) with the amount of mutation that has accumulated between the two species since common ancestry, and assuming a divergence time of 5 Myr BP, an estimate of the mutation rate can be obtained:
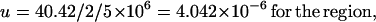
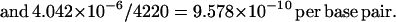
These rates are similar to reports from other loci (4, 5).
The simulations used to generate the maximum likelihood estimate of θ also yield estimates of the age of specific mutations, and these can be used to bracket the age of the origin of population subdivision between Africans and non-Africans. The mutation causing the fixed difference probably arose in the presence of some population subdivision and then became fixed in the population in which it arose. The alternative scenario, in which a polymorphism arose in a well mixed population before population subdivision, requires both fixation in one population and loss in the other. The maximum likelihood coalescent analysis returned an estimated age for the fixed difference of 0.48 N generations. By equating the time of the base of the tree (4.75 N generations) with 1.86 Myr BP, we find that the estimated time for the mutation at position 544 corresponds to 189 Kyr BP (SD = 79.6 Kyr BP). The same analysis and reasoning, applied to the most recent mutation shared by haplotypes A, B, and C, provide an estimated age of 1.04 N generations, or 410 Kyr BP (SD = 200 Kyr BP). These estimated times are early but not inconsistent with recent reports that describe population structure as having arisen greater than 100 Kyr BP, and perhaps as great as 200 Kyr BP (4–6, 8, 9). Most of this range of dates is older than the estimated ages of the first modern human fossils. If the morphological transition to modern humans occurred more recently than the origin of population structure, it necessarily follows that genes associated with the transformation to modern humans spread via gene flow between populations. This is an interesting and plausible scenario, particularly to the extent that those genes were favored by natural selection.
Any one locus may have a history dominated by natural selection or, in the absence of natural selection, by population level forces that act on all genes. To discern the action of these two categories of forces, we compared PDHA1 with other X and autosomal loci. The PDHA1 data do indicate a relatively deep gene tree; however, this was not due to a high mutation rate as our estimate of 9.7 × 10−10 per base per year is lower than the estimated rate (by the same method) for the β-globin locus (1.34 × 10−9), which does not have as deep a tree within humans (estimated depth, 800 Kyr BP) (4). The effective population size estimate for PDHA1 (18,184) is higher than reported for β-globin (11,661) but similar to that reported from a study of segregating Alu insertions (18,000) (31). The maximum likelihood estimate of 3_Nu_ (4.41, or 0.0011/bp) is very similar to per base pair estimates of from other genes (32), although it is not lower as might be expected of an X chromosome gene.
A direct test of natural selection was made by using the HKA method (33) in a comparison between PDHA1 and the β-globin locus (4). The test is based on the neutral expectation that different human loci should reveal similar levels of polymorphism, as compared with the level of divergence from chimpanzees. The results are shown in Table 2 for the three sample groupings listed in Table 1. The outcomes for both the entire Old World sample and the African sample were close to the neutral expectation. This test takes full account of the different modes of inheritance (X versus autosome) of the two genes. Their conformity suggests that the relatively ancient depth of the PDHA1 gene tree is not extreme with respect to the variance that arises under the coalescent process. Similarly, if the PDHA1 gene tree is deep by chance, then it is understandable that the estimate of effective population size (Table 1), which is tightly mathematically linked to the estimate of gene tree depth, is larger than estimates from other loci. We also assessed Tajima’s D statistic (34), which describes the departure of polymorphism frequencies from those expected under neutrality and constant population size. The value of D for the entire PDHA1 sample (0.78, Table 1) is not significantly different from zero, and it is similar to the values for other autosomal and X chromosome loci, including β-globin (1.062) (4), dystrophin (0.962) (5), elastin (0.387) (35), and lipoprotein lipase (0.909) (36). Like these other genes, PDHA1 does not appear to have an overall history of recent population expansion, which is expected to generate negative D values (34, 35). Rather the positive D values result from a relative excess of intermediate frequency polymorphisms, which occur on deep gene tree branches. For the non-African samples, D is negative (although it does not approach statistical significance), consistent with population expansion. However, in this case there are only two polymorphic sites.
Table 2.
