A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17 (original) (raw)
. Author manuscript; available in PMC: 2006 Oct 22.
Published in final edited form as: Nat Immunol. 2005 Oct 2;6(11):1133–1141. doi: 10.1038/ni1261
Abstract
Interleukin 17 (IL-17) has been linked to autoimmune diseases, although its regulation and function have remained unclear. Here we have evaluated in vitro and in vivo the requirements for the differentiation of naive CD4 T cells into effector T helper cells that produce IL-17. This process required the costimulatory molecules CD28 and ICOS but was independent of the cytokines and transcription factors required for T helper type 1 or type 2 differentiation. Furthermore, both IL-4 and interferon-γ negatively regulated T helper cell production of IL-17 in the effector phase. In vivo, antibody to IL-17 inhibited chemokine expression in the brain during experimental autoimmune encephalomyelitis, whereas overexpression of IL-17 in lung epithelium caused chemokine production and leukocyte infiltration. Thus, IL-17 expression characterizes a unique T helper lineage that regulates tissue inflammation.
CD4 T helper (TH) lymphocytes are essential regulators of immune responses and inflammatory diseases. After being activated by professional antigen-presenting cells (APCs), TH cells differentiate into effector cells specialized in cytokine secretion and function. Effector TH cells have been classified as type 1 (TH1) and type 2 (TH2) based on their cytokine expression profiles and immune regulatory function1. TH1 cells produce interferon-γ (IFN-γ) and mediate cellular immunity, whereas TH2 cells produce interleukin 4 (IL-4), IL-5 and IL-13 and mediate humoral immunity and allergic responses. TH cell differentiation is regulated by the interaction of naive CD4 T cells with innate immune cells that express specific peptide–major histocompatibility complex class II complexes, costimulatory molecules and inflammatory cytokines.
Naive T cell activation normally requires two signals: T cell receptor (TCR) signals, and costimulation through several accessory molecules. The main costimulatory molecule on TH cells is CD28 (ref. 2), which interacts with CD80 (B7-1) and CD86 (B7-2) expressed on mature dendritic cells and other APCs. The inducible costimulator ICOS is another member of the CD28 ‘superfamily’ that also regulates naive CD4 T cell activation and effector differentiation3. In addition to TCR and costimulatory molecules, IL-12 produced by activated APCs is critical in TH1 differentiation4. Additional cytokines in the IL-12 family, IL-23 and IL-27, are also important for TH cell differentiation and function5,6. IL-23, in particular, is crucial in the pathogenesis of experimental autoimmune encephalomyelitis (EAE) and collagen-induced arthritis7,8. Other cytokines also influence the development of effector functions of TH cells; for example, IL-4 produced by activated T cells (and perhaps by other innate cells as well) is crucial in driving TH2 differentiation9.
TH differentiation and effector cytokine expression are mediated by several key transcription factors. IL-12 regulates TH1 differentiation through activation of the transcription factor STAT4 (refs. 4,5). The transcription factor T-bet is a ‘master regulator’ of TH1 differentiation through the potentiation of IFN-γ production and suppression of TH2-associated cytokine expression4. IL-4, in contrast, drives TH2 differentiation through the actions of STAT6 (ref. 9) and GATA-3, which is a ‘master regulator’ of TH2 differentiation through potentiation of IL-4 and suppression of IFN-γ10. In addition, c-Maf has been identified as a TH2-specific transcription factor that binds to the Il4 proximal promoter11. Studies of _Maf_-knockout mice and mice with overexpression of a Maf transgene, furthermore, have indicated that c-Maf selectively regulates Il4 expression11,12.
IL-17 (also called IL-17A) has been associated with many inflammatory diseases such as rheumatoid arthritis, asthma, lupus and allograft rejection13–15. The IL-17 receptor (IL-17R) is distributed ubiquitously in various tissues14, and its engagement activates both transcription factor NF-κB and kinase Jnk pathways16. Many in vitro studies have indicated a proinflammatory function for IL-17 (ref. 15). In particular, IL-17 has been linked to tissue neutrophil recruitment through the induction of granulocyte colony-stimulating factor and IL-8 (ref. 15), and IL-17R-deficient mice have impaired host defense against microbacterial infection because of a substantial reduction in granulocyte colony-stimulating factor and macrophage inflammatory protein 2 in the lung17. IL-17 is also important in contact, delayed-type and airway hypersensitivities, as shown in a study using IL-17-deficient mice18. In related reports, IL-1_7_-deficient mice19, as well as wild-type mice that received an IL-17R antagonist20, have shown resistance to an arthritis-like disease.
IL-17 expression is generally thought to be restricted to T cells. In humans, IL-17 is expressed by activated CD4 T cells and by TH1 and TH0 cells but not by TH2 cells21, whereas in mice, IL-17 expression is strongly induced by IL-23 in memory T cells22. Notably, IL-23 (but not IL-12) deficiency is associated with resistance to EAE and collagen-induced arthritis7,8, a phenotype that correlates with a defect in IL-17 expression8,23. IL-23 also selectively expands IL-17-expressing T cell populations, which may also coexpress another IL-17 family cytokine, IL-17F, as well as tumor necrosis factor (TNF) and IL-6 (ref. 23). These data suggest that a unique cytokine requirement is needed for generating T cells that express IL-17.
Here we have analyzed the regulation and function of IL-17 in vitro and in vivo. We found that IL-17 was expressed by a distinct lineage of TH effector cells; generation of these cells required CD28 and ICOS costimulation and was independent of the cytokine and transcription programs normally associated with TH1 and TH2 differentiation. In vivo, IL-17 potently regulated chemokine expression by tissue cells, and IL-17 overexpression in the lung caused airway inflammation. In contrast, IL-17-specific inhibition attenuated immune infiltration in the brain in an EAE model. We conclude from these data that IL-17 is expressed by a previously unknown subset of TH cells and is crucial in regulating tissue inflammatory reactions.
RESULTS
Generation of IL-17-expressing TH cells requires CD28 and ICOS
We hypothesized that antigen-specific naive TH precursor cells differentiate into IL-17-expressing effector cells during immune responses. To test this, we immunized C57BL/6 (B6) mice with myelin oligodendrocyte glycoprotein (MOG) peptide and, after 7 d, isolated spleen and lymph node cells and restimulated them with MOG peptide in vitro for cytokine production. We detected IL-17, IFN-γ and TNF in supernatants only after restimulation (Fig. 1a). In contrast, nonimmunized mice or those receiving only complete Freund’s adjuvant (CFA) produced neither IL-17 nor IFN-γ. We obtained similar results by intracellular cytokine staining of MOG-restimulated spleen and lymph node cells. The numbers of IL-17-producing CD4 cells were substantially and reproducibly increased after MOG immunization and did not express IFN-γ (Fig. 1b). These results indicated that naive MOG-specific TH cells differentiated into IL-17-producing effector cells in vivo after immunization with MOG peptide.
Figure 1.
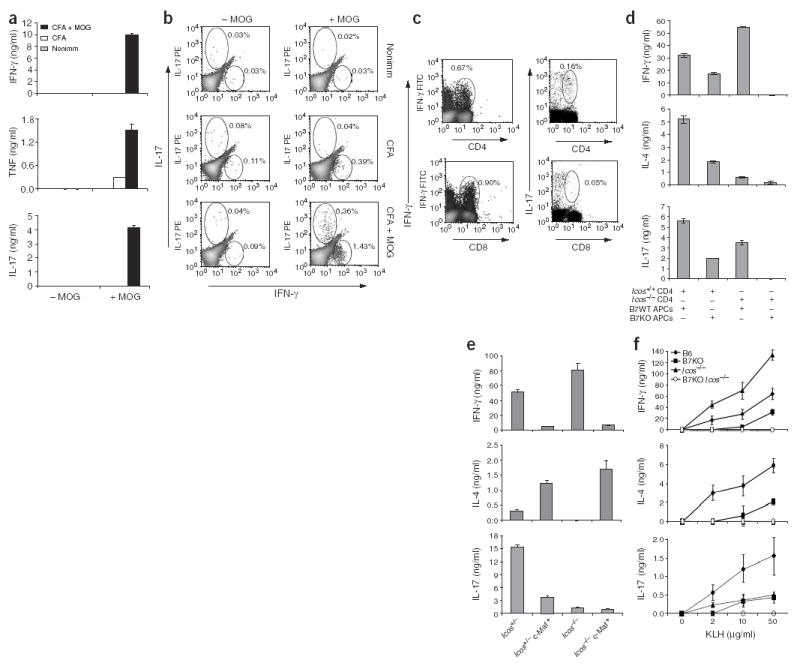
The generation of IL-17-producing T cells requires CD28 and ICOS costimulation. (a,b) B6 mice were immunized with MOG(35–55) in CFA or CFA alone; 7 d later, lymph node and spleen cells from immunized mice (two mice in each group) were collected. (a) ELISA of cytokines in supernatants of lymph node and spleen cells from nonimmunized (Nonimm) and immunized mice stimulated in triplicate with MOG peptide for 72 h in vitro. (b) Intracellular staining for IL-17, IFN-γ or TNF after MOG restimulation. Data are of gated CD4+ cells; numbers beside outlined areas indicate percent positive cells in that area. (c) Intracellular detection of IL-17 and IFN-γ in conjunction with anti-CD4 or anti-CD8. B6 mice were immunized for 7 d with KLH in CFA and then spleen and lymph node cells were collected and restimulated with PMA and ionomycin. Numbers beside outlined areas indicate percent positive cells in that area. (d) ELISA of cytokine production by CD4 T cells isolated from Icos-sufficient (Icos+/+) or Icos-deficient (_Icos_−/−) OTII mice and activated for 4 d with splenic APCs from B6 mice (B7 WT) or mice doubly deficient in B7.1 and B7.2 (B7 KO) in the presence of OVA peptide; activated cells were then washed and restimulated for 24 h with anti-CD3. (e) ELISA of cytokine production by naive T cells isolated from Icos+/1 and _Icos_−/−mice with or without the Maf transgene and activated for 4 d with anti-CD3, APCs and IL-2. (f) ELISA of cytokine production by spleen cells isolated from B6, B7-deficient (B7 KO), _Icos_−/− or B7-deficient _Icos_−/− mice (three in each group, analyzed individually) immunized with KLH in CFA; spleen cells were restimulated ex vivo with various doses of KLH. Data are representative of at least two independent experiments with similar results.
IFN-γ and TNF are also expressed by effector CD8 T cells24. To determine if CD8 T cells could also be stimulated to produce IL-17, we immunized B6 mice with keyhole limpet hemocyanin (KLH) protein, which elicits both CD4 and CD8 responses. We isolated spleen cells and, as with the CD4 T cells, stained them to assess intracellular expression of cytokines. In contrast to antigen-specific CD4 T cells, the KLH-specific CD8 T cells expressed IFN-γ but much less IL-17 (Fig. 1c).
To further characterize the requirements for IL-17 expression, we next analyzed the function of CD28 and ICOS in the generation of IL-17-producing CD4 effector cells. First, we isolated ovalbumin (OVA)–specific TCR-transgenic CD4 T cells (OTII cells) from wild-type or ICOS-deficient mice and cultured them for 4 d with OVA peptide–loaded spleen cells from wild-type B6 mice or B7-deficient mice (_Cd80_−/−_Cd86_−/−). Only OTII T cells positive for the variable α2-chain (Vα2+) remained in culture and had upregulated the activation marker CD44 and downregulated the cell surface marker CD62L (data not shown). We then washed the activated OTII cells and restimulated them for 24 h with antibody to CD3 (anti-CD3), after which we assessed cytokine production in cell supernatants by ELISA. Supernatants from cells deficient in CD28 costimulation (wild-type CD4 T cells plus B7-deficient APCs) contained much less IL-4, IFN-γ and IL-17 than did wild-type CD4 T cells and wild-type APCs (Fig. 1d). In contrast, T cells deficient in ICOS costimulation (ICOS-deficient CD4 T cells and wild-type APCs) produced both less IL-4 and IL-17 and more IFN-γ than did wild-type CD4 T cells and wild-type APCs (Fig. 1d). Finally, deficiencies in both B7 and ICOS caused the most profound cytokine defect, as T cells did not produce IFN-γ, IL-4, or IL-17 after restimulation (Fig. 1d). Consistent with that, naive ICOS-deficient (_Icos_−/−) CD4 T cells, when activated with anti-CD3 and wild-type APCs, demonstrated defects in the production of IL-4 and IL-17 (Fig. 1e). ICOS is thus differentially required for the development of TH cells into different effector subsets.
To understand the function of CD28 and ICOS in IL-17 regulation in vivo, we used wild-type B6 mice and mice deficient in B7, ICOS or both and immunized the mice with KLH in CFA. Consistent with the in vitro results, during ex vivo recall responses, T cells from mice deficient in B7 produced much less IFN-γ, IL-17 and IL-4, whereas ICOS deficiency resulted in the production of less IL-4 and IL-17 but not IFN-γ (Fig. 1f). T cells from mice deficient in expression of both B7 and ICOS produced none of the cytokines found after stimulation of cells from mice with single deficiencies in response to KLH restimulation. Thus, both the in vitro and in vivo data indicated that CD28 and ICOS had specific and overlapping functions in the development of antigen-specific TH subsets from naive T cells and that the generation of IL-17-expressing TH cells was regulated by both CD28 and ICOS. Additionally, in the absence of both CD28 and ICOS costimulation, T cells did not differentiate into effector cells capable of expressing IFN-γ, IL-4 or IL-17.
Regulation of IL-17 effector cell differentiation
Many studies have demonstrated that cytokine environment is critical for the development of polarized TH effector subsets with unique functions1. To determine which cytokines were required for the development of IL-17-expressing T cells, we incubated OTII cells for 5 d with OVA peptide–loaded B6 splenocytes in the absence or presence of anti-IL-4, anti-IFN-γ or IL-23, restimulated the cells with anti-CD3 and then evaluated cytokine expression by ELISA (Fig. 2a). IL-17 expression was increased substantially when anti-IFN-γ was added during TH differentiation, suggesting that IFN-γ negatively regulates the generation of IL-17-producing cells. IL-4 production and TH2 differentiation were also increased in the presence of anti-IFN-γ. Additionally, anti-IL-4 inhibited TH2 differentiation and, when added in combination with anti-IFN-γ, further increased IL-17 expression. Finally, the addition of IL-23 together with anti-IL-4 and anti-IFN-γ led to the most IL-17 production, which we confirmed by intracellular cytokine staining. In contrast, there were very few IL-17-expressing Vα2+ OTII cells after activation in neutral conditions (Fig. 2a). Finally, the combination of anti-IFN-γ, anti-IL-4 and IL-23 greatly facilitated the differentiation of TH cells into IL-17-expressing cells, resulting in expression of IL-17 by more than 20% of the OTII T cells (Fig. 2a). These in vitro experiments indicated that the differentiation of IL-17-producing cells occurred efficiently only in the absence of TH1 and TH2 cell development.
Figure 2.
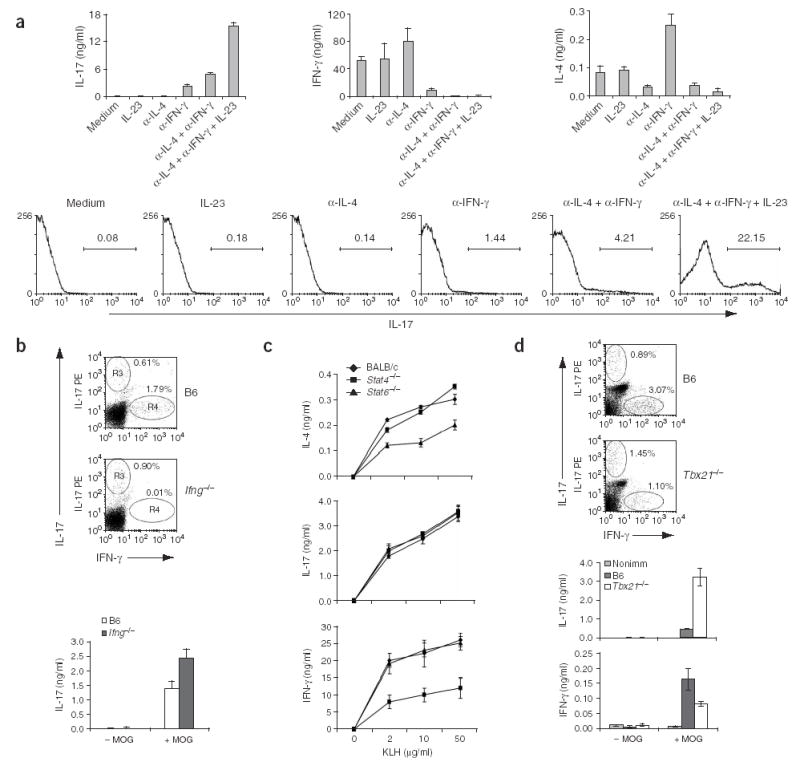
Regulation of IL-17 expression by cytokines and transcriptional factors. (a) ELISA (top) and intracellular staining (bottom) to assess cytokine production by OTII cells. Naive T cells isolated from OTII mice were cultured for 5 d together with APCs from B6 mice along with 10 μg/ml of OVA peptide in presence or absence of anti-IL-4 (α-IL-4), anti-IFN-γ (α-IFN-γ) or IL-23. Cells were restimulated for 24 h with anti-CD3 for ELISA or for 5 h with PMA and ionomycin for intracellular staining. (b) Flow cytometry (dot plots) and ELISA (graph) of spleen and lymph node cells collected from MOG-immunized B6 and _Ifng_−/− mice (two per group), analyzed with a CD4+ gate for expression IL-17 and IFN-γ after MOG restimulation. (c) ELISA of cytokine expression by spleen and lymph node cells from BALB/c, STAT4-deficient (_Stat4_−/−) and STAT6-deficient (_Stat6_−/−) mice immunized with KLH in CFA, analyzed 7 d later after ex vivo restimulation of cells with KLH. (d) Intracellular staining (dot plots) or ELISA (graphs) of cytokine production by draining lymph node cells and splenocytes from T-bet-knockout mice (_Tbx21_−/−) and B6 mice immunized with MOG in CFA at the tail base and analyzed 7 later. Numbers above bracketed lines (a) or beside oval areas (b,d) indicate percent of cells in that area. Data are representative of at least two independent experiments with similar results.
We further analyzed the development of IL-17-expressing cells in vivo. We immunized mice deficient in IFN-γ with MOG peptide in CFA and assessed cytokine expression by intracellular cytokine staining and ELISA after MOG restimulation of T cells ex vivo. In cells from IFN-γ-deficient mice, both the number of IL-17-expressing cells and the amount of IL-17 secretion after MOG restimulation were moderately but consistently increased (Fig. 2b), supporting the conclusion that TH cell effector differentiation to IL-17-producing TH cells in vivo is negatively regulated by IFN-γ. Using the same immunization protocol, we found that IL-4 was not detectable after restimulation and there was no difference in IL-17 expression in IFN-γ-knockout mice after treatment with either anti-IL-4 or a control antibody (data not shown).
Because TH1 and TH2 differentiation are regulated by complex transcriptional mechanisms, we next evaluated STAT4-deficient and STAT6-deficient mice and BALB/c control mice that we immunized with KLH in CFA. As before, we restimulated cells from the spleen and lymph node ex vivo with KLH. Unlike IL-4 and IFN-γ, whose production is greatly reduced in T cells from STAT6-deficient and STAT4-deficient mice, respectively, there was no difference in IL-17 production compared with that of control mice (Fig. 2c). To analyze the involvement of T-bet in IL-17 expression, we immunized B6 or T-bet-deficient mice with MOG and measured IL-17 and IFN-γ expression ex vivo. Although both the number of IFN-γ-expressing cells and the production of IFN-γ were much lower in the absence of T-bet, the number of IL-17 cells and the production of IL-17 much higher in T-bet-deficient mice after immunization (Fig. 2d). To assess the involvement of c-Maf, the TH2 transcription factor important for IL-4 expression, in IL-17 expression, we purified naive CD4+ T cells from mice with or without a c-Maf transgene on an Iocs+/−or _Icos_−/−background and activated them for 4 d with anti-CD3 and wild-type APCs. After restimulation, cells overexpressing c-Maf produced much more IL-4, but their expression of IL-17 as well as IFN-γ was much less (Fig. 1e). Thus, c-Maf seems to negatively regulate IL-17 and IFN-γ expression. In summary, our in vitro and in vivo experiments indicated that the cytokine and transcription programs regulating the differentiation of TH cells into IL-17-expressing cells were distinct from those known to be critical for TH1 and TH2 cells.
Regulation of IL-17 expression in effector or memory T cells
IFN-γ expression by TH1 cells can be activated by concomitant stimulation with cytokines (IL-12 plus IL-18) and TCR stimulation25. To study mechanisms that may regulate IL-17 expression in the effector phase, we sorted CD4+CD62Llo CD4 T cells, treated them with various stimuli and then measured IL-17 production. After TCR and CD28 stimulation for 36 h, we detected IL-17 production in culture supernatants (Fig. 3a). We also evaluated the effect of IFN-γ and IL-4 on IL-17 production. The addition of exogenous IFN-γ or IL-4 alone did not affect the amount of IL-17 produced, whereas in combination they caused a moderate reduction in IL-17 (Fig. 3a). As effector or memory T cells in these conditions might produce large amounts of these two cytokines as well, we tested the effect of blocking with neutralizing antibodies. Treatment with anti-IL-4, anti-IFN-γ or both greatly increased IL-17 production (Fig. 3a). We also found that B1S1 (ref. 26), a negative regulator of T cell activation, inhibited TCR-driven IL-17 expression (data not shown). In addition, because IL-23 induces IL-17 expression by effector and memory T cells22, we evaluated the effects of adding IFN-γ or IL-4 on IL-17 expression induced by IL-23. IFN-γ had no effect, whereas IL-4 strongly inhibited IL-23-dependent IL-17 expression (Fig. 3b).
Figure 3.
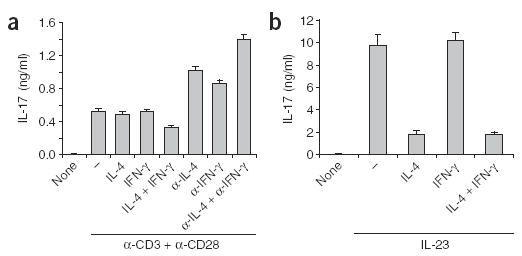
Regulation of IL-17 production by effector and memory T cells. (a) ELISA of IL-17 production by enriched CD62L−CD4+ cells treated for 36 h with various stimuli (horizontal axis) in the presence or absence of plate-bound anti-CD3 and anti-CD28. (b) ELISA of IL-17 production by sorted CD62L−CD4+ cells preactivated with plate-bound anti-CD3, washed and then treated for 24 h with various stimuli (horizontal axis) plus IL-23. None, absence of anti-CD3 plus anti-CD28; –, without other stimuli. Data are representative of two experiments.
IL-17 regulates chemokine expression
Although it is known that the TH1 cytokine IFN-γ regulates cell-mediated immunity and TH2 cytokines (IL-4 and IL-13) regulate humoral responses, the function of IL-17 is not understood as well. Published data have indicated that IL-17 is a proinflammatory mediator on multiple cell types in vitro14 and acts in synergy with TNF to regulate inflammatory gene expression27. To better understand the direct action of IL-17 in immune responses, we did gene expression profiling analysis with an IL-17 recombinant protein that contains a human immunoglobulin G1 (IgG1) tag (IL-17–Ig)28. Treatment of mouse embryonic fibroblasts (MEFs) with IL-17–Ig induced IL-6 production that was inhibited by anti-IL-17 (Supplementary Fig. 1 online). Next, we treated MEFs for 6 h with human IgG or IL-17–Ig and then prepared total RNA for DNA microarray analysis. We used a combined data processing strategy that takes into account both ‘fold changes’ and the λ-value (which is a monotonically increasing function of likelihood ratio describing how likely the gene is differentially expressed for each spot on the array; we arbitrarily chose ‘fold changes’ of more than 1.5 or λ-values greater than 19.5 as cutoffs). Approximately 60 genes were upregulated by IL-17, many encoding proinflammatory molecules, including chemokines and matrix metalloproteinases (Table 1). We found upregulation of expression of genes encoding chemokines CCL2, CCL7, CXCL1 and CCL20, as well as matrix metalloproteinases 3 and 13, and subsequently confirmed that finding by RT-PCR (Fig. 4). Thus, IL-17 seems to directly induce inflammatory responses in primary fibroblasts.
Supplementary Figure 1. Anti-IL-17 antibody specifically blocks IL-17 biological activity.
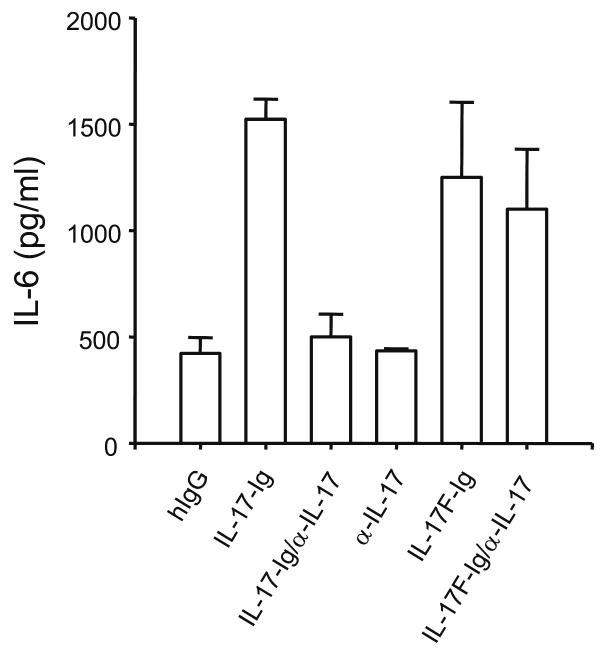
MEF were treated with 500 ng/ml IL-17-Ig, IL-17F-Ig or human IgG in the presence of a rat Ig control or anti-IL-17 for 24 hrs. IL-6 production was measured by ELISA.
Table 1.
Upregulation of genes in MEFs after treatment with IL-17–Ig
| Accession codea | Gene symbolb | ‘Fold increase’c |
|---|---|---|
| NM_011333 | Ccl2 (small inducible cytokine A2) | 3.22 |
| NM_013654 | Ccl7 (small inducible cytokine A7) | 2.40 |
| NM_016960 | Ccl20 (small inducible cytokine subfamily A20) | 2.32 |
| NM_009142 | Cx3cl1 (small inducible cytokine subfamily D1) | 1.76 |
| NM_008176 | Cxcl1 (Gro1 oncogene) | 3.85 |
| NM_008607 | Mmp13 (matrix metalloproteinase 13) | 2.87 |
| NM_010809 | Mmp3 (matrix metalloproteinase 3) | 2.89 |
Figure 4.
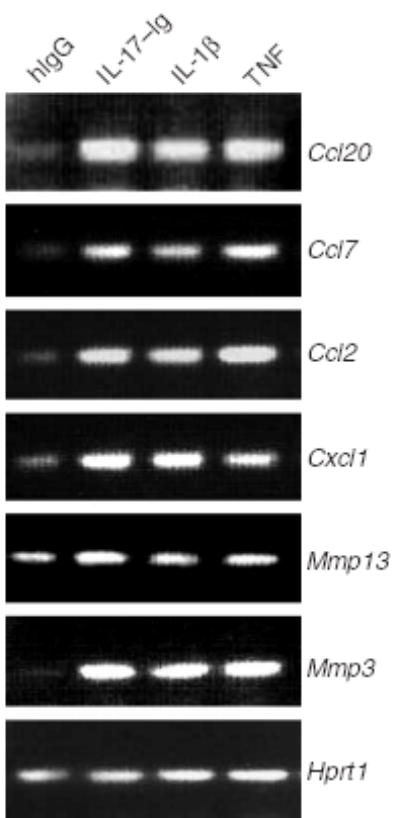
IL-17 regulates genes encoding inflammatory molecules. RT-PCR of the expression of chemokines and matrix metalloproteinases (genes, right margin) in MEFs stimulated for 6 h with human IgG (hIgG), IL-17–Ig, IL-1β or TNF.
IL-17 has been suggested as being important for EAE pathogenesis23. To examine the mechanisms whereby IL-17 regulates central nervous system (CNS) inflammation, we used anti-IL-17, which inhibited IL-6 induction in MEFs treated with IL-17 but not in those treated with IL-17F (Supplementary Fig. 1 online). As T cells reportedly infiltrate the CNS by day 7 after MOG immunization23, we immunized B6 mice twice with MOG peptide and treated them intraperitoneally with anti-IL-17 or control rat IgG starting on day 9 and continuing every other day for a total of three injections. This treatment regimen resulted in a considerably delayed onset of disease compared with that of control mice (day 18 versus day 12; Fig. 5a). At 6 d after the last antibody administration, the mice treated with anti-IL-17 began to develop signs of EAE. Consistent with the clinical signs, there was a characteristic mononuclear cell infiltrate in the white matter of spinal cords of mice treated with rat IgG, whereas there was no obvious cellular infiltration in mice treated with anti-IL-17 (data not shown). To further test the effect of anti-IL-17 after the onset of EAE, we delayed the antibody treatment until the mice used in the experiment developed signs of EAE. Even at this late stage, anti-IL-17 reversed the progression of EAE and resulted in reduced numbers of CD4 T cells and CD11b+ macrophages in the CNS (Supplementary Fig. 2 online).
Figure 5.
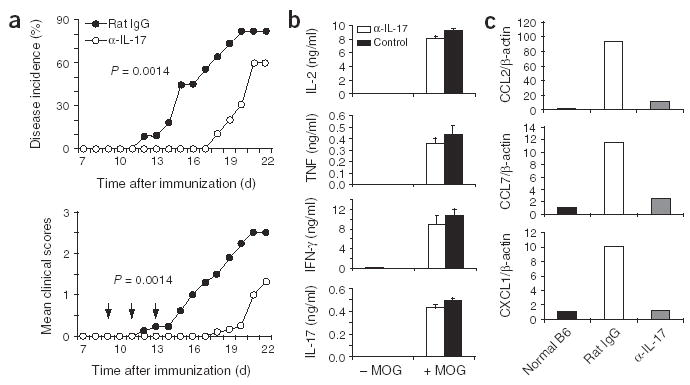
IL-17 regulates chemokine expression in brain tissue during EAE. Anti-IL-17 or control rat IgG (100 μg/mouse) was administered intraperitoneally to mice with EAE on days 9, 11 and 13 after the first MOG immunization. (a) EAE incidence is reduced by treatment with anti-IL-17 (α-IL-17). Data are representative of two independent experiments; anti-IL-17, n = 10 mice, and control, n = 11 mice. Mean clinical scores of sick mice are for nine and six mice for the control and anti-IL-17 groups, respectively. (b) ELISA of proinflammatory cytokines in spleen and lymph node cells from control or anti-IL-17-treated mice (at least three in each group) restimulated for 72 h in triplicate in vitro with MOG and analyzed on day 18 after MOG immunization. Data combine more than three mice in each group. (c) Taqman PCR expression of chemokines CCL2, CCL7 and CXCL1 in brains from normal B6 mice and mice subjected to EAE and treated with rat IgG or anti-IL-17. Results are normalized to β-actin expression and were analyzed in triplicate. Expression in normal mice was considered to be 1.
Supplementary Figure 2. Blockage of IL-17 after disease onset reduces inflammation during EAE.
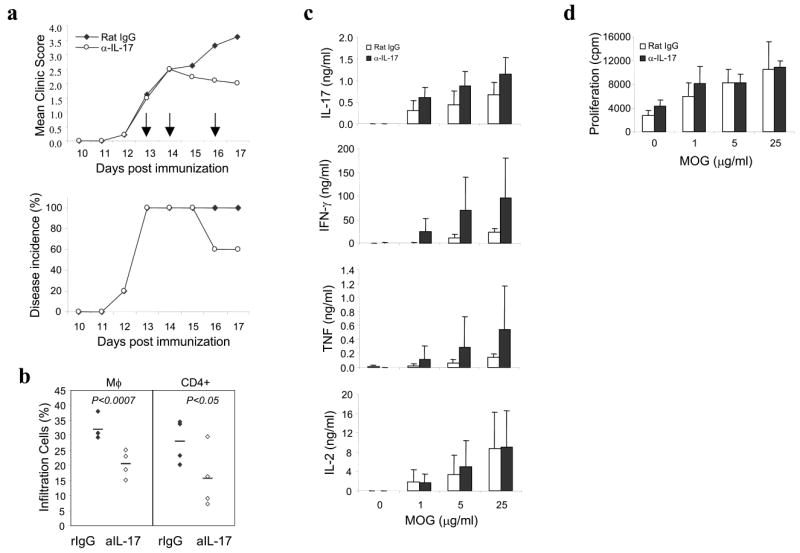
(a) Anti-IL-17 treatment attenuates EAE incidence and severity. C57BL/6 mice were immunized with MOG/CFA and administered with pertussis toxin (see “Experimental Procedures”). Three doses of anti-IL-17 or rat IgG was administered after the disease onset (clinic score >1) on day 13, 14 and 16 post first immunization shown as arrows (n=5 in each group). (b) Less infiltration of CD4 T cells and macrophages were observed in anti-IL17 treatment group than rat IgG group. On day 17, mononuclear cells isolated from brains and spinal cords were analyzed for surface markers CD11b and CD4. Student _t_-test is shown as P values. (c) Splenocytes were ex vivo restimulated with MOG for 48 hours and cytokines expression were measured by ELISA. (d) Splenocyte proliferation in anti-IL-17 treatment group was similar with control group.
To determine if treatment with anti-IL-17 attenuated the activation of MOG-specific T cells in EAE, we obtained splenocytes from control mice and mice treated with anti-IL-17 and restimulated the cells ex vivo with MOG peptide. T cell proliferation and expression of proinflammatory cytokines TNF, IFN-γ and IL-17 were not inhibited by treatment with anti-IL-17 (Fig. 5b and Supplementary Fig. 2 online). Thus, these data demonstrated that blocking IL-17 in the effector phase of EAE does not result in reduced generation or tolerance of autoreactive T cells.
We then determined the basis for lack of recruitment of autoreactive T cells and macrophages into the CNS in the mice treated with anti-IL-17. During EAE, several chemokines are induced considerably in the CNS29. Monocyte chemotactic peptide 1 and its receptor CCR2 have been suggested to be prominent during EAE initiation30. We therefore used real-time RT-PCR to assess chemokine expression in brain tissue after neutralization of IL-17. The expression of chemokines CCL2, CCL17 and CXCL1 was inhibited considerably after IL-17 blockade (Fig. 5c). These data suggest that IL-17 derived from infiltrating TH cells probably mediates the inflammatory reactions in the CNS through regulation of chemokine induction.
Il17 transgene overexpression causes inflammation in lungs
To further understand the action of IL-17 in vivo, we generated transgenic mice that expressed Il17 in lung epithelial cells using the Cc10 promoter (Fig. 6a). We obtained two independent transgenic lines in which IL-17 production in bronchoalveolar lavage fluid could be detected by ELISA (Fig. 6b) and IL-17 mRNA could be detected by RT-PCR in total lung RNA (Fig. 6c). The independent lines had similar phenotypes in lung pathology; however, male transgenic mice of one line developed disease much more slowly than their female littermates.
Figure 6.
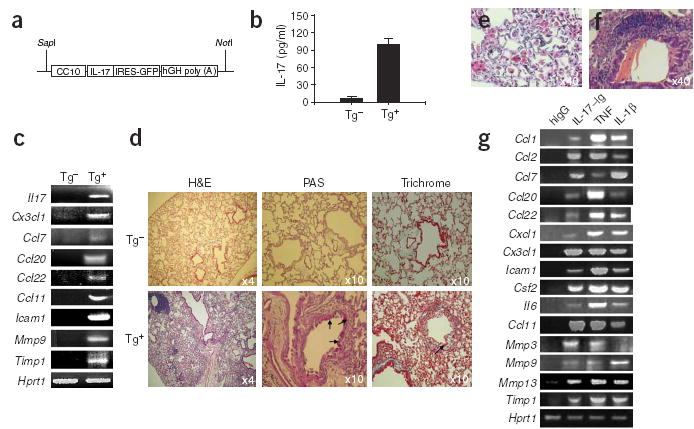
Generation and analysis of _Cc10-Il17_–transgenic mice. (a) CC10–IL-17 construct. hGH, human growth hormone. (b) ELISA of IL-17 in bronchoalveolar lavage fluid collected from transgene-positive mice (Tg+) and transgene-negative littermates (Tg−). (c) RT-PCR of mRNA (genes, left margin) in lungs obtained from 5-month-old transgene-positive and transgene-negative mice. (d–f) Histological comparison of lung tissue. Lungs from 10-month-old transgene-positive and transgene-negative mice (d) or 10-month-old (e) or 5-month-old (f) transgene-positive mice are stained with hematoxylin and eosin (H&E); with periodic acid Schiff (PAS) to compare mucus production in airway epithelium; and with Masson’s trichrome staining to compare airway collagen deposition. Arrows, blue-staining collagen. (g) RT-PCR of expression of genes (left margin) in MLE12 cells treated for 6 h with various stimuli (above lanes). Csf2 encodes granulocyte colony-stimulating factor.
We did histological analysis to determine the consequences of chronic overexpression of IL-17. At 3 months of age, _Il17_-transgenic mice had hypertrophic lung epithelium in the bronchus and bronchiole and obvious alveolar wall thickening compared with that of littermate control mice (data not shown). After 3 months of age, large, eosinophilic and sometimes multinucleated macrophages were frequently present in the lung parenchyma of the _Il17_-transgenic mice; between 5 and 10 months of age, _Il17_-transgenic mice had focal accumulation and infiltration of mononuclear cells, including nodular collections of mononuclear CD4 lymphocytes (Fig. 6d,e and data not shown). Long, thin, needle-like crystals in the bronchioles (Fig. 6f) and mucus production by epithelial cells, visualized by periodic acid Schiff staining, were present in _Il17_-transgenic but not control mice (Fig. 6d). Masson’s trichrome staining also indicated substantial amounts of collagen on the subepithelium of bronchioles adjacent to lymphocyte aggregates in _Il17_-transgenic mice (Fig. 6d). In contrast, control littermates had only loosely packed collagen.
We compared cytokine and chemokine gene expression profiles in lung tissues of _Il17_-transgenic mice and their littermate controls. We did not find upregulation of several TH2-associated genes (Il4, Il5, Il9 and Il13; data not shown). However, the expression of several chemokines (CCL7, CCL22, CCL20, CCL11 and CX3CL1) was greatly increased (Fig. 6c). We also found increases in the matrix metalloproteinase inhibitor TIMP-1 and matrix metalloproteinase 9, which has been noted in patients with airway inflammation31,32. To determine if the induction of these inflammatory genes was directly caused by signals produced by IL-17, we treated MLE12 cells (a mouse lung epithelial cell line) with IL-17–Ig and then measured expression of those same genes. We noted a very similar expression profile of the genes discussed above in MLE12 cells after 6 h of treatment with IL-17–Ig, as well as treatment with TNF and IL-1β (Fig. 6g). Other chemokines such as CCL1, CCL2 and CXCL1 were also upregulated by IL-17. These data indicate that IL-17 acts directly on epithelial cells to induce chemokine expression and that chronic IL-17 overproduction results in leukocyte infiltration and airway changes.
DISCUSSION
Activation of CD4 T lymphocytes by APCs in the presence of specific cytokines causes differentiation into distinct effector TH subsets (or lineages) that produce cytokines with immune regulatory functions. Here we have characterized the regulation of IL-17 expression by CD4 T cells and the function of IL-17 in producing inflammatory reactions in vivo. IL-17 was produced by a unique subset of TH cells during immune and autoimmune responses and IL-17 had a critical function in vivo in regulating chemokine expression and tissue inflammation. Thus, these IL-17-producing effector T cells are important in inflammatory responses (Supplementary Fig. 3 online).
Supplementary Figure 3. Summary of TH differentiation.
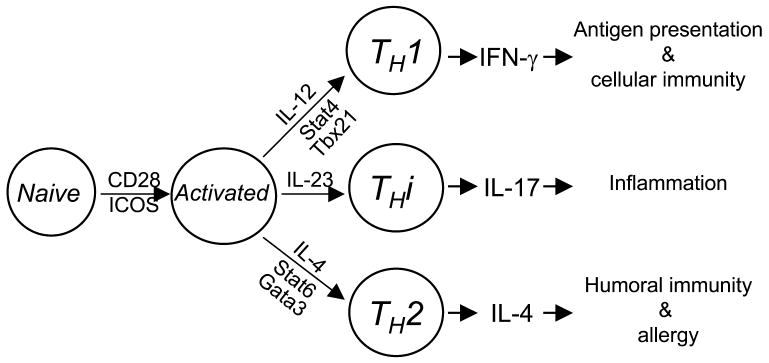
THi is a novel arm of TH cells. Cytokine and transcription factor for TH progenitor to differentiate into THi cells are distinct from those required by conventional TH1 and TH2.
Although IL-17 has emerged as a crucial regulator in immune responses and diseases, its regulation is still poorly understood. Consistent with published reports8,23,33, we found that IL-17 was produced mainly by a subset of antigen-specific effector CD4 T cells in immune and autoimmune responses. Furthermore, differentiation of naive CD4 T cells to IL-17-producing effector T cells was mediated by the innate immune system through CD28 and ICOS costimulation. Although deficiency of B7-1 and B7-2 led to a reduction in all cytokines, ICOS stimulation seemed to be required for production of IL-4 and IL-17 but not of IFN-γ. Compound deficiency in both CD28 and ICOS signals resulted in decreases in all TH subsets both in vitro and in vivo, supporting the idea that costimulation is required for proper activation of all T cell subsets for entry into effector differentiation programs.
Based on our analysis, differentiation of IL-17-producing effector T cells is independent of the mechanisms required for TH1 or TH2 cell development. IL-17 expression by CD4 T cells does not require IFN-γ or the transcription factors T-bet or STAT4, all of which are essential regulators of TH1 differentiation. In contrast, our data have shown that IL-4 negatively regulates the development of IL-17-producing effector T cells and IL-17 expression. We also found that overexpression of c-Maf, a TH2-acting transcription factor, greatly inhibited IL-17 as well as IFN-γ expression. Thus, our data indicate that IL-17-producing effector T cells are a previously unknown subset of CD4 TH cells.
The biological function of IL-17 also supports the conclusion that IL-17-producing effector T cells are a distinct TH subset. IFN-γ produced by TH1 cells is well known for its importance in antigen presentation and cellular responses to intracellular bacteria and viruses. Likewise, IL-4 and other cytokines derived from TH2 cells have crucial functions in humoral immunity and allergic reactions. IL-17, in contrast, is important in tissue inflammation. We analyzed cellular responses to IL-17 and found that IL-17 induced expression of chemokines and matrix metalloproteinases in fibroblasts and lung epithelial cells, similar to the actions of TNF and IL-1. Those data are consistent with many reports in the literature supporting the idea of a proinflammatory function in vitro for IL-17 (ref. 15). We also found that blocking IL-17 resulted in attenuation and delay of EAE and reversed the progression of active EAE. Notably, the effect of anti-IL-17 could not be attributed to reduced T cell priming or cytokine expression but instead acted through the inhibition of chemokine upregulation in the CNS tissue. Consistent with that, there were fewer CD4 T cells and macrophages in the CNS of mice treated with anti-IL-17. Those data indicate that IL-17 is important in chemokine expression and tissue inflammation in EAE.
We also developed two transgenic mouse lines that chronically overexpressed IL-17 in lung epithelial cells. We found substantial inflammation and other airway changes such as mucus production and subepithelial collagen deposition in the transgenic mice, but no apparent neutrophilia34,35. Instead, macrophages were the initial and dominant inflammatory cell type in the lung parenchyma starting as early as 3 months of age. Those findings, although different from the widely accepted idea regarding neutrophil recruitment as a chief function for IL-17, are in agreement with the hypothesis that IL-17 is critical in autoimmune diseases such as EAE and collagen-induced arthritis, in which macrophages are the main inflammatory cells. Furthermore, whereas mucus production and Charcot-Leyden-like crystals are associated with IL-13-transgenic mice36, we did not find evidence of TH2 cytokine expression in lungs of _Il17_-transgenic mice. In contrast, several chemokines were increased in _Il17_-transgenic mice, and they are probably the direct targets of IL-17, as treatment of a lung epithelial cell line with IL-17 resulted in upregulation of the genes encoding those chemokines. Thus, overexpression of IL-17 in lungs resulted in chemokine induction in epithelial cells and caused lung inflammation.
We have demonstrated here that IL-17 is crucial in inflammatory responses. Notably, the differentiation of IL-17-producing effector T cells and their IL-17 expression were negatively regulated by TH1 and TH2 cytokines. IL-12, IL-4 and IFN-γ strongly inhibited the generation and population expansion of IL-17-expressing cells and their cytokine expression, which may serve as a protective strategy to ‘fine-tune’ the expression IL-17 so it does not cause excessive inflammation. Thus, balanced differentiation of TH cells is crucial for immunity and host protection. Abnormal expression of TH1 or TH2 cytokines will affect IL-17 expression and, in turn, may contribute to autoimmune disease.
In summary, we have characterized a previously unknown lineage of TH cells. These IL-17-producing effector T cells produce IL-17 that regulates inflammatory chemokine expression and responses. Further research on this subset of TH cells should demonstrate greater complexity of TH cell regulation and function in immune responses. Additional research may also show that modulation of IL-17-producing effector T cells therapeutically is an important method for treating T cell–mediated chronic inflammatory diseases.
METHODS
Generation of IL-17 recombinant protein
The cDNA sequences encoding the mature form of mouse IL-17 were amplified by PCR and were sequenced before being subcloned into the DES-Ig vector consisting of an insect expression plasmid pMT/BiP/V5-His A (Invitrogen) backbone and a human IgG1 Fc tag28. The primers used for cDNA amplification, with _Bgl_II and _Spe_I sites, respectively, were as follows: Il17 forward, 5′-AGATCTGCGGCTACAGTGAAGGCA-3′, and reverse, 5′-ACTAGTGGCTGCCTGGCGGACAAT-3′. According to the manufacture’s protocol, the DES–IL-17–Ig construct was transfected together with a hygromycin-resistance plasmid (Invitrogen) into the drosophila cell line S2 to generate a stable cell line. After CuSO4 induction, IL-17–Ig fusion protein produced by the stable cell line was further purified with a protein A–agarose column (Sigma-Aldrich).
Ex vivo T cell responses
Mixed T cells and APCs from draining lymph nodes and spleen were prepared 7–11 d after immunization. Viable cells (3.75 × 106) were cultured in complete medium with or without MOG peptide (amino acids 35–55) at various concentrations. Supernatants from activated cells were collected 72 h later and TNF, IFN-γ and IL-17 were measured by ELISA (BD Pharmingen). For intracellular cytokine staining, spleen and lymph node cells from immunized mice were stimulated for 24 h with peptide antigen, and GolgiPlug (BD Pharmingen) was added in the last 5 h or GolgiPlug plus 500 ng/ml of ionomycin and 50 ng/ml of phorbol 12-myristate 13-acetate (PMA; Sigma-Aldrich) were added for 5 h. Cells were permeabilized with the Cytofix/Cytoperm Plus Kit (BD Pharmingen) according to the manufacturer’s protocol.
TH differentiation
Naive T cells, isolated from OTII mice by AutoMACS (Miltenyi Biotec) selection of CD4 T cells following the manufacturer’s instruction, were cultured at a ratio of 1:1 with irradiated B6 splenic APC samples depleted of T cells, along with 10 μg/ml of OVA peptide (amino acids 323–339; ISQAVHAAHAEINEAGR) in the presence or absence of 100 ng/ml of IL-23 (R&D systems), 10 μg/ml of anti-IL-4 (11B11) or 10 μg/ml of anti-IFN-γ (XMG1.2) or ‘cocktails’ of these reagents. The medium was changed when necessary but the concentrations of IL-23, anti-IL-4 and/or anti-IFN-γ were maintained. Then, 5 d after being activated, cells were restimulated with 500 ng/ml of ionomycin and 50 ng/ml of PMA, after which cells producing IL-17 and IFN-γ were analyzed by intracellular staining as described above.
DNA microarray protocol and data analysis
Mouse arrays consisted of 16,463 oligonucleotide probes in the Mouse Genome Set Version 2.0 (QIAGEN/Operon). A Bio-Rad Versarray Chipwriter Pro with Telechem SMP3 spotting pins was used to spot these probes onto Corning GAPS2 substrates. Each oligonucleotide was spotted in duplicate on each array, such that the top and bottom halves of the array were identical. Probes were labeled by reverse transcription of total RNA with oligo(dT) primer (BD Bioscience) in the presence of indocarbocyanine- or indodicarbocyanine-labeled nucleotides to produce labeled cDNA. Labeled cDNA was then hybridized to arrays with Roche DIG hybridization buffer at 37 °C. A Packard BioChip ScanArray 5000 was used for scanning and MolecularWare’s AnalyzerDG software was used for image analysis. Preprocessing of array data involved background subtraction followed by median normalization. Intensity measurements from three arrays, each containing two duplicates per probe, were combined for a total of six replicates per time point. A STATistical method that has been described37 was then used on these replicates to determine likelihood of differential expression for the transcript associated with each probe. A threshold on this differential expression score was then applied across all time points to yield a subset of genes that were differentially expressed in at least one time point, and the resulting data were analyzed further by cluster analysis with the J-Express computer program.
RT-PCR
Total RNA was extracted from homogenized brain tissue, lung tissue, MEFs or MLE12 cells (CRK-2100; American Type Culture Collection) with TRIzol reagent (Invitrogen) following the manufacturer’s instructions. The cDNA was generated with an oligo(dT) primer (Invitrogen) and, for real-time PCR, was analyzed by Taqman PCR with the Brillian QPCR kit (Stratagene) based on expression of the reference gene (Actb, encoding β-actin). The following primer pairs were used: Actb, forward, 5′-TCCTTCGTTGCCGGTCC AC-3′, and reverse, 5′-ACCAGCGCAGCGATATCGTC-3′; Mmp3 (matrix metalloproteinase 3), forward, 5′-TGCAGTTGGAGAACATGGAGACTT-3′, and reverse, 5′-GTAGAGCTGCACATTGGTGATGTCT-3′; Mmp9, forward, 5′-TGTACACAGGCAAGACCGTGCTG-3′, and reverse, 5′-CTCATGGTCCAC CTTGTTCACCTC-3′; Mmp13, forward, 5′-TTACCAGTCTCCGAGGAGAAA CTA-3′, and reverse, 5′-GTCTTCCCCGTGTTCTCAAAGTGA-3′; Ccl2, forward, 5′-CTCAGCCAGATGCAGTTAACGCCC-3′, and reverse, 5′-GGTGCTG AAGACCTTAGGGCAGAT-3′; Ccl7, forward, 5′-CTCATAGCCGCTGCTTT CAGCATC-3′, and reverse, 5′-GTCTAAGTATGCTATAGCCTCCTC-3′; Cxcl1, forward, 5′-CGCTTCTCTGTGCAGCGCTGCTGCT-3′, and reverse, 5′-AAGC CTCGCGACCATTCTTGAGTG-3′; Cxcl3, forward 5′-TGCAGGGCTTACGGC TAAGCCTCAG-3′, and reverse 5′-GGCATGGATGGGTTCCTCTATCTT-3′; Ccl20, forward, 5′-ATGGCCTGCGGTGGCAAGCGTCTG-3′, and reverse, 5′-TAGGCTGAGGAGGTTCACAGCCCT-3′; Ccl11, forward, 5′-GCGCTTCTATT CCTGCTGCTCACGG-3′, and reverse, 5′-GTGGCATCCTGGACCCACTTCT TC-3′; Icam1 (intercellular adhesion molecule), forward, 5′-CAGGTCCAATTC ACACTGAATGCC-3′, and reverse, 5′-GTACACATTCCTGGTGACATTCCCA-3′; Timp1, forward, 5′-GCTTCCAGTAAGGCCTGTAGCTGT-3′, and reverse, GACCTGATCCGTCCACAAACAGTG-3′; Hprt1 (hypoxanthine guanine phosphoribosyl transferase), forward, 5′-GCTGGTGAAAAGGACCTCTGC-3′, and reverse, 5′-CACAGGACTAGAACACCTGC-3′.
Generation of _Il17_-transgenic mice
For the generation of _Il17-_transgenic mice, the Cc10 promoter sequence (a gift from J. Elias, Yale University School of Medicine, New Haven, Connecticut) was cloned into pBluescript II with both flanking _Hin_dIII sites. Full-length cDNA encoding mouse Il17 was first cloned into retrovirus vector containing an internal ribosomal entry site–green fluorescent protein (IRES-GFP) cassette (provided by K. Murphy, Washington University, St. Louis, Missouri) with _Bgl_II and _Xho_I. The entire IL-17_–_IRES–GFP construct was cut out with _Bgl_II and _Bam_HI restriction enzymes and was ligated into pBluescript II containing the Cc10 promoter. Human growth hormone intronic and polyadenylation sequences were then inserted into the CC10–IL-17_–_IRES–GFP–pBluescript II construct using the _Bam_HI and _Not_I restriction enzyme sites. The Il17 transgene construct was isolated by digestion with _Not_I and _Sap_I and was microinjected into B6 mice at the Transgenic Mouse Facility at University of Washington (Seattle, Washington). Two _Il17_-transgenic founders were obtained and were bred with B6 mice.
EAE induction in mice
Female B6 mice were purchased from Charles River and Jackson laboratory. For the induction of EAE, mice were immunized with the MOG peptide (amino acids 35–55; MEVGWYRSPFS ROVHLYRNGK) emulsified in CFA as described26,38. Anti-IL-17 or control rat IgG (100 μg/mouse each time) was administered at various times after MOG immunization. Signs of EAE were assigned scores on a scale of 1–5 as follows: 0, none; 1, limp tail or waddling gait with tail tonicity; 2, wobbly gait; 3, hindlimb paralysis; 4, hindlimb and forelimb paralysis; 5, death. All animal experiments used protocols approved by the Institutional Animal Care and Use Committees of the University of Washington (St. Louis, Missouri) and MD Anderson Cancer Center (Houston, Texas).
Acknowledgments
We thank L. Glimcher and I.-C. Ho for _Maf_-transgenic mice; L. Glimcher and C. Wilson for T-bet-deficient mice; J. Elias for the Cc10 promoter construct; A. Farr for guidance in animal work; and the Dong lab for help and discussions. Supported by the National Institutes of Health (C.D.), Arthritis Foundation (S.H.C., R.N. and C.D.), Cancer Research Institute (C.D.) and MD Anderson Cancer Center (C.D.).
Footnotes
COMPETING INTERESTS STATEMENT
The authors declare that they have no competing financial interests.
References
- 1.Mosmann TR, Coffman RL. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol. 1989;7:145–173. doi: 10.1146/annurev.iy.07.040189.001045. [DOI] [PubMed] [Google Scholar]
- 2.Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell costimulation. Annu Rev Immunol. 1996;14:233–258. doi: 10.1146/annurev.immunol.14.1.233. [DOI] [PubMed] [Google Scholar]
- 3.Dong C, Nurieva RI. Regulation of immune and autoimmune responses by ICOS. J Autoimmun. 2003;21:255–260. doi: 10.1016/s0896-8411(03)00119-7. [DOI] [PubMed] [Google Scholar]
- 4.Szabo SJ, Sullivan BM, Peng SL, Glimcher LH. Molecular mechanisms regulating Th1 immune responses. Annu Rev Immunol. 2003;21:713–758. doi: 10.1146/annurev.immunol.21.120601.140942. [DOI] [PubMed] [Google Scholar]
- 5.Trinchieri G, Pflanz S, Kastelein RA. The IL-12 family of heterodimeric cytokines: new players in the regulation of T cell responses. Immunity. 2003;19:641–644. doi: 10.1016/s1074-7613(03)00296-6. [DOI] [PubMed] [Google Scholar]
- 6.Robinson DS, O’Garra A, Steinman L, Gijbels K. Further checkpoints in Th1 development: CD4+ T-cell subsets in autoimmunity. Immunity. 2002;16:755–758. doi: 10.1016/s1074-7613(02)00331-x. [DOI] [PubMed] [Google Scholar]
- 7.Cua DJ, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- 8.Murphy CA, et al. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med. 2003;198:1951–1957. doi: 10.1084/jem.20030896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Glimcher LH, Murphy KM. Lineage commitment in the immune system: the T helper lymphocyte grows up. Genes Dev. 2000;14:1693–1711. [PubMed] [Google Scholar]
- 10.Zheng W, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell. 1997;89:587–596. doi: 10.1016/s0092-8674(00)80240-8. [DOI] [PubMed] [Google Scholar]
- 11.Ho IC, Hodge MR, Rooney JW, Glimcher LH. The proto-oncogene c-maf is responsible for tissue-specific expression of interleukin-4. Cell. 1996;85:973–983. doi: 10.1016/s0092-8674(00)81299-4. [DOI] [PubMed] [Google Scholar]
- 12.Kim JI, Ho IC, Grusby MJ, Glimcher LH. The transcription factor c-Maf controls the production of interleukin-4 but not other Th2 cytokines. Immunity. 1999;10:745–751. doi: 10.1016/s1074-7613(00)80073-4. [DOI] [PubMed] [Google Scholar]
- 13.Aggarwal S, Gurney AL. IL-17: prototype member of an emerging cytokine family. J Leukoc Biol. 2002;71:1–8. [PubMed] [Google Scholar]
- 14.Moseley TA, Haudenschild DR, Rose L, Reddi AH. Interleukin-17 family and IL-17 receptors. Cytokine Growth Factor Rev. 2003;14:155–174. doi: 10.1016/s1359-6101(03)00002-9. [DOI] [PubMed] [Google Scholar]
- 15.Kolls JK, Linden A. Interleukin-17 family members and inflammation. Immunity. 2004;21:467–476. doi: 10.1016/j.immuni.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 16.Schwandner R, Yamaguchi K, Cao Z. Requirement of tumor necrosis factor receptor-associated factor (TRAF)6 in interleukin 17 signal transduction. J Exp Med. 2000;191:1233–1240. doi: 10.1084/jem.191.7.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ye P, et al. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med. 2001;194:519–527. doi: 10.1084/jem.194.4.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakae S, et al. Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity. 2002;17:375–387. doi: 10.1016/s1074-7613(02)00391-6. [DOI] [PubMed] [Google Scholar]
- 19.Nakae S, Nambu A, Sudo K, Iwakura Y. Suppression of immune iInduction of collagen-induced arthritis in IL-17-deficient mice. J Immunol. 2003;171:6173–6177. doi: 10.4049/jimmunol.171.11.6173. [DOI] [PubMed] [Google Scholar]
- 20.Bush KA, Farmer KM, Walker JS, Kirkham BW. Reduction of joint inflammation and bone erosion in rat adjuvant arthritis by treatment with interleukin-17 receptor IgG1 Fc fusion protein. Arthritis Rheum. 2002;46:802–805. doi: 10.1002/art.10173. [DOI] [PubMed] [Google Scholar]
- 21.Yao Z, et al. Human IL-17: a novel cytokine derived from T cells. J Immunol. 1995;155:5483–5486. [PubMed] [Google Scholar]
- 22.Aggarwal S, Ghilardi N, Xie MH, De Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003;278:1910–1914. doi: 10.1074/jbc.M207577200. [DOI] [PubMed] [Google Scholar]
- 23.Langrish CL, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seder RA, Ahmed R. Similarities and differences in CD4+ and CD8+ effector and memory T cell generation. Nat Immunol. 2003;4:835–842. doi: 10.1038/ni969. [DOI] [PubMed] [Google Scholar]
- 25.Yang J, Murphy TL, Ouyang W, Murphy KM. Induction of interferon-γ production in Th1 CD4+ T cells: evidence for two distinct pathways for promoter activation. Eur J Immunol. 1999;29:548–555. doi: 10.1002/(SICI)1521-4141(199902)29:02<548::AID-IMMU548>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 26.Prasad DV, Richards S, Mai XM, Dong C. B7S1, a novel B7 family member that negatively regulates T cell activation. Immunity. 2003;18:863–873. doi: 10.1016/s1074-7613(03)00147-x. [DOI] [PubMed] [Google Scholar]
- 27.Ruddy MJ, et al. Functional cooperation between interleukin-17 and tumor necrosis factor-α is mediated by CCAAT/enhancer-binding protein family members. J Biol Chem. 2004;279:2559–2567. doi: 10.1074/jbc.M308809200. [DOI] [PubMed] [Google Scholar]
- 28.Sun M, et al. Characterization of mouse and human B7–H3 genes. J Immunol. 2002;168:6294–6297. doi: 10.4049/jimmunol.168.12.6294. [DOI] [PubMed] [Google Scholar]
- 29.Ransohoff RM. The chemokine system in neuroinflammation: an update. J Infect Dis. 2002;186:S152–S156. doi: 10.1086/344266. [DOI] [PubMed] [Google Scholar]
- 30.Huang D, Wang J, Kivisakk P, Rollins BJ, Ransohoff RM. Absence of monocyte chemoattractant protein 1 in mice leads to decreased local macrophage recruitment and antigen-specific T helper cell type 1 Immune response in experimental auto-immune encephalomyelitis. J Exp Med. 2001;193:713–726. doi: 10.1084/jem.193.6.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vignola AM, et al. Sputum metalloproteinase-9/tissue inhibitor of metalloproteinase-1 ratio correlates with airflow obstruction in asthma and chronic bronchitis. Am J Respir Crit Care Med. 1998;158:1945–1950. doi: 10.1164/ajrccm.158.6.9803014. [DOI] [PubMed] [Google Scholar]
- 32.Beeh KM, Beier J, Kornmann O, Buhl R. Sputum matrix metalloproteinase-9, tissue inhibitor of metalloprotinease-1, and their molar ratio in patients with chronic obstructive pulmonary disease, idiopathic pulmonary fibrosis and healthy subjects. Respir Med. 2003;97:634–639. doi: 10.1053/rmed.2003.1493. [DOI] [PubMed] [Google Scholar]
- 33.Infante-Duarte C, Horton HF, Byrne MC, Kamradt T. Microbial lipopeptides induce the production of IL-17 in Th cells. J Immunol. 2000;165:6107–6115. doi: 10.4049/jimmunol.165.11.6107. [DOI] [PubMed] [Google Scholar]
- 34.Linden A. Role of interleukin-17 and the neutrophil in asthma. Int Arch Allergy Immunol. 2001;126:179–184. doi: 10.1159/000049511. [DOI] [PubMed] [Google Scholar]
- 35.Stark MA, et al. Phagocytosis of apoptotic neutrophils regulates granulopoiesis via IL-23 and IL-17. Immunity. 2005;22:285–294. doi: 10.1016/j.immuni.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 36.Zhu Z, et al. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J Clin Invest. 1999;103:779–788. doi: 10.1172/JCI5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ideker T, Thorsson V, Siegel AF, Hood LE. Testing for differentially-expressed genes by maximum-likelihood analysis of microarray data. J Comput Biol. 2000;7:805–817. doi: 10.1089/10665270050514945. [DOI] [PubMed] [Google Scholar]
- 38.Dong C, et al. ICOS co-stimulatory receptor is essential for T-cell activation and function. Nature. 2001;409:97–102. doi: 10.1038/35051100. [DOI] [PubMed] [Google Scholar]