A direct role for GRASP65 as a mitotically regulated Golgi stacking factor (original) (raw)
Abstract
Cell-free assays that mimic the disassembly and reassembly cycle of the Golgi apparatus during mitosis implicated GRASP65 as a mitotically regulated stacking factor. We now present evidence that GRASP65 is directly involved in stacking Golgi cisternae. GRASP65 is the major phosphorylation target in rat liver Golgi membranes of two mitotic kinases, cdc2–cyclin B and polo-like kinases, which alone will unstack Golgi membranes, generating single cisternae. Mitotic cells microinjected with antibodies to GRASP65 fail to form proper Golgi stacks after cell division. Beads coated with GRASP65 homodimers form extensive aggregates consistent with the formation of trans oligomers. These can be disaggregated using purified cdc2–cyclin B1 and polo-like kinases, and re-aggregated after dephosphorylation of GRASP65. Together, these data demonstrate that GRASP65 has the properties required to bind surfaces together in a mitotically regulated manner.
Keywords: cdc2 kinase/Golgi/phosphorylation/polo-like kinase/stacking
Introduction
The central and unique feature of the Golgi apparatus in most eukaryotic cells is the stack of flattened cisternal membranes with dilated rims (Rambourg and Clermont, 1997). The stack carries out post-translational processing of newly synthesized proteins as they pass through this organelle after assembly in the endoplasmic reticulum (ER) (Farquhar and Palade, 1981). Processing enzymes include those involved in modifying bound oligosaccharides, and these enzymes are arranged across the stack (in the cis to trans direction) in the order in which they function (Kornfeld and Kornfeld, 1985). The stack probably improves the efficiency of post-translational modification by concentrating subsets of these enzymes in individual cisternal compartments so that they can work more optimally, and it may improve the rate of transport through the stack by facilitating the speed of transfers between cisternae (Shorter and Warren, 1999). The mechanism that organizes Golgi cisternae into stacks is, however, poorly understood and is of central importance to studies of Golgi biogenesis.
Proteins involved in stacking Golgi cisternae were first identified using cell-free systems that exploited the mitotic fragmentation of Golgi membranes (Rabouille et al., 1995b). Fragmentation of the Golgi apparatus in animal cells occurs at the onset of mitosis as part of a process that is thought to aid the partitioning of these membranes between daughter cells. At the end of mitosis, reassembly of stacked cisternal structures occurs in each daughter cell (Souter et al., 1993). Mimicking this process in the test-tube led to the identification of GRASP65, a protein that was exposed and accessible to the alkylating reagent _N_-ethylmaleimide (NEM) only after unstacking of Golgi membranes in the presence of mitotic cytosol (MC) from HeLa cells (Barr et al., 1997). Antibodies to GRASP65 inhibited re-stacking of mitotic Golgi fragments (MGFs), as did a soluble form of GRASP65, though not after pre-treatment with NEM (Shorter et al., 1999). This soluble form was generated by removing the N-terminal signal for addition of myristic acid which helps anchor GRASP65 to Golgi membranes, as do certain members of the p24 family of putative cargo receptors and perhaps other proteins (Kuo et al., 2000; Barr et al., 2001). GRASP65 has also been implicated in cisternal stacking in vivo by recent studies on apoptosis (Lane et al., 2002). Cleavage of GRASP65 by caspase-3 correlates with Golgi fragmentation, and this is inhibited, at least partially, by expression of a caspase-resistant form of GRASP65.
GRASP65 is located in cis Golgi membranes, whereas a second member of the GRASP family, GRASP55, is located more towards the middle of the Golgi stack (Shorter et al., 1999). These locations argue that the GRASP family of proteins help determine stacking of different cisternal layers (Pfeffer, 2001). Both GRASPs are bound to members of the golgin family of coiled-coil proteins. GRASP65 is bound to the C-terminal domain of GM130 via a PDZ-like domain, whereas GRASP55 is bound to golgin-45 (Barr et al., 1998; Short et al., 2001). GM130 is thought to provide the base of a tether that anchors COPI vesicles to Golgi membranes. Giantin (another golgin) in the vesicle is linked to GM130 by a bridging molecule, p115 (Lowe et al., 1998; Sonnichsen et al., 1998). This tether is also involved in Golgi stacking, at least in vitro, helping to bring cisternal membranes together for subsequent binding by GRASP65 (Shorter and Warren, 1999).
GRASP proteins are targets of mitotic kinases. GRASP65 is phosphorylated both in vivo and in vitro by cdc2/B1 (cdc2 complexed with cyclin B1) and polo-like (plk) kinases (Lin et al., 2000). GRASP55 is phosphorylated in vitro by ERK2 (Jesch et al., 2001). What remains unclear is the consequence of these phosphorylation events. Mitotic fragmentation involves vesiculation and tubulation of cisternae as well as cisternal unstacking (Misteli and Warren, 1995). It is not clear whether GRASP phosphorylation leads primarily to cisternal unstacking or affects one or more of the other processes. It is also unclear whether GRASPs play a direct role in cisternal stacking or an indirect role via other, as yet uncharacterized, stacking factors.
To address these issues, we have used our cell-free assay and recombinant mitotic kinases to dissect out the role played by GRASP65 phosphorylation. We then used mitotic cells microinjected with antibodies to GRASP65 to test the role of GRASP65 in the reassembly of Golgi stacks after cell division. We have also used recombinant GRASP65 alone to determine whether it has the capacity to bind surfaces together. Our data show that GRASP65 has the properties required of a mitotically regulated stacking protein.
Results
GRASP65 is the major target of cdc2/B1 and plk kinases on Golgi membranes
Our earlier work had shown that GRASP65 is a prominent phosphorylation target on rat liver Golgi (RLG) membranes for mitotic kinases from HeLa cell cytosol (Barr et al., 1997). Subsequent work by Erikson and colleagues using cells transfected with cDNA encoding GRASP65 identified cdc2/B1 and plk as the two mitotic kinases responsible for almost all of this phosphorylation (Lin et al., 2000). The question then was whether endogenous GRASP65 in Golgi membranes was the main target of these two kinases.
Purified RLG membranes were incubated with MC from HeLa cells in the presence of [γ-32P]ATP, and labeled proteins were visualized by SDS–PAGE and autoradiography. As shown in Figure 1A (lane 5), 8–10 proteins were clearly labeled that were not labeled using interphase cytosol (IC, lane 4). A similar number were detected when RLG membranes were treated with purified, recombinant cdc2/B1 and plk kinases, and most were identical to those phosphorylated by MC (Figure 1A, compare lanes 3 and 5). Importantly, the most strongly labeled protein had a mol. wt of 65–70 kDa, the same as that for GRASP65. Essentially all of this phosphorylated protein could be removed by antibodies to GRASP65, which confirmed its identity (Figure 1B; Supplementary figure 1 available at The EMBO Journal Online) and showed that it was the major phosphorylated protein in Golgi membranes. Note that GM130 co-immunoprecipitated with GRASP65, consistent with their known interaction with each other (Barr et al., 1997, 1998).
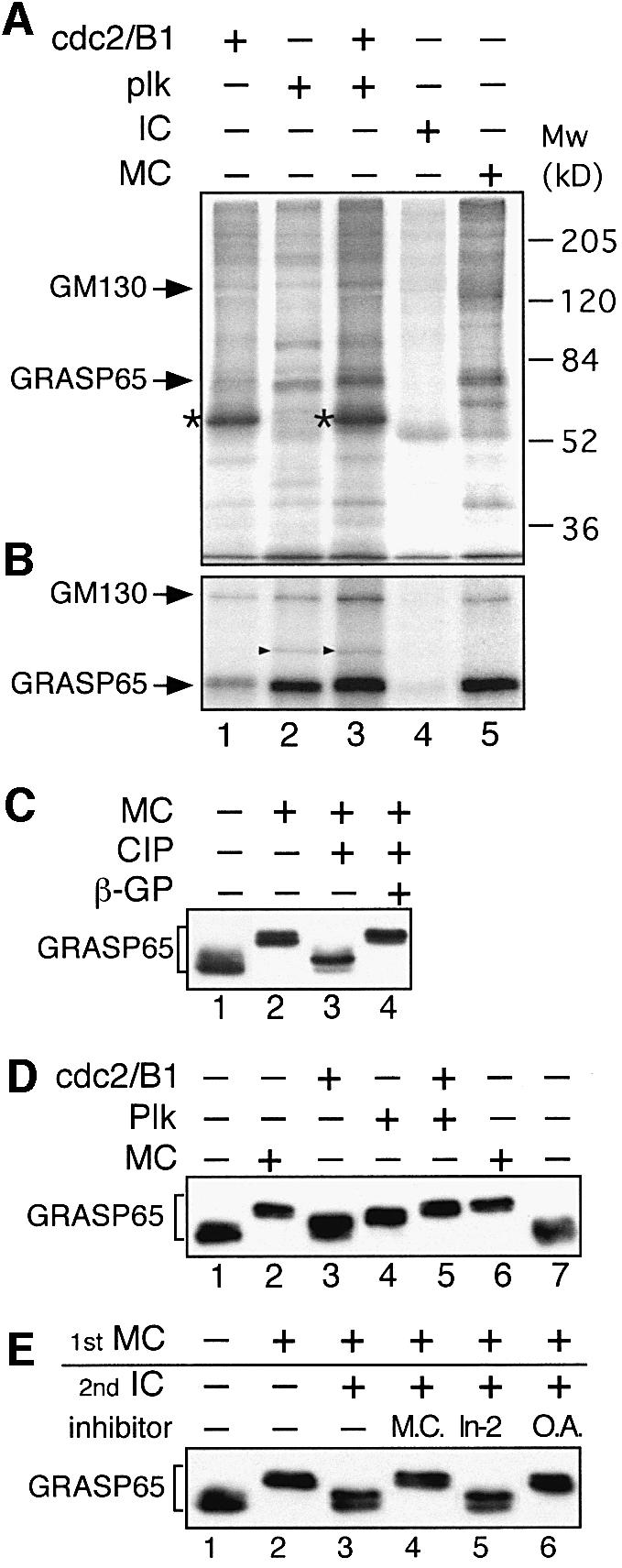
Fig. 1. GRASP65 is the major target of cdc2/B1 and plk kinases on Golgi membranes. (A) Membranes were incubated in the presence of [γ-32P]ATP with purified cdc2/B1 (lane 1), plk (lane 2), cdc2/B1 and plk (lane 3), interphase cytosol (IC, lane 4) or mitotic cytosol (MC, lane 5). Kinases and MC were adjusted to equivalent levels of kinase activity. The membranes were isolated and analyzed by SDS–PAGE followed by autoradiography. Arrows indicate phosphorylated GM130 and GRASP65. The strongly labeled protein in lanes 1 and 3 (asterisks) is cyclin B1. (B) Membrane samples from (A) were solubilized and GRASP65 immunoprecipitated followed by SDS–PAGE and autoradiography. Phosphorylated GM130 co-precipitated with phosphorylated GRASP65. The additional band in lanes 2 and 3 is GST–plk (arrowheads) which co-precipitated with GRASP65. (C) Detection of phosphorylated GRASP65 by a bandshift assay. RLG membranes were treated with either buffer (lane 1) or MC (lanes 2–4). Membranes were isolated and treated with alkaline phosphatase (CIP) in the absence (lane 3) or presence (lane 4) of β-glycerophosphate (β-GP), and analyzed by immunoblotting. (D) RLG membranes were incubated with the indicated kinases and immunoblotted for GRASP65. Note that the combination of cdc2/B1 and plk kinases phosphorylated GRASP65 to a similar extent as MC. (E) Phosphorylation and dephosphorylation