Statistical tests of natural selection
| Locus | Measure | Total | African | NonAfrican |
|---|---|---|---|---|
| β-Globin | No. of polymorphic sites | 28 (31.2)* | 24 (27.2) | 20 (15.3) |
| DNA length | 2843 | 2689 | 3000 | |
| Divergence | 29.8 (26.6) | 25.7 (22.6) | 32.2 (37.0) | |
| DNA length | 2150 | 1992 | 2241 | |
| PDHA1 | No. of polymorphic sites | 25 (21.8) | 24 (20.9) | 2 (6.7) |
| DNA length | 4153 | 4101 | 4212 | |
| Divergence | 45.6 (48.8) | 44.1 (47.2) | 49.2 (44.5) | |
| DNA length | 4141 | 4109 | 4171 | |
| _X_2 | 0.7194 | 0.7661 | 3.965 | |
| Probability | 0.396 | 0.381 | 0.046† |
Unlike the situation among the African samples, the low level of PDHA1 variation among the non-African samples, and the fixed difference between African and non-African samples, is not consistent with reports from other genes. When β-globin and PDHA1 were compared via the HKA test for just the non-African samples, the result was significant (Table 2), suggesting that natural selection has contributed to the low level of PDHA1 variation among non-Africans. Also, it is a common observation that human populations tend not to have private alleles, let alone to exhibit a complete absence of shared haplotypes (25). For example, at β-globin, the two most frequent haplotypes were common in all sampled populations. A measure of the proportion of genetic variation that is due to population divergence, applied to African/non-African population pairs in the β-globin data, was quite low (Fst mean value of 0.096). This is slightly lower than an overall mean reported for DNA markers among human populations (0.139) (25). In contrast, Fst for PDHA1, between pooled African and non-African samples, was 0.617.
The combination of low non-African variation and zero shared haplotypes between African and non-African samples suggests that the founders that gave rise to the non-African populations were relatively few, or that following splitting there was a bottleneck in the non-African population. But given the disparity between PDHA1 and other loci, such a bottleneck was probably not manifest at the population level, but rather was imposed by natural selection limiting just the spread of PDHA1 sequences. Under this view, the site of selection is probably near but not within the sequenced region. The fixed difference gives rise to a synonymous polymorphism (GCA and GCG codons for alanine) and so is probably not under selection. PDHA1 is a subunit of the pyruvate dehydrogenase enzyme, and a number of mutations have been described that disrupt gene function and cause disease (37), but no reports have been found that inform on whether, or to what extent, there exist geographically restricted functional variants.
CONCLUSIONS
Apart from the fixed difference and the low variation among non-Africans, the PDHA1 data are consistent with other studies of X chromosomal and autosomal genes that show an abundance of highly polymorphic and thus fairly ancient variation (4, 5, 36). These DNA sequence results do not suggest an expanding human population and thus are in conflict with mitochondrial DNA patterns and recent reports on microsatellite variation (38–40). One possible resolution between these two interpretations is that recent expansion of human populations, for example within the past 50,000 years, may explain much of the pattern found within these other data sets. Yet such an expansion would not have left much of an imprint within the slower mutation process of single DNA sequences. Also, in the case of the mitochondria, natural selection has probably played a role in shaping the variation (35).
For PDHA1 overall, the simplest historical model that is consistent with the data is a mixed one, with demographic factors dominating within Africa and locus specific forces having shaped variation outside of Africa. It is important to note that the evidence of selection outside of Africa (Table 2) and the fixed difference between African and non-African samples do not appear to have had a large effect on the analyses for the entire data set. Parameter and time estimates for the African samples alone are similar to those for the data as a whole (Table 1).
Our estimate of the timing of the onset of population subdivision between African and non-African populations is about 200 Kyr BP. This estimate is similar to that reported for a haplotype analysis of β-globin (4), although it is higher than estimates in other reports, including a study of segregating Alu insertions (137 Kyr BP) (8); mitochondrial mismatch distributions (100 Kyr BP) (9); protein polymorphism (110 Kyr BP) (41); and microsatellite loci (156 Kyr BP) (7). However, all of these estimates are of effective dates because they do not provide independent assessments of gene flow. Thus any gene flow between populations that occurred after population splitting will cause estimated dates of effective splitting to be more recent than the actual dates of splitting.
Considering both the haplotype analyses that could distinguish gene flow (i.e., the analyses of β-globin and PDHA1) and the other analyses that could not distinguish gene flow, it appears that the onset of population structure occurred before the estimated dates of the earliest modern human fossils. It is likely, therefore, that the transformation from archaic H. sapiens to anatomically modern H. sapiens occurred in a geographically subdivided ancestral population. In this light, it is noteworthy that variation among present-day African populations at PDHA1 and other loci (4, 11, 24, 25) reveals little population structure. The relative lack of ancient population structure among current African populations adds further weight to the findings of population subdivision.
The possibility of population separation before the appearance of modern H. sapiens does not require independent evolution of the same modern characteristics in separate populations. So long as some gene flow occurred, genes for traits that were favored by natural selection—including those associated with the transformation to modern humans—would have spread preferentially between populations. If we consider a general model that includes population subdivision, some gene flow, and natural selection, then it is expected that those genes associated with the transformation to modern humans should reveal a relative absence of evidence of population structure.
It must be emphasized that present day genetic interpretations of historical demographic processes cannot discern the location or the proximity of historical populations. Similarly, inferences regarding historical gene flow cannot distinguish the degree to which the movement of individuals, versus the movement and splitting of populations, have generated present day patterns. In particular, the present evidence for ancient structure in human populations should not be interpreted to mean that our ancestors were necessarily very geographically widespread or very numerous. Indeed, evidence of ancient population structure notwithstanding, demographic interpretations must still be constrained by the fairly low estimates of historical effective population size, as suggested by PDHA1 and many other loci.
Acknowledgments
We thank Eldredge Birmingham, Laurent Excoffier, Trefor Jenkins, Lynne Jorde, Connie Kolman, and Jeff Rogers for providing DNA samples. Bob Griffiths kindly provided the source code, and advice, for the genetree analyses. Henry Harpending, Rosalind Harding, John Wakeley, Dan Lieberman, Ozzie Pearson, Bob Griffiths, and three anonymous reviewers provided critical comments. This work was supported by the National Institutes of Health (Grant R55GM54684).
ABBREVIATIONS
Kyr BP
thousand years before present
Myr BP
million years before present
PDHA1
pyruvate dehydrogenase E1 α subunit
TMRCA
time to most recent common ancestor
Footnotes
Data deposition: The sequences reported in this paper have been deposited in the GenBank database (accession nos. AF125053–AF125089).
A Commentary on this article begins on page 2852.
References
- 1.Bräuer G. In: The Human Revolution: Behavioral and Biological Perspectives on the Origins of Modern Humans. Mellars P, Stringer C, editors. Princeton, NJ: Princeton Univ. Press; 1989. pp. 123–154. [Google Scholar]
- 2.Valladas H, Reyss J L, Joron J L, Valladas G, Bar-Yosef O, Vandermeersch B. Nature (London) 1988;331:614–616. [Google Scholar]
- 3.Klein R G. The Human Career: Human Biological and Cultural Origins. Chicago: Univ. Chicago Press; 1989. [Google Scholar]
- 4.Harding R M, Fullerton S M, Griffiths R C, Bond J, Cox M J, Schneider J A, Moulin D S, Clegg J B. Am J Hum Genet. 1997;60:772–789. [PMC free article] [PubMed] [Google Scholar]
- 5.Zietkiewicz E, Yotova V, Jarnik M, Korab-Laskowska M, Kidd K K, Modiano D, Scozzari R, Stoneking M, Tishkoff S, Batzer M, et al. J Mol Evol. 1998;47:146–155. doi: 10.1007/pl00006371. [DOI] [PubMed] [Google Scholar]
- 6.Tishkoff S A, Dietzsch E, Speed W, Pakstis A J, Kidd J R, Cheung K, Bonné-Tamir B, Santachiara-Benerecetti A S, Moral P, Krings M, et al. Science. 1996;271:1380–1387. doi: 10.1126/science.271.5254.1380. [DOI] [PubMed] [Google Scholar]
- 7.Goldstein D B, Linares A R, Cavalli-Sforza L L, Feldman M W. Proc Natl Acad Sci USA. 1995;92:6723–6727. doi: 10.1073/pnas.92.15.6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stoneking M, Fontius J J, Clifford S L, Soodyall H, Arcot S S, Saha N, Jenkins T, Tahir M A, Deininger P L, Batzer M A. Genome Res. 1997;7:1061–1071. doi: 10.1101/gr.7.11.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harpending H C, Sherry S T, Rogers A R, Stoneking M. Curr Anthropol. 1993;14:483–496. [Google Scholar]
- 10.Cann R L, Stoneking M, Wilson A C. Nature (London) 1987;325:31–36. doi: 10.1038/325031a0. [DOI] [PubMed] [Google Scholar]
- 11.Vigilant L, Stoneking M, Harpending H, Hawkes K, Wilson A C. Science. 1991;253:1503–1507. doi: 10.1126/science.1840702. [DOI] [PubMed] [Google Scholar]
- 12.Stringer C B, Andrews P. Science. 1988;239:1263–1268. doi: 10.1126/science.3125610. [DOI] [PubMed] [Google Scholar]
- 13.Wolpoff M H. In: The Human Revolution: Behavioral and Biological Perspectives on the Origins of Modern Humans. Mellars P, Stringer C, editors. Princeton, NJ: Princeton Univ. Press; 1989. pp. 123–154. [Google Scholar]
- 14.Foley R, Lahr M M. Cambridge Archaeol J. 1997;7:3–36. [Google Scholar]
- 15.Harpending H C, Batzer M A, Gurven M, Jorde L B, Rogers A R, Sherry S T. Proc Natl Acad Sci USA. 1998;95:1961–1967. doi: 10.1073/pnas.95.4.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hey J. Trends Genet. 1998;14:303–304. doi: 10.1016/s0168-9525(98)01521-2. [DOI] [PubMed] [Google Scholar]
- 17.Koike K, Urata Y, Matsuo S, Koike M. Gene. 1990;91:307–311. doi: 10.1016/0378-1119(90)90241-i. [DOI] [PubMed] [Google Scholar]
- 18.Hey J, Wakeley J. Genetics. 1997;145:833–846. doi: 10.1093/genetics/145.3.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Griffiths R C, Tavaré S. Stochastic Models. 1998;14:273–295. [Google Scholar]
- 20.Griffiths R C, Tavaré S. Math Comput Modelling. 1996;23:141–158. [Google Scholar]
- 21.Griffiths R C, Tavaré S. Theor Pop Biol. 1994;46:131–159. [Google Scholar]
- 22.Kingman J F C. Stochastic Process Appl. 1982;13:235–248. [Google Scholar]
- 23.Hey J. Genetics. 1991;128:831–840. doi: 10.1093/genetics/128.4.831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hammer M F. Nature (London) 1995;378:376–378. doi: 10.1038/378376a0. [DOI] [PubMed] [Google Scholar]
- 25.Cavalli-Sforza L L, Menozzi P, Piazza A. The History and Geography of Human Genes. Princeton, NJ: Princeton Univ. Press; 1994. [Google Scholar]
- 26.Hammer M F, Spurdle A B, Karafet T, Bonner M R, Wood E T, Novelletto A, Malaspina P, Mitchell R J, Horai S, Jenkins T, et al. Genetics. 1997;145:787–805. doi: 10.1093/genetics/145.3.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stephens J C. Mol Biol Evol. 1985;2:539–556. doi: 10.1093/oxfordjournals.molbev.a040371. [DOI] [PubMed] [Google Scholar]
- 28.Gillespie J H, Langley C H. J Mol Evol. 1979;13:27–34. doi: 10.1007/BF01732751. [DOI] [PubMed] [Google Scholar]
- 29.Nei M. Molecular Evolutionary Genetics. New York: Columbia Univ. Press; 1987. [Google Scholar]
- 30.Wood B. Nature (London) 1992;355:783–790. doi: 10.1038/355783a0. [DOI] [PubMed] [Google Scholar]
- 31.Sherry S T, Harpending H C, Batzer M A, Stoneking M. Genetics. 1997;147:1977–1982. doi: 10.1093/genetics/147.4.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li W H, Sadler L. Genetics. 1991;129:513–523. doi: 10.1093/genetics/129.2.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hudson R R, Kreitman M, Aguadé M. Genetics. 1987;116:153–159. doi: 10.1093/genetics/116.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tajima F. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hey J. Mol Biol Evol. 1997;14:166–172. doi: 10.1093/oxfordjournals.molbev.a025749. [DOI] [PubMed] [Google Scholar]
- 36.Clark A, Weiss K M, Nickerson D A, Taylor S L, Buchanan A, Stengård J, Salomaa V, Vartiainen E, Perola M, Boerwinkle E, et al. Am J Hum Genet. 1998;63:595–612. doi: 10.1086/301977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Robinson B H, MacKay N, Chun K, Ling M. J Inherit Metab Dis. 1996;19:452–462. doi: 10.1007/BF01799106. [DOI] [PubMed] [Google Scholar]
- 38.Di Rienzo A, Donnelly P, Toomajian C, Sisk B, Hill A, Petzl-Erler M L, Haines G K, Barch D H. Genetics. 1998;148:1269–1284. doi: 10.1093/genetics/148.3.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kimmel M, Chakraborty R, King J P, Bamshad M, Watkins W S, Jorde L B. Genetics. 1998;148:1921–1930. doi: 10.1093/genetics/148.4.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jorde L B, Rogers A R, Bamshad M, Watkins W S, Krakowiak P, Sung S, Kere J, Harpending H C. Proc Natl Acad Sci USA. 1997;94:3100–3103. doi: 10.1073/pnas.94.7.3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nei M, Roychoudhury A K. Am J Hum Genet. 1974;26:421–443. [PMC free article] [PubMed] [Google Scholar]
- 42.Watterson G A. Theor Pop Biol. 1975;7:256–275. doi: 10.1016/0040-5809(75)90020-9. [DOI] [PubMed] [Google Scholar]
- 43.Hudson R R. In: Oxford Surveys in Evolutionary Biology. Harvey P H, Partridge L, editors. Vol. 7. New York: Oxford Univ. Press; 1990. pp. 1–44. [Google Scholar]
- 44.Savatier P, Trabuchet G, Faure C, Chebloune Y, Gouy M, Verdier G, Nigon V M. J Mol Biol. 1985;182:21–29. doi: 10.1016/0022-2836(85)90024-5. [DOI] [PubMed] [Google Scholar]
- 45.Jorde L B, Bamshad M H, Rogers A R. Am J Hum Genet. 1995;57:523–538. doi: 10.1002/ajmg.1320570340. [DOI] [PMC free article] [PubMed] [Google Scholar]