Treatment of Chlamydia trachomatis with a small molecule inhibitor of the Yersinia type III secretion system disrupts progression of the chlamydial developmental cycle (original) (raw)
. Author manuscript; available in PMC: 2006 Oct 18.
Summary
The obligate intracellular bacterium Chlamydia trachomatis possesses a biphasic developmental cycle that is manifested by differentiation of infectious, metabolically inert elementary bodies (EBs) to larger, metabolically active reticulate bodies (RBs). The cycle is completed by asynchronous differentiation of dividing RBs back to a population of dormant EBs that can initiate further rounds of infection upon lysis of the host cell. Chlamydiae express a type III secretion system (T3SS) which is presumably employed to establish and maintain the permissive intracellular niche by secretion of anti-host proteins. We hypothesize that T3SS activity is essential for chlamydial development and pathogenesis. However, the lack of a genetic system has confounded efforts to establish any role of the T3SS. We therefore employed the small molecule Yersinia T3SS inhibitor ′N′-(3,5-dibromo-2-hydroxybenzylidene)-4-nitrobenzohydrazide, designated compound 1 (C1), to examine the inter-dependence of the chlamydial T3SS and development. C1-treatment inhibited C. trachomatis but not T4SS-expressing Coxiella burnetii development in a dose-dependent manner. Although chlamydiae remained viable and metabolically active, they failed to divide significantly and RB to EB differentiation was inhibited. These effects occurred in the absence of host cell cytotoxicity and were reversible by washing out C1. We further demonstrate that secretion of T3S substrates is perturbed in C1-treated chlamydial cultures. We have therefore provided evidence that C1 can inhibit C. trachomatis development and T3SS activity and present a model in which progression of the C. trachomatis developmental cycle requires a fully functional T3SS.
Keywords: Chlamydia, type III secretion, Compound 1
Introduction
The genus Chlamydia contains a group of obligate intracellular parasites that exhibit diverse host range and tissue specificity. Three species, C. trachomatis, C. pneumoniae, and C. psittaci, are significant human pathogens. Despite differences in disease manifestation, intimate association with eukaryotic host cells is essential for the propagation of all Chlamydia spp. These Gram-negative bacteria develop entirely within specialized, membrane-bound, parasitophorous vacuoles termed inclusions. During development, all species display a similar bi-phasic developmental cycle alternating between morphologically and functionally distinct developmental forms (Moulder, 1991). The Chlamydia developmental cycle can also be segmented based on temporal gene expression patterns that roughly correspond to alterations in developmental forms (Belland et al., 2003; Shaw et al., 2000). The cycle is initiated when environmentally stable, yet metabolically inert, infectious particles termed elementary bodies (EBs) invade target cells and begin to differentiate into vegetative, yet non-infectious, developmental forms termed reticulate bodies (RBs). Early-cycle gene products include genes such as dnaE and rpoB whose products are involved in synthesis of macromolecules important for metabolic activity. RBs multiply within expanding inclusions but microscopic analyses suggest that they likely remain tethered to the inclusion membrane. Mid-cycle is devoid of EBs and expressed genes products include structural components (ompA) or catabolic enzymes (eno) essential for actively dividing bacteria. The developmental cycle is completed when an unidentified signal induces asynchronous differentiation of RBs back into EBs. EBs accumulate within the lumen of the inclusion until released by exocytosis or host cell rupture to initiate subsequent rounds of infection. Examples of late-cycle gene products include histone-like (HctA and HctB) and cysteine-rich proteins (OmcA and OmcB) which are required for production of EBs. For C. trachomatis serovar L2 these stages correspond roughly to 0-2 hr (early-cycle), 8-10 hr (mid-cycle) and 16+ hr (late-cycle) (Shaw et al., 2000).
Chlamydiae employ a type III secretion system (T3SS) throughout the developmental cycle (Fields et al., 2003). Although de novo expression of apparatus components occurs during mid-cycle (Fields et al., 2003; Shaw et al., 2000), EBs contain preformed channels that mediate secretion of substrates during early-cycle development. The full set of chlamydial type III substrates and their respective impact on Chlamydia pathogenesis remains largely unknown. The identity of some substrates has begun to emerge yet most have not been functionally characterized (Subtil et al., 2005). T3S substrates with presumed or deduced function include components of the extended T3SS apparatus such as Chlamydia outer proteins CopN (Fields and Hackstadt, 2000) and CopB (Fields et al., 2005), as well as host-interactive proteins such as translocated actin recruiting phosphprotein (Tarp) (Clifton et al., 2004) and Chlamydia protein associating with death domains (CADD) (Stenner-Liewen et al., 2002; Subtil et al., 2005). Current evidence indicates that members of the Chlamydia Inc protein family represent another group of T3SS substrates (Fields et al., 2003; Subtil et al., 2001). Secreted Inc proteins accumulate in inclusion membranes where they can contribute to pathogenesis directly by interacting with host proteins or more indirectly by contributing to structural integrity or maturation of the inclusion (Rockey et al., 2002). Incs are represented in all described stages of temporal gene expression (Belland et al., 2003; Shaw et al., 2000).
The T3S mechanism is a common virulence determinant in Gram negative pathogens where contributions to pathogenesis are manifested primarily via the action of specific anti-host factors secreted by respective bacteria (Hueck, 1998). Generation of type III null mutants in these pathogens has demonstrated that the T3S mechanism is essential for full virulence but has not provided a complete understanding of how these complex machines function. The availability of synthetic small molecule libraries has resulted in new tools to dissect the molecular mechanisms of T3SS activity. Kauppi et al. have identified a small molecule inhibitor of the Yersinia T3SS (Kauppi et al., 2003). ′N′-(3,5-dibromo-2-hydroxybenzylidene)-4-nitrobenzohydrazide, now designated as compound 1 (C1), was shown to inhibit secretion of Yop effector proteins (Nordfelth et al., 2005). This inhibition was not due to effects on protein expression. Instead C1 was shown to efficiently interfere with T3S activity in a dose-dependent and reversible manner.
Although chlamydiae are clearly using the T3SS to secrete host-interactive proteins, the importance of this secretion mechanism in chlamydial pathogenesis has remained untested due to the lack of a tractable genetic system for Chlamydia spp. We hypothesize that C1 could be used to circumvent this confounding aspect of Chlamydia biology. We report herein that C1 efficiently interferes with progression of the C. trachomatis developmental cycle at multiple steps. This effect is reversible and could be manifested through an effect on T3S. Taken together our data suggest an essential nature of T3S in C. trachomatis and raise the intriguing possibility that T3S activity and chlamydial development are intimately linked processes.
Results
C1 treatment inhibits C. trachomatis development
The C. trachomatis developmental cycle is manifested by the differentiation of EBs into RBs, expansion of RB numbers and completed subsequent conversion of a subset of RBs back into EBs. Calculation of infectious forming units (IFUs) represents a quantitative and reproducible means to evaluate progression through this cycle. We therefore chose to initially test for a C1-dependent effect on chlamydial development by measuring the capacity of Chlamydia to generate progeny EBs (Fig. 1). HeLa monolayers were initially infected in the absence of C1 to ensure equal invasion efficiency. Following removal of the inoculum, cultures were supplemented with medium containing either DMSO (Mock) or C1 and maintained 24 hr prior to disruption of infected cells for IFU assay on fresh HeLa monolayers. We observed a dose-dependent decrease in recoverable IFUs after treatment with C1. Inhibition ranged from a ca. 86% reduction in IFUs in the presence of 5.0 μM C1 to no detectible IFUs at the 50 μM C1 dose. This decrease was unlikely due to host cell cytotoxicity since potential toxicity assayed via measurements of released LDH revealed no difference when compared to controls (data not shown). As an additional control, Vero cells infected with the obligate intracellular bacterium C. burnetii were similarly treated with C1. Even in the presence of 50 μM C1, no significant decrease (3.3 x 104 +/− 1414.2 compared to 6.75 × 104 +/− 10606.6 for mock-treated control) in C. burnetii focus forming units (FFUs) was detected. Identical IFU patterns were detected when C. trachomatis was cultivated in Vero instead of HeLa cells and treated with C1 (data not shown). Hence the observed decrease in Chlamydia IFUs is most likely due to a direct effect on the bacteria.
Fig. 1.
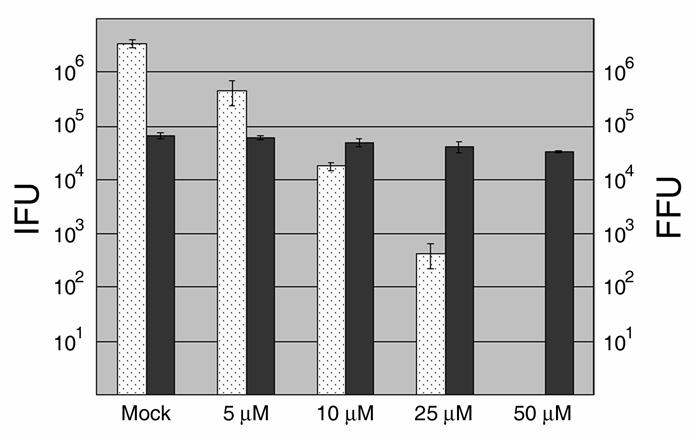
Dose-dependent inhibition of C. trachomatis but not C. burnetii development by C1. HeLa (C. trachomatis) or Vero (C. burnetii) cells were infected at an MOI of 0.5-1 with ca. 106 C. trachomatis L2 or 104 C. burnetii, respectively. Immediately after infection, DMSO was added as a control (mock) or C1 was added to 5, 10, 25, or 50 μM final. Cultures were maintained for 24 hr (Chlamydia) or 96 hrs (Coxiella) then disrupted for second passage on fresh cell culture monolayers lacking C1. Bacteria were stained with _Chlamydia_- or _Coxiella_-specific antibodies and C. trachomatis IFUs (white bars) or C. burnetii FFUs (dark grey bars) were enumerated by direct count of duplicate samples via indirect immunofluorescence microscopy. Data are represented as mean ± standard deviation.
Parallel cultures were processed and examined by indirect immunofluorescence to ascertain where the developmental cycle might be blocked (Fig. 2). Representative epi-fluorescence microscopy fields of view are shown in which chlamydiae were detected with antibodies specific for whole Chlamydia, and host cell nuclei were visualized via staining with DAPI. A dose-dependent decrease in inclusion size was detected that corresponded well with IFU data. In all cases, chlamydiae were localized to the perinuclear region of infected cells. Synchronization of cultures by infecting at 4°C prior to addition of C1 did not alter these results (data not shown). A noticeable alteration in inclusion content was not appreciable until C1 was present at 10 μM where inclusions were smaller in size compared to mock-treated controls. This effect was even more dramatic at 25 μM and 50 μM. More striking was the decrease in abundance of detectable Chlamydia observed in these treatments, indicating a lack of replication. These data therefore indicate that C1-treatment can effectively interfere with Chlamydia development and that this effect can be manifested at an early stage. Since 50 μM C1 seemed to be most effective at blocking development, we chose this concentration for subsequent experiments.
Fig. 2.
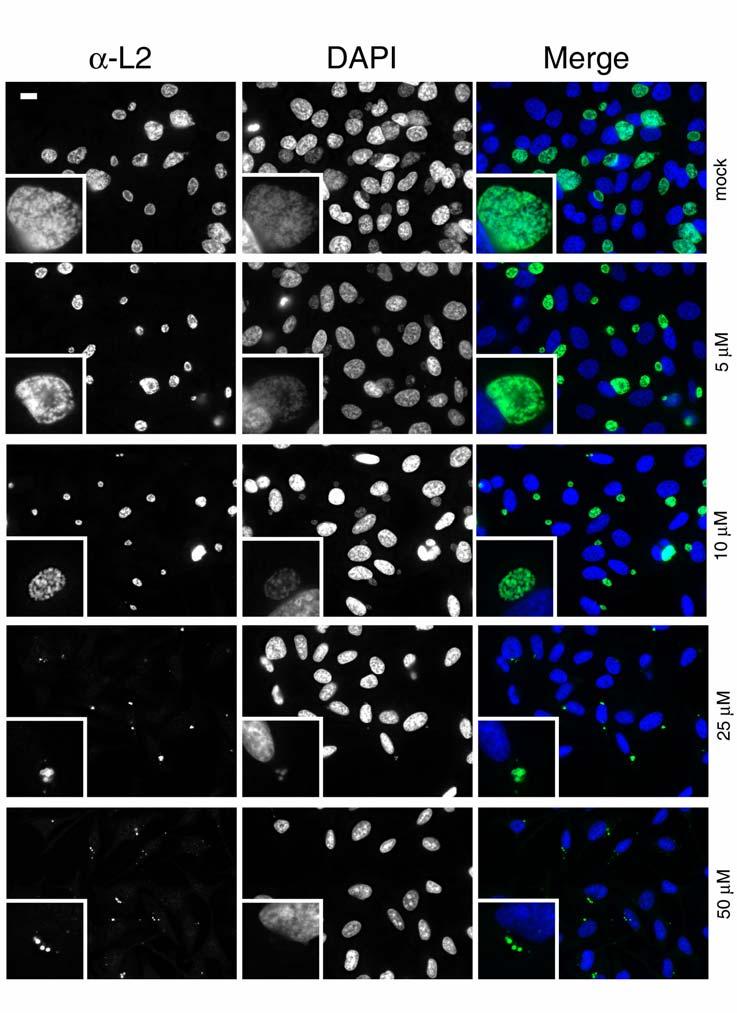
Indirect immunofluorescence analysis of C1-treated cultures. C. trachomatis L2-infected and DMSO (mock) or C1-treated cultures were examined to assess development of chlamydial inclusions. All cultures were fixed at 24 hr p.i., and chlamydiae were detected by probing with α-L2 followed by Alexa 488-conjugated secondary antibodies. Host-cell nuclei were visualized via DAPI staining of DNA. Epi-fluorescence images were acquired at 90X magnification and relative magnification of insets was maintained for each treatment. Bar = 5 μm.
C1 treated Chlamydia remain metabolically active
Our microscopy analysis did not provide a definitive indication of whether C1-inhibited chlamydiae remained viable. We therefore extended our investigation to test whether metabolic activity consistent with differentiation to RBs was apparent. We first tested whether C1-treated cultures contained transcriptionally active chlamydiae. Whole-culture RNA and DNA was harvested from mock or C1-treated cultures at 20 hr p.i. DNA was used in QC-PCR studies as previously described (Fields and Hackstadt, 2000; Shaw et al., 2000) to quantitate relative C. trachomatis genome equivalents (data not shown). RNA, normalized for genome equivalents, was added to RT-PCR reactions and mRNA for four representative genes from each temporal gene class were amplified with specific primer sets. Messages for all tested early- and mid-cycle genes were readily detected, yet dnaE, eno, and _ftsW_-specific signals were noticeably reduced in C1-treated samples (Fig. 3A). Compared to mock-treated controls, low levels of message for the late-cycle genes hctB omcA and omcB were detected in C1-treated samples. _hctA_- specific signals were below detection in C1-treated material.
Fig. 3.
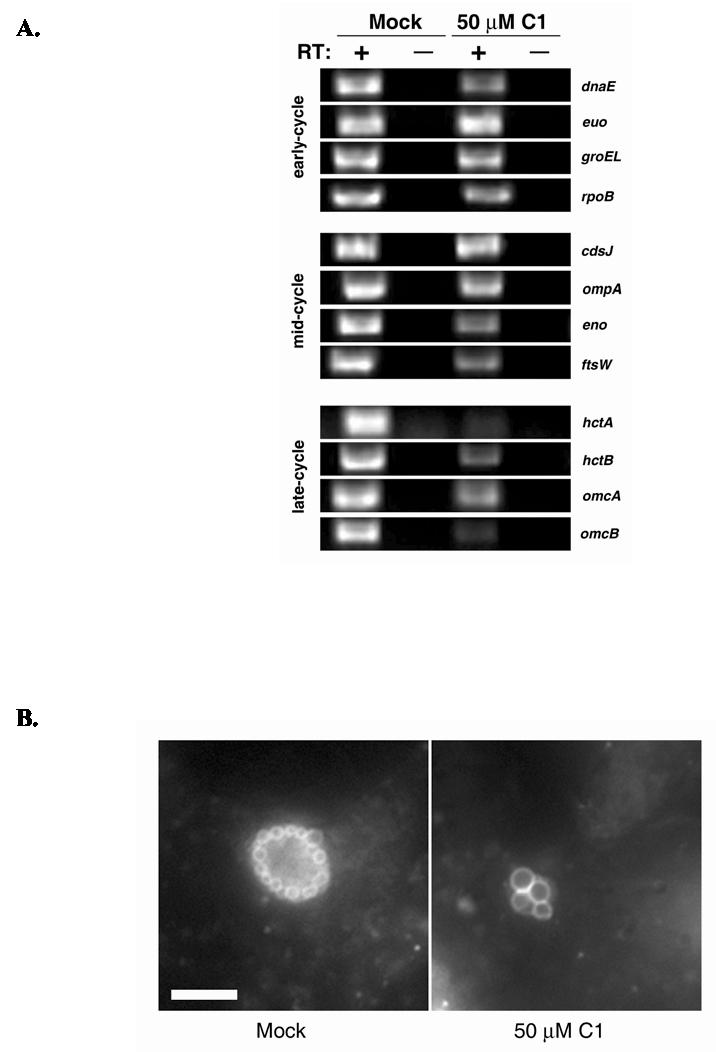
C1-treated Chlamydia are metabolically active. (A) C. trachomatis L2-infected cultures were either treated with DMSO (mock) or 50 μM C1 for 20 hr. Whole culture DNA and RNA was harvested and chlamydial genome content normalized by QC-PCR prior to RT-PCR analysis. RNA corresponding to ca. 105 chlamydial genome equivalents was added to RT-PCR reactions in the presence (+) or absence (−) of reverse transcriptase (RT) and primers specific for representative genes from early-, mid-, and late-cycle development. Amplification products were resolved in 2.0% (wt/vol) agarose gels and visualized by staining with ethidium bromide. B) C. trachomatis L2-infected HeLa cultures were labeled with C6-NBD-Ceramide and subjected to 1 hr of back-exchange. Uptake of fluorescent sphingomyelin by chlamydiae was detected in DMSO (mock) or C1-treated cultures by immunofluorescence microscopy at 90X magnification. Bar = 5 μm.
Trafficking of sphingomyelin from the host cell exocytic pathway to the inclusion requires chlamydial protein synthesis and the lipid intercalates into the membranes of RBs but not EBs (Hackstadt et al., 1995; Hackstadt et al., 1996). Hence, sphingomyelin acquisition by chlamydiae has been employed as an indicator of chlamydial metabolic activity and viability (Hackstadt et al., 1995). Staining with the fluorescent shingomyelin precursor C6-NBD-Ceramide was therefore employed to assess the viability of intracellular chlamydiae. We tested whether C1-inhibited C. trachomatis could acquire sphingomyelin by pulsing cultures at 18 hr p.i. with C6-NBD-Ceramide and examination of live cells via epi-fluorescence microscopy immediately after 1 hr of back extraction. Similar to the mock-treated control, signal was detected in C1-treated Chlamydia. The spherical staining patterns were consistent with previous studies (Hackstadt et al., 1995) in which the fluorescent lipid analog accumulated in the envelope of RBs. Consistent with data in Fig. 2, C1-treated RBs were at least twice the diameter of those found in mock-treated cultures. Taken together, the RT-PCR and sphingomyelin trafficking data demonstrate that C1-treated Chlamydia differentiate into RBs that remain viable and metabolically active.
C1-mediated growth inhibition is reversible
Nordfelth et al. (Nordfelth et al., 2005) reported that C1-mediated inhibition of Yersinia Yop secretion was reversed after removal of the compound. Since our data indicated that chlamydiae remain viable when cultivated in the presence of C1, we tested whether C1-mediated inhibition was reversible in wash-out experiments (Fig. 4). Duplicate HeLa monolayers were infected with C. trachomatis L2 and cultured in the presence of DMSO (mock) or ampicillin (amp) as controls, or increasing concentrations of C1. Amp treatment results in the reversible inhibition of chlamydial development by inducing a persistent state (Mpiga and Ravaoarinoro, 2006) and was used to gauge the amplitude of IFU recovery in wash-out treatments. At 24 hr p.i., media were removed from one replicate, cells washed, and fresh media lacking drugs added. Progeny EBs were quantitated from all cultures via IFU assay after an additional 20 hr. Compared to data in Fig. 1, IFU levels were higher overall in cultures maintained for the entire time-course in DMSO or low concentrations of C1 and reflected the increased time for development. However, the dose-dependent decrease in IFUs with increasing C1 levels was repeated. As expected, no IFUs were detected in ampicillin-treated cultures. IFUs were more abundant from wash-out cultures with the most dramatic increases occurring at 25 and 50 μM C1. Interestingly, significant numbers of IFUs were detected in 50 μM wash-out cultures but not from those cultures maintained in 50 μM C1. Levels of recovered IFUs were similar to those detected in Amp wash-out treatments. These data indicate that the inhibitory effect of C1 can be long-lived and is reversible when C1 is removed.
Fig. 4.
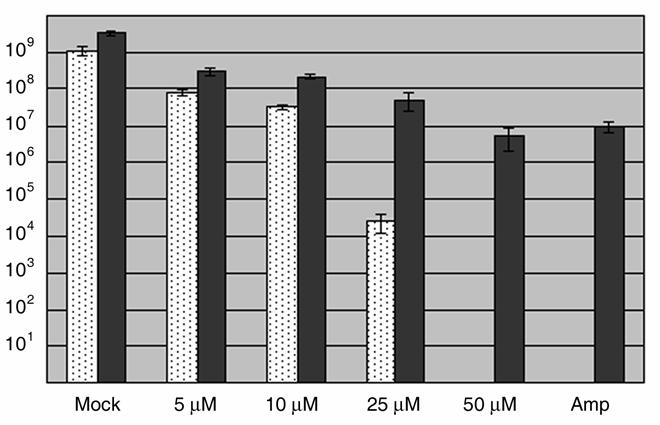
C1-mediated inhibition of Chlamydia development is reversible. Duplicate C. trachomatis L2-infected HeLa monolayers were either mock-treated with DMSO, treated with 5, 10, 25, and 50 μM C1 or with 20 μg ml−1 ampicillin (Amp) for 24 hr. C1 or Amp was then washed out of one replicate and all cultures were maintained for an additional 20 hr prior to cell lysis and re-plating for IFU quantitation on fresh HeLa monolayers. Dark grey bars correspond to cultures where C1 was washed out and white bars to cultures continuously maintained in C1. IFUs were directly enumerated in duplicate by indirect immunofluorescence microscopy. Data are represented as mean ± standard deviation.
C1 inhibits the Chlamydia T3SS
Compound 1 has been characterized as an inhibitor of the Yersinia T3SS (Kauppi et al., 2003). The molecular mechanism of C1-mediated inhibition remains unknown, yet this molecule does block secretion of Yersinia Yop effectors by interfering with the activity of the T3S apparatus (Nordfelth et al., 2005). Components of the T3SS apparatus are highly conserved among bacterial pathogens, raising the possibility that C1 could also inhibit the Chlamydia T3SS. We examined whether known C. trachomatis type III substrates accumulate within C1-treated bacteria to test this possibility. C. trachomatis L2 cultures were treated with DMSO as a control or C1 added to 15 μM immediately after infection, RBs were purified 15 hr p.i., and proteins resolved for immunoblot analysis (Fig. 5). Even though 15 μM C1 does not completely inhibit chlamydial growth, this level was chosen to increase overall abundance of proteins by allowing some development of chlamydiae. Moreover, treatment with 50 μM C1 caused lysis of RBs during the purification procedure (data not shown). Material was normalized for overall Chlamydia content via immunoblot using antibodies prepared against whole, purified C. trachomatis L2 (α-L2). Examination of individual proteins in mock and C1-treated RBs revealed that the T3S apparatus component CdsJ was detectible in equal abundance. Apparent abundance of secreted apparatus components CopN and CopB2 appeared only slightly increased in the presence of C1. In contrast, signal corresponding to the T3SS substrates CADD, IncA, and Tarp were significantly elevated in C1-treated samples. These data are consistent with a block of T3S resulting in accumulation of substrates within bacteria. To rule out the possibility that synthesis of these proteins was merely elevated in C1-treated chlamydiae we performed immunoblot analysis of whole-culture material harvested 15 hr p.i. after mock or L2 infection. L2-infected cultures were untreated or treated with 15 μM C1 (Fig. 5B). Loading was normalized to CdsJ content and levels of CADD and Tarp were analyzed by immunoblot. IncA levels were below detection in both untreated and C1-treated cultures via this assay (not shown). CADD and Tarp levels were less abundant in C1-treated compared to the C1-untreated control. Hence, increased expression is not a likely explanation for increased levels of detected protein in purified chlamydiae.
Fig. 5.

C1 treatment causes accumulation of T3SS substrates in RBs. DMSO (mock) or C1 (added to15 μM final) was added to HeLa monolayers immediately after infection at an MOI of 1 with C. trachomatis L2. (A) RBs were purified to homogeneity from disrupted cultures at 15 hr p.i. and proteins concentrated via TCA precipitation. SDS-PAGE-resolved proteins were probed in immunoblots with _Chlamydia_-specific antibodies (α-L2) generated against whole bacteria or individual proteins CdsJ, CopN, CopB2, CADD, IncA, and Tarp. (B) Whole-culture material from similarly treated cultures that were mock infected (M) or L2 infected and untreated (−C1) or C1 treated (+C1) was resolved via SDS-PAGE and probed with antibodies specific for CdsJ, CADD, Tarp. Respective proteins were visualized by probing with horseradish peroxidase-conjugated (A) or alkaline phosphatase-conjugated (B) secondary antibodies and development with appropriate chemical substrates.
We noticed that HeLa cells often contained multiple inclusions at 24 hr p.i. when cultures were maintained at lower concentrations of C1 (10 μM) that allowed some chlamydial development (data not shown). The T3SS substrate IncA (Subtil et al., 2001) is a mid-cycle gene product that has been shown to mediate homotypic fusion of C. trachomatis inclusions when multiple chlamydiae enter the same cell (Hackstadt et al., 1999; Suchland et al., 2000). We chose to assay IncA localization in C1-treated cultures as an additional test of whether C1 inhibits chlamydial T3SS activity (Fig. 6A). HeLa monolayers were infected with C. trachomatis L2 6 hr prior to addition of DMSO or C1 to 50 μM to allow inclusions to develop yet block T3SS activity prior to de novo IncA synthesis. As an additional control, a treatment containing 100 μg/ml of ampicillin was included to evaluate the ability of persistent Chlamydia to secrete IncA. At 20 hr p.i., cultures were prepared for indirect immunofluorescence analysis, and IncA (green) localization was examined by laser-scanning confocal microscopy. Antibodies specific for the cytoplasmic chlamydial protein Hsp60 (red) were also employed to ascertain position of bacteria. As expected IncA was secreted to the inclusion membrane in mock treated cultures, and IncA-specific signal was detected as the rim-like pattern of staining typical of the Inc class of proteins. Likewise, ampicillin-treated cultures revealed a rim-like IncA signal circumscribing inclusions containing one or two large, aberrant chlamydiae. Minimal signal was detected co-localizing with Hsp60-specific staining in both cases. However, IncA-specific signal co-localized with chlamydiae but was not detectible in the inclusion membrane of C1-treated cultures. Hence, the T3SS-dependent secretion of IncA was inhibited.
Fig. 6.

IncA secretion to the inclusion membrane is inhibited in C1 treated Chlamydia cultures. (A) HeLa monolayers were infected with C. trachomatis L2 at an MOI of ca. 5 and cultivated for 6 hr prior to addition of DMSO (−C1), 50 μM C1 (+C1), or 100 μg/ml ampicillin (AMP). At 20 hr p.i., cultures were paraformaldehyde fixed, permeablized and probed with Hsp60- and IncA-specific antibodies. Samples were stained with either Alexa 594-conjugated (Hsp60) or Alexa 488-conjugated (IncA) secondary antibodies and visualized via laser-scanning confocal microscopy. Bars = 5 μm. (B) Whole-culture lysates were prepared from similarly treated cultures infected at an MOI of 5, proteins resolved via SDS-PAGE, and probed in immunoblots with α-RFX5, α-Caspase-1, or α-Actin as a loading control. Respective proteins were visualized by probing with horseradish peroxidase-conjugated secondary antibodies and development with chemiluminescent reagent.
We generated whole-culture lysates from similarly treated cultures to further test the specificity of C1 activity on the C. trachomatis T3SS. Levels of host Caspase-1 and transcription factor RFX-5 were then assessed by immunoblot (Fig. 6B). The chlamydial protease CPAF gains access to the host cytoplasm by a T3SS-independent mechanism where it degrades host proteins including the transcription factor RFX5 (Zhong et al., 2001). In contrast, C. trachomatis induces the cleavage of pro-caspase-1 to the p20 active form (Lu et al., 2000), and similar activation of Caspase-1 is T3SS-dependent in other pathogens such as Salmonella and Shigella spp. (Hueck, 1998). Levels of detected RFX-5 were similarly decreased in infected cultures regardless of the presence of C1. Accumulation of the p20 fragment of Caspase-1 was detected only in the mock-treated, L2-infected cultures. Hence, C1 specifically interfered with only the T3SS-related process.
C1 inhibits RB to EB differentiation
The presence of EBs was not visually apparent in cultures treated later (6 hr p.i.) with C1 for analysis of IncA localization. Since C1 interfered with C. trachomatis development when added immediately after infection, we wondered whether it could also interfere with late events, namely differentiation of RBs back into EBs. Significant conversion of C. trachomatis L2 RBs to EBs occurs at ca. 18 hr p.i. (Moulder, 1991). Therefore C. trachomatis L2-infected HeLa cultures were maintained until 15 hr p.i. prior to treatment with DMSO or 50 μM C1. Since some EBs may form prior to this time-point, additional cultures were treated similarly with inhibitory concentrations of chloramphenicol (Cm) as a control. C1 treatment resulted in a greater than 3 log10 reduction in recovery of progeny EBs compared to the mock-treated control (Fig. 7). This was ca. 1 log10 higher (5338.2 +/− 718.9 compared to 466 +/− 179 IFUs) than levels recovered from Cm-treated cultures. Hence C1-mediated inhibition of type III secretion is also able to interfere with conversion of RBs back into infectious EB forms.
Fig. 7.
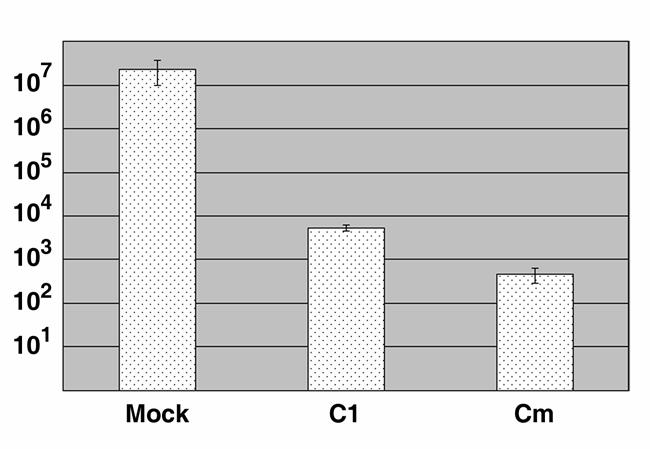
Addition of C1 during mid-cycle development inhibits differentiation of RBs into EBs. C. trachomatis L2-infected HeLa monolayers were cultivated for 15 hr prior to addition of DMSO (mock), C1 to 50 μM, or chloramphenicol (Cm) to 200 μg ml−1. Cultures were disrupted 24 hr later and EB levels determined by IFU assay performed on fresh HeLa monolayers. IFUs were directly enumerated in duplicate by indirect immunofluorescence microscopy, and data are represented as mean ± standard deviation.
Discussion
T3SSs are present in a diverse group of Gram-negative pathogens including Chlamydia spp. where they mediate specific delivery of anti-host effector proteins into target eukaryotic cells. In bacterial systems where genetic manipulation is possible, it is apparent that these specialized secretion systems are essential for full virulence of respective pathogens (Hueck, 1998). Like other Chlamydia species, the minimal genome of C. trachomatis contains comparatively few predicted coding sequences (ca. 894 predicted proteins) (Stephens et al., 1998). By analogy with other T3SSs and based on the observation that Chlamydia dedicates significant coding capacity to type III-related products (Fields and Hackstadt, 2002), it is intuitive that the T3SS would also be essential for chlamydial pathogenesis. In the absence of a tractable genetic system this question has not been formally addressed. However the use of T3SS-specific small molecule inhibitors represents a unique and powerful mechanism to address contributions of T3S to the biology of Chlamydia. We have employed the T3SS inhibitor designated compound 1 originally described as an inhibitor of Yersinia T3SS activity (Kauppi et al., 2003; Nordfelth et al., 2005). Although the mechanism of C1 activity has not been elucidated, C1 effects are dominant over regulatory mutants and do not alter substrate expression levels, strongly suggesting that the basal secretion apparatus is directly targeted by C1. C1 is membrane permeable and would therefore be predicted to gain unimpeded access to intra-inclusion chlamydiae. Indeed, our working concentration of 50 μM C1 is similar to the 40 μM employed in Yersinia studies (Nordfelth et al., 2005). The overall type III secretion mechanism is well conserved (Hueck, 1998) and deduced Chlamydia T3SS apparatus components are similar in primary sequence to those found in Yersinia spp. The C. trachomatis serovar D core apparatus component CdsV, for example, displays ca. 72% overall sequence similarity to YscV of Yersinia (Hsia et al., 1997). Although predicted chlamydial T3SS accessory proteins are comparatively less similar in primary sequence to their Yersinia counterparts, essential regions of proteins tend to be maintained. CopN for example is only 50% similar to YopN, yet residues found to be essential for YopN function in Yersinia (Ferracci et al., 2005)are absolutely conserved in CopN, reinforcing the concept of functional conservation. Perhaps the most convincing indication of functional conservation between the Yersinia and Chlamydia T3SS is the observation that chlamydial proteins can be recognized and secreted by the heterologous Yersinia T3SS (Fields and Hackstadt, 2000; Fields et al., 2003; Fields et al., 2005). This degree of conservation increases the probability that C1 has a similar effect on Chlamydia compared to Yersinia.
Treatment with the T3SS inhibitor C1 had a significant and reproducible detrimental effect on Chlamydia growth and development. We detected a nearly nine log10 reduction of Chlamydia IFUs after maintaining cultures with 50 μM C1 for 44 hr (Fig. 4). Importantly, no effects on recoverable FFUs were detected in C1 treated C. burnetii cultures. C. burnetii is also an obligate intracellular pathogen which resides within a membrane-bound vacuole (Hackstadt, 1998). C. burnetii deploys anti-host proteins via expression of a type IV secretion system instead of a T3SS (Seshadri et al., 2003). Hence the efficacy of C1 is probably not derived from specific toxicity on host cells but may well be confined to investigation of T3SSs. Direct examination of cultures by indirect immunofluorescence microscopy 24 hr p.i. indicated that lack of recoverable IFUs did not reflect a deficiency in accumulated RBs to differentiate into EBs but was instead due to an arrest in initial growth. Developmental forms detected after early addition of C1 probably corresponded to RBs since our RT-PCR and sphingomyelin trafficking data are consistent with the existence of metabolically active chlamydiae. Moreover, if EBs were still present they would be detected in subsequent IFU studies. As seen for in vitro grown Yersinia (Nordfelth et al., 2005), C1 effects on Chlamydia were reversible (Fig. 4). It is not surprising that inhibited Chlamydia would remain viable within and recoverable from host cells. _Chlamydia_-dependent segregation of inclusions from the host endocytic pathway occurs rapidly. Previous studies have shown that inclusions containing chloramphenicol-inhibited Chlamydia fuse with lysosomes slowly (Scidmore et al., 2003), and when reversibly inhibited viable chlamydiae can be recovered after greater than 20 hr of chloramphenicol treatment (Scidmore et al., 1996).
As seen for Yersinia, our data indicate that C1 inhibits the Chlamydia T3SS. However, in the absence of a genetic system, we remain unable to exclude the possibility that C1 may affect chlamydial processes unrelated to type III secretion. Although our data indicate that spurious toxicity of C1 on chlamydiae is unlikely, we cannot completely rule out this possibility. Hence, the link between C1-mediated inhibition of development and type III secretion remains correlative. It is never the less tempting to speculate that type III secretion is essential for progression of the C. trachomatis developmental cycle. We envision at least two possibilities for the manifested effect of type III secretion on C. trachomatis development. Although not mutually exclusive, the T3SS could influence cycle progression indirectly through protein secretion or directly by sensing environmental alterations and transducing signals leading to cycle progression. First T3SS substrates may be required to alter the intracellular compartment such that it is hospitable for chlamydial growth. In the absence of these secreted proteins chlamydiae would fail to grow. In this sense, C1-inhibited Chlamydia are at least superficially similar to persistent states induced by addition of β-lactam antibiotics or IFNγ (Hogan et al., 2004) in that they remain viable and metabolically active but do not complete development. However, C1-induced effects are clearly distinct from defined persistence models. Although C1-treated chlamydiae were enlarged compared to mock controls, they did not display the aberrant chlamydial morphology seen in ampicillin treated cultures (Fig. 6A). Secondly, our RT-PCR analysis of selected gene expression indicated an expression pattern that is not consistent with known forms of persistence. The gene for Euo, for example, is typically up-regulated in all persistence models (Hogan et al., 2004). Finally, ampicillin treatment also did not inhibit the ability of IncA to reach the inclusion membrane. We therefore propose that the effect of C1 on chlamydial development and type III secretion is probably not via induction of characterized persistent states of growth.
We were also able to inhibit conversion of established RBs to EBs by late addition of C1 (Fig. 7). Based on transcriptome analyses (Belland et al., 2003), all necessary gene products would presumably have been synthesized and available for secretion by this time. Indeed, we were able to detect EBs in C1- and Cm-treated cultures (Fig. 7), indicating that some conversion had already begun. Hence, lack of secreted anti-host or structural proteins probably does not completely explain our observed effects of C1 on chlamydial development.
A second, and more intriguing, possibility is that the signals governing development may originate or be transduced through the T3S apparatus. The bi-phasic developmental cycle common to all Chlamydia spp. is rapidly becoming more thoroughly described. For example, temporal gene expression patterns have been elucidated that correspond with developmental phase-specific activities (Belland et al., 2003; Shaw et al., 2000). Multiple sigma factors are poised throughout development to receive signals that would presumably lead to expression of distinct gene subsets required for cycle progression (Belland et al., 2003; Douglas and Hatch, 2000). It is also becoming clear that complex regulation of the histone-like protein HctA is required to orchestrate critical events in the differentiation of developmental forms (Grieshaber et al., 2004; Grieshaber et al., 2006). Yet there is only one apparent two-component regulatory system (Koo and Stephens, 2003) available to sense environmental cues to control all of these events. Precedent in other T3SS-expressing pathogens indicate these complex apparatus are capable of i) sensing a variety of environmental cues and ii) inducing alterations in gene expression through feed-back regulatory mechanisms (Hueck, 1998). Interestingly, Chlamydia possess core components of a seemingly functional partner switching mechanism containing a switch protein kinase and antagonist protein (Douglas and Hatch, 2000; Hua et al., 2006) homologous to that characterized in regulation of Bacillus subtilis spore formation (Hilbert and Piggot, 2004). Interestingly, a similar system is employed to regulate the Bordetella spp. T3SS (Kozak et al., 2005; Mattoo et al., 2004). As suggested by Hua et al. (Hua et al., 2006), type III activity and partner switching may be linked to govern aspects of Chlamydia development.
The developmental cycle has long been thought to be dependent on or involve an intimate association with eukaryotic-derived membranes. Indeed, there is significant circumstantial evidence to support this hypothesis. First, immediately subsequent to invasion, EBs are contained within tightly-associated, membrane-bound vacuoles. Second, microscopic data indicate that although mature C. trachomatis inclusions posses spacious, fluid-filled lumens, RBs remain intimately associated with the inclusion membrane during vegetative growth. Third, RBs differentiation to EBs seems to occur within the lumen of the inclusion and after apparent detachment from the inclusion membrane (Moulder, 1991). The mechanisms of described T3SSs in other Gram negative pathogens involve intimate association with host cell membranes and have been described as “contact dependent”. In all probability this is also true for Chlamydia systems. In support of this notion, injectisome filaments have been visualized protruding through inclusion membranes (Matsumoto, 1981; Nichols et al., 1985). The membrane-dependent connection between T3SS and chlamydial development reinforces the possibility that these two complex processes may be linked. Complex regulatory cascades have been described in other bacteria that govern the initiation of T3SS activity (Hueck, 1998). As in these systems, it is unclear how Chlamydia type III activity would be turned off during normal conversion of RBs to EBs. Clearly, this is an area requiring further investigation.
The literature is rich with examples of _Chlamydia_-mediated effects on host cells (Fields and Hackstadt, 2002), yet the mechanisms of most are not known. Clearly not all processes are type III dependent. The host-interactive chlamydial protease CPAF contains an apparent Sec-dependent secretion signal and has been predicted to gain access to the host cytoplasm by a type III-independent mechanism (Heuer et al., 2003; Subtil et al., 2005). Our data in which C1 treatment does not prevent RFX-5 degradation are consistent with this assertion and indicate that C1-treatment does not interfere with type II secretion. Activation of Caspase-1 by Salmonella and Shigella spp. is T3SS-dependent and two T3S substrates SipB and IpaB, respectively, have been found to interact with pro-caspase-1 (Hueck, 1998). C. trachomatis infection also induces the cleavage of pro-caspase-1 and chlamydiae express two possible homologs of SipB and IpaB. Both C. trachomatis CopB and CopB2 are secreted by a T3SS mechanism and we have speculated that either could mediate _Chlamydia_-induced activation of Caspase-1 (Fields et al., 2005). Our observation that C1 interferes with cleavage of pro-caspase-1 while permitting degradation of RFX5 is consistent with a specific effect of C1 on the C. trachomatis T3SS.
We anticipate that C1 will be efficacious in discerning which _Chlamydia_-mediated effects on host cells may be type III-dependent as an initial step in experimental designs for the characterization of molecular mechanisms. For example, our IncA-related data are in agreement with the notion that IncA-mediated homotypic fusion of inclusions is a type III-mediated process (Subtil et al., 2001). In contrast, our observation that chlamydiae are able to accumulate labeled sphingomyelin not only indicated that the bacteria were viable but that interruption of the host exocytic pathway is either type III-independent or the necessary _C. trachomatis_-derived components are in place prior to C1 exerting its effect (see below). It may therefore not be productive to screen type III substrates for this activity.
One factor confounding the efficacy of C1 in this approach was that we were unable to inhibit invasion of HeLa cells or early tyrosine phosphorylation of the T3SS substrate Tarp by pretreatment of C. trachomatis EBs with even higher levels of C1 (data not shown). T3S substrates (Tarp in particular) are most likely involved in invasion since the reported cytochalasin D sensitivity of chlamydial entry correlates with Tarp activity. Since the mechanism by which C1 inhibits T3SS has not been elucidated, it is difficult to predict why C1 was not effective in inhibiting Tarp secretion at these early times. The accumulation of Tarp in purified C1-treated RBs (Fig. 5) indicates that Tarp secretion can be impacted at later times. We speculate that the inability of C1 to inhibit Tarp phosphorylation may reflect an inability to completely neutralize T3SS activity fast enough. Tarp is translocated into host cells within minutes of attachment of EBs with the plasma membrane where it mediates rearrangement of the host actin cytoskeleton (Clifton et al., 2004). Alternatively C1 activity could require metabolic activity which is lacking in EBs or if C1 activity depended on the inhibition of de novo assembly of the T3S apparatus then the preformed secretion channels found in EBs would not be susceptible to C1. A more interesting possibility is that Chlamydia spp. possess functionally distinct T3SSs. In addition to homologs of the Yersinia T3SS, Chlamydial genomes possess homologs of flagellar apparatus components (Stephens et al., 1998). Since chlamydiae are not motile it is logical to predict that these components are part of the T3SS. However, these components are not expressed at the same time that cds genes are (Belland et al., 2003). It is therefore possible that Chlamydia express functionally distinct T3SSs by alteration in the content of apparatus components. Only one of these types of secretion channels may be susceptible to C1 inhibition. Clearly, more experiments are required to address this intriguing possibility. In all of our experiments, Chlamydia were able to traffic to the peri-nuclear region of infected cells. This process is rapid (Clausen et al., 1997; Grieshaber et al., 2003), dependent on de novo gene expression, and not reversible once established (Scidmore et al., 1996). Given the inability of C1 to inhibit early events, it is therefore, unclear whether the intracellular trafficking or other early processes might be type III-dependent. Other experimental approaches will be required to address this issue.
In aggregate, our data support a working model in which the T3SS is essential for Chlamydia pathogenesis on multiple levels. Secretion of effector proteins would modulate host-cell processes necessary to establish and maintain Chlamydia's specialized environmental niche while the apparatus itself continually gauges environmental conditions and conveys that information to developmental cycle machinery. The identity of those environmental signals remains elusive but probably involves contact with eukaryotic membranes.
Experimental procedures
Cell culture and organisms
C. trachomatis serovar L2 (LGV-434; American Type Culture Collection [ATCC]) was cultivated in HeLa 229 epithelial cells (CCL 2.1; ATCC) and C. burnetii (Nine Mile strain in phase II, kindly provided by R.A. Heinzen; Rocky Mountain Laboratories) in Vero 76 cells (African green monkey kidney epithelial cells; CCL-81; ATCC). Cultures were maintained at 37°C in the presence of 5% CO2/95% humidified air in RPMI-1640 (C. trachomatis) and DMEM (C. burnetii) supplemented with 10% (vol/vol) fetal bovine serum. C. trachomatis cultures were also maintained with 10 μg ml−1 gentamicin (Invitrogen, Carlsbad, CA). Where appropriate, media were supplemented with ′N′-(3,5-dibromo-2-hydroxybenzylidene)-4-nitrobenzohydrazide (C1; ChemBridge Corp., San Diego, CA) solublized in dimethyl sulfoxide (DMSO, Sigma, St. Louis, MO), 20 μg ml−1 for ampicillin (Sigma), or 200 μg ml−1 for chloramphenicol (Sigma).
Infection procedures
Infections with C. trachomatis serovar L2 were initiated with MD-76 (Mallinckrodt, Inc., St. Louis, MO) density gradient-purified EBs in Hank's balanced salt solutions (HBSS) (Invitrogen) at 37°C for 1 hr as previously described (Caldwell et al., 1981). Inocula were replaced with fresh medium and unless otherwise noted, DMSO or C1 was added immediately. At 24 post infection (p.i.) the infected cells were lysed and re-plated onto fresh HeLa monolayers for detection of progeny EBs by inclusion forming units (IFUs) as described (Furness et al., 1960). For experiments requiring the purification of RBs, cultures were harvested at 15 hr p.i. and chlamydiae were purified by density-gradient centrifugation as described (Caldwell et al., 1981).
In focus forming unit (FFU) experiments with Coxiella burnetii, Vero cells were cultivated in 24-well tissue culture plates and infected with C. burnetii in K-36 buffer (0.1 M KCl, 0.015 M NaCl, 0.05M potassium phosphate, pH 7.0) at MOI of 0.5 at 37°C for 1 hour. After infection the inoculum was removed and infected cells were either mock (DMSO) treated or treated with increasing doses of C1 in DMEM medium supplemented with 10% FBS. 96 hours post infection the mock as well as the C1 treated cells were harvested, lysed, and re-plated onto fresh monolayer of Vero cells for FFUs counts as previously described (Coleman et al., 2004). After 4 days of incubation the infected cells were fixed with methanol and the FFUs were stained with polyclonal rabbit antiserum recognizing Nine Mile/phase II C. burnetii (Heinzen et al., 1996) followed by Alexa-488-conjugated goat anti-rabbit secondary antibody (Invitrogen).
Analysis of gene expression
C. trachomatis genome-equivalents were enumerated in material harvested from infected HeLa-cell monolayers by quantitative-competitive polymerase chain reaction (QC-PCR) essentially as described (Fields and Hackstadt, 2000; Shaw et al., 2000). Briefly, HeLa monolayers were infected at a multiplicity of infection (MOI) of 5 in HBSS at 37°C for 60 min. Inocula were replaced with media containing 50 μM C1 or DMSO and cultivated at 37°C + 5% CO2. Whole-culture DNA and RNA were sequentially harvested using Trizol® Reagent (Invitrogen) according to the manufacturer's protocol 20 hr p.i. QC-PCR was performed by amplifying groEL as described (Shaw et al., 2000) from isolated culture DNA and exogenously added competitor. Genomes were quantitated by comparing integrated density values of agarose-gel-resolved PCR products as done previously (Shaw et al., 2000).
Gene expression was evaluated by reverse transcription PCR (RT-PCR) of whole-culture RNA using the Access RT-PCR kit (Promega, Madison, WI) according to the manufacturer's instructions. Specific messages were amplified from RNA, normalized corresponding to genome content (ca. 105 chlamydial genomes) of original samples, using custom primers reported by Shaw et al. (Shaw et al., 2000). Reverse transcription was carried out for 45 min at 48°C followed by 2 min at 94°C and the resulting cDNA was amplified for 40 cycles consisting of 30 sec at 94°C, 30 sec at 55°C, and 30 sec 72°C each. RT-PCR products were resolved by electrophoresis in 2.0 % (wt/vol) agarose gels and DNA visualized by staining with ethidium bromide.
Immunodetection
For immunoblot analysis, whole-culture proteins were concentrated by addition of trichloroacetic acid (Sigma) to 10% (wt/vol) and pellets were subsequently solublized in equal volumes of electrophoresis sample buffer (2.3% [wt/vol] sodium dodecyl sulfate [SDS], 5% [vol/vol] β-mercaptoethanol, 25% [vol/vol] glycerol, and 60 mM Tris pH 6.8). Proteins were resolved in polyacrylamide gels (12% [vol/vol] polyacrylamide) by SDS-PAGE (Laemmli, 1970) and transferred to Immobilon-P (Millipore, Corp., Bedford, MA) in carbonate buffer (10 mM NaHCO3, 3 mM Na2CO3, 10% methanol, pH 9.9). Whole-cell Chlamydia proteins were detected by probing with α-L2 (Heinzen et al., 1996), and specific proteins detected with α-RFX-5 (Abcam, Cambridge, MA), α-Caspase-1 (gift from D. Miller, Merck Laboratories, Rahway, HJ), α-β-Actin (Sigma), α-Tarp (Clifton et al., 2004), α-CopN (Fields and Hackstadt, 2000), α-CdsJ (Fields et al., 2003) α-CADD, α-CopB2 (Fields et al., 2005), or α-IncA (Scidmore-Carlson et al., 1999) followed by horseradish peroxidase-conjugated secondary antibodies (Sigma). Visualization was achieved by development with ECL Plus chemiluminescent substrate (GE Healthcare, Buckinghamshire, UK) and exposure to CL-Exposure film (Pierce Biothechnology, Inc., Rockford, IL).
Indirect immunofluorescence was employed to assess chlamydial development and protein localization in infected host cells. HeLa-cell monolayers grown as described (Hackstadt et al., 1996) on 12-mm-diameter glass coverslips, infected with C. trachomatis L2, and cultivated in the presence or absence of C1. At indicated times p.i., infected cultures were fixed and permeablized with methanol (Fig. 2) or fixed with 2% paraformaldehyde and permeablized with a PBS solution containing 0.1% (vol/vol) Triton X-100 and 3% (wt/vol) BSA (Fig. 6A). All samples were blocked with 5% (wt/vol) BSA in PBS supplemented with 0.05% (vol/vol) Tween-20 (Sigma), and probed with antibodies specific for Hsp60 (Yuan et al., 1992), whole C. trachomatis L2, or IncA. Proteins were visualized by probing with Alexa Fluor 488- or Alexa Fluor 594-conjugated secondary antibodies (Invitrogen). HeLa and Chlamydia DNA was visualized by staining with 4', 6-diamidino-2-phenyl-indole, dihydrochloride (DAPI; Invitrogen). Images were acquired by epi-fluorescence microscopy using a 60X apochromat objective on a TE2000U inverted photomicroscope (Nikon) equipped with a Retiga EXi 1394, 12 bit monochrome CCD camera and MetaMorph imaging software. For protein co-localization experiments, fluorescent images were acquired using a Zeiss Axiovert Zoom LSM 510 confocal microscope. Micrographs were processed using Adobe Photoshop 6.0 (Adobe Systems).
Lipid trafficking
Fluorescent [_N_-[7-(4-nitrobenzo-2-oxa-1,3-diazole)]-6-aminocaproyl-D_erythro_-sphingosine (C6-NBD-Ceramide) was complexed with 0.034% (wt/vol) defatted bovine serum albumin (dfBSA, Sigma) in serum-free RPMI as described previously (Pagano and Martin, 1988). A 5 μM C6-NBD-Ceramide solution was added to _C. trachomatis_-infected HeLa cells, incubated at 4°C for 30 min, washed with 10 mM HEPES-buffered calcium- and magnesium-free Puck's saline, pH 7.4 (HCMF), and incubated for 1 hr in RPMI + 0.34% (wt/vol) dfBSA to ‘back-exchange’ excess probe from plasma membrane. Where appropriate, C1 was maintained at 50 μM during staining and back-exchange procedures. Cultures were rinsed in HCMF solution and examined by fluorescent microscopy as described (Hackstadt et al., 1995).
Acknowledgments
The author's gratefully acknowledge Dr. Robert Heinzen for kindly providing C. burnetii, Dr. Harlan Caldwell for the gift of Hsp60-specific antibodies, and Dr. Douglas Miller for the gift of Caspase-1 specific antibodies. We would also like to thank Dr. Greg Plano for critical review of the manuscript. This work was supported by the Public Health Service Grant AI065530 from the National Institutes of Health to K.A.F.
References
- Belland RJ, Zhong G, Crane DD, Hogan D, Sturdevant D, Sharma J, Beatty WL, Caldwell HD. Genomic transcriptional profiling of the developmental cycle of Chlamydia trachomatis. Proc Natl Acad Sci U S A. 2003;100:8478–8483. doi: 10.1073/pnas.1331135100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell HD, Kromhout J, Schachter J. Purification and partial characterization of the major outer membrane protein of Chlamydia trachomatis. Infect Immun. 1981;31:1161–1176. doi: 10.1128/iai.31.3.1161-1176.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen JD, Christiansen G, Holst HU, Birkelund S. Chlamydia trachomatis utilizes the host cell microtubule network during early events of infection. Mol Microbiol. 1997;25:441–449. doi: 10.1046/j.1365-2958.1997.4591832.x. [DOI] [PubMed] [Google Scholar]
- Clifton DR, Fields KA, Grieshaber SS, Dooley CA, Fischer ER, Mead DJ, Carabeo RA, Hackstadt T. A chlamydial type III translocated protein is tyrosine-phosphorylated at the site of entry and associated with recruitment of actin. Proc Natl Acad Sci U S A. 2004;101:10166–10171. doi: 10.1073/pnas.0402829101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman SA, Fischer ER, Howe D, Mead DJ, Heinzen R. Temporal analysis of Coxiella burnetii morphological differentiation. J Bacteriol. 2004;186:7344–7352. doi: 10.1128/JB.186.21.7344-7352.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas AL, Hatch TP. Expression of the transcripts of the sigma factors and putative sigma factor regulators of Chlamydia trachomatis L2. Gene. 2000;247:209–214. doi: 10.1016/s0378-1119(00)00094-9. [DOI] [PubMed] [Google Scholar]
- Ferracci F, Schubot FD, Waugh DS, Plano GV. Selection and characterization of Yersinia pestis YopN mutants that constitutively block Yop secretion. Mol Microbiol. 2005;57:970–987. doi: 10.1111/j.1365-2958.2005.04738.x. [DOI] [PubMed] [Google Scholar]
- Fields KA, Hackstadt T. Evidence for the secretion of Chlamydia trachomatis CopN by a type III secretion mechanism. Mol Microbiol. 2000;38:1048–1060. doi: 10.1046/j.1365-2958.2000.02212.x. [DOI] [PubMed] [Google Scholar]
- Fields KA, Hackstadt T. The Chlamydial Inclusion: Escape from the Endocytic Pathway. Annu Rev Cell Dev Biol. 2002;14:14. doi: 10.1146/annurev.cellbio.18.012502.105845. [DOI] [PubMed] [Google Scholar]
- Fields KA, Mead DJ, Dooley CA, Hackstadt T. Chlamydia trachomatis type III secretion: evidence for a functional apparatus during early-cycle development. Mol Microbiol. 2003;48:671–683. doi: 10.1046/j.1365-2958.2003.03462.x. [DOI] [PubMed] [Google Scholar]
- Fields KA, Fischer ER, Mead DJ, Hackstadt T. Analysis of putative Chlamydia trachomatis chaperones Scc2 and Scc3 and their use in the identification of type III secretion substrates. J Bacteriol. 2005;187:6466–6478. doi: 10.1128/JB.187.18.6466-6478.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furness G, Graham DM, Reeve P. The titration of trachoma and inclusion blennorrhoea viruses in cell cultures. J. Gen. Microbiol. 1960;23:613–619. doi: 10.1099/00221287-23-3-613. [DOI] [PubMed] [Google Scholar]
- Grieshaber NA, Fischer ER, Mead DJ, Dooley CA, Hackstadt T. Chlamydial histone-DNA interactions are disrupted by a metabolite in the methylerythritol phosphate pathway of isoprenoid biosynthesis. Proc Natl Acad Sci U S A. 2004;101:7451–7456. doi: 10.1073/pnas.0400754101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieshaber NA, Grieshaber SS, Fischer ER, Hackstadt T. A small RNA inhibits translation of the histone-like protein Hc1 in Chlamydia trachomatis. Mol Microbiol. 2006;59:541–550. doi: 10.1111/j.1365-2958.2005.04949.x. [DOI] [PubMed] [Google Scholar]
- Grieshaber SS, Grieshaber NA, Hackstadt T. Chlamydia trachomatis uses host cell dynein to traffic to the microtubule-organizing center in a p50 dynamitin-independent process. J Cell Sci. 2003;116:3793–3802. doi: 10.1242/jcs.00695. [DOI] [PubMed] [Google Scholar]
- Hackstadt T, Scidmore MA, Rockey DD. Lipid metabolism in Chlamydia trachomatis-infected cells: directed trafficking of Golgi-derived sphingolipids to the chlamydial inclusion. Proc Natl Acad Sci U S A. 1995;92:4877–4881. doi: 10.1073/pnas.92.11.4877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackstadt T, Rockey DD, Heinzen RA, Scidmore MA. Chlamydia trachomatis interrupts an exocytic pathway to acquire endogenously synthesized sphingomyelin in transit from the Golgi apparatus to the plasma membrane. Embo J. 1996;15:964–977. [PMC free article] [PubMed] [Google Scholar]
- Hackstadt T. The diverse habitats of obligate intracellular parasites. Curr Opin Microbiol. 1998;1:82–87. doi: 10.1016/s1369-5274(98)80146-x. [DOI] [PubMed] [Google Scholar]
- Hackstadt T, Scidmore-Carlson M, Shaw E, Fischer E. The Chlamydia trachomatis IncA protein is required for homotypic vesicle fusion. Cellular Microbiology. 1999;1:119–130. doi: 10.1046/j.1462-5822.1999.00012.x. [DOI] [PubMed] [Google Scholar]
- Heinzen RA, Scidmore MA, Rockey DD, Hackstadt T. Differential interaction with endocytic and exocytic pathways distinguish parasitophorous vacuoles of Coxiella burnetii and Chlamydia trachomatis. Infect Immun. 1996;64:796–809. doi: 10.1128/iai.64.3.796-809.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuer D, Brinkmann V, Meyer TF, Szczepek AJ. Expression and translocation of chlamydial protease during acute and persistent infection of the epithelial HEp-2 cells with Chlamydophila (Chlamydia) pneumoniae. Cell Microbiol. 2003;5:315–322. doi: 10.1046/j.1462-5822.2003.00278.x. [DOI] [PubMed] [Google Scholar]
- Hilbert DW, Piggot PJ. Compartmentalization of gene expression during Bacillus subtilis spore formation. Microbiol Mol Biol Rev. 2004;68:234–262. doi: 10.1128/MMBR.68.2.234-262.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan RJ, Mathews SA, Mukhopadhyay S, Summersgill JT, Timms P. Chlamydial persistence: beyond the biphasic paradigm. Infect Immun. 2004;72:1843–1855. doi: 10.1128/IAI.72.4.1843-1855.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsia RC, Pannekoek Y, Ingerowski E, Bavoil PM. Type III secretion genes identify a putative virulence locus of Chlamydia. Mol Microbiol. 1997;25:351–359. doi: 10.1046/j.1365-2958.1997.4701834.x. [DOI] [PubMed] [Google Scholar]
- Hua L, Hefty PS, Lee YL, Lee YM, Stephens RS, Price CW. Core of the partner switching signalling mechanism is conserved in the obligate intracellular pathogen Chlamydia trachomatis. Mol Microbiol. 2006;59:623–636. doi: 10.1111/j.1365-2958.2005.04962.x. [DOI] [PubMed] [Google Scholar]
- Hueck CJ. Type III protein secretion systems in bacterial pathogens of animals and plants. Microbiol Mol Biol Rev. 1998;62:379–433. doi: 10.1128/mmbr.62.2.379-433.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppi AM, Nordfelth R, Uvell H, Wolf-Watz H, Elofsson M. Targeting bacterial virulence: inhibitors of type III secretion in Yersinia. Chem Biol. 2003;10:241–249. doi: 10.1016/s1074-5521(03)00046-2. [DOI] [PubMed] [Google Scholar]
- Koo IC, Stephens RS. A developmentally regulated two-component signal transduction system in Chlamydia. J Biol Chem. 2003;278:17314–17319. doi: 10.1074/jbc.M212170200. [DOI] [PubMed] [Google Scholar]
- Kozak NA, Mattoo S, Foreman-Wykert AK, Whitelegge JP, Miller J. Interactions between partner switcher orthologs BtrW and BtrV regulate type III secretion in Bordetella. J Bacteriol. 2005;187:5665–5676. doi: 10.1128/JB.187.16.5665-5676.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lu H, Shen C, Brunham RC. Chlamydia trachomatis infection of epithelial cells induces the activation of caspase-1 and release of mature IL-18. J Immunol. 2000;165:1463–1469. doi: 10.4049/jimmunol.165.3.1463. [DOI] [PubMed] [Google Scholar]
- Matsumoto A. Electron microscopic observations of surface projections and related intracellular structures of Chlamydia organisms. J Electron Microsc (Tokyo) 1981;30:315–320. [PubMed] [Google Scholar]
- Mattoo S, Yuk MH, Huang L, Miller J. Regulation of type III secretion in Bordetella. Mol Microbiol. 2004;52 doi: 10.1111/j.1365-2958.2004.04053.x. [DOI] [PubMed] [Google Scholar]
- Moulder JW. Interaction of chlamydiae and host cells in vitro. Microbiol Rev. 1991;55:143–190. doi: 10.1128/mr.55.1.143-190.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mpiga P, Ravaoarinoro M. Chlamdyia trachomatis persistence: an update. Microbiol Res. 2006;161:9–19. doi: 10.1016/j.micres.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Nichols BA, Setzer PY, Pang F, Dawson CR. New view of the surface projections of Chlamydia trachomatis. J Bacteriol. 1985;164:344–349. doi: 10.1128/jb.164.1.344-349.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordfelth R, Kauppi AM, Norberg HA, Wolf-Watz H, Elofsson M. Small molecule inhibitors specifically targeting type III secretion. Infect Immun. 2005;73:3104–3114. doi: 10.1128/IAI.73.5.3104-3114.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano RE, Martin OC. A series of flourescent N-acylsphingosines: synthesis, physical properties, and studies in cultured cells. Biochemistry. 1988;27:4439–4445. doi: 10.1021/bi00412a034. [DOI] [PubMed] [Google Scholar]
- Rockey DD, Scidmore MA, Bannantine JP, Brown WJ. Proteins in the chlamydial inclusion membrane. Microbes Infect. 2002;4:333–340. doi: 10.1016/s1286-4579(02)01546-0. [DOI] [PubMed] [Google Scholar]
- Scidmore M, Fischer E, Hackstadt T. Restricted fusion of Chlamydia trachomatis vesicles with endocytic compartments during the initial stages of infection. Infect Immun. 2003;71:973–984. doi: 10.1128/IAI.71.2.973-984.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scidmore MA, Rockey DD, Fischer ER, Heinzen RA, Hackstadt T. Vesicular interactions of the Chlamydia trachomatis inclusion are determined by chlamydial early protein synthesis rather than route of entry. Infect Immun. 1996;64:5366–5372. doi: 10.1128/iai.64.12.5366-5372.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scidmore-Carlson MA, Shaw EI, Dooley CA, Fischer ER, Hackstadt T. Identification and characterization of a Chlamydia trachomatis early operon encoding four novel inclusion membrane proteins. Mol Microbiol. 1999;33:753–765. doi: 10.1046/j.1365-2958.1999.01523.x. [DOI] [PubMed] [Google Scholar]
- Seshadri R, Paulsen I, Eisen J, Read T, Nelson K, Nelson W, Ward N, Tettelin H, Davidsen T, Beanan M, Deboy R, Daugherty S, Brinak L, Mapupu R, Dodson R, Khouri H, Lee K, Carty H, Scanlan D, Heinzen R, Thompson H, Samuel J, Fraser C, Heidelberg J. Complete genome sequence of the Q-fever pathogen Coxiella burnetii. Proc Natl Acad Sci U S A. 2003;100:5455–5460. doi: 10.1073/pnas.0931379100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw EI, Dooley CA, Fischer ER, Scidmore MA, Fields KA, Hackstadt T. Three temporal classes of gene expression during the Chlamydia trachomatis developmental cycle. Mol Microbiol. 2000;37:913–925. doi: 10.1046/j.1365-2958.2000.02057.x. [DOI] [PubMed] [Google Scholar]
- Stenner-Liewen F, Liewen H, Zapata J, Pawlowski K, Godzik A, Reed J. CADD, a Chlamydia protein that interacts with death receptors. J Biol Chem. 2002;277:9633–9636. doi: 10.1074/jbc.C100693200. [DOI] [PubMed] [Google Scholar]
- Stephens RS, Kalman S, Lammel C, Fan J, Marathe R, Aravind L, Mitchell W, Olinger L, Tatusov RL, Zhao Q, Koonin EV, Davis RW. Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science. 1998;282:754–759. doi: 10.1126/science.282.5389.754. [DOI] [PubMed] [Google Scholar]
- Subtil A, Parsot C, Dautry-Varsat A. Secretion of predicted Inc proteins of Chlamydiapneumoniae by a heterologous type III machinery. Mol Microbiol. 2001;39:792–800. doi: 10.1046/j.1365-2958.2001.02272.x. [DOI] [PubMed] [Google Scholar]
- Subtil A, Delevoye C, Balana ME, Tastevin L, Perrinet S, Dautry-Varsat A. A directed screen for chlamydial proteins secreted by a type III mechanism identifies a translocated protein and numerous other new candidates. Mol Microbiol. 2005;56:1636–1647. doi: 10.1111/j.1365-2958.2005.04647.x. [DOI] [PubMed] [Google Scholar]
- Suchland RJ, Rockey DD, Bannantine JP, Stamm WE. Isolates of Chlamydia trachomatis that occupy nonfusogenic inclusions lack IncA, a protein localized to the inclusion membrane. Infect Immun. 2000;68:360–367. doi: 10.1128/iai.68.1.360-367.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Lyng K, Zhang YX, Rockey DD, Morrison RP. Monoclonal antibodies define genus-specific, species-specific, and cross-reactive epitopes of the chlamydial 60-kilodalton heat shock protein (hsp60): specific immunodetection and purification of chlamydial hsp60. Infect Immun. 1992;60:2288–2296. doi: 10.1128/iai.60.6.2288-2296.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong G, Fan P, Ji H, Dong F, Huang Y. Identification of a chlamydial protease-like activity factor responsible for the degradation of host transcription factors. J Exp Med. 2001;193:935–942. doi: 10.1084/jem.193.8.935. [DOI] [PMC free article] [PubMed] [Google Scholar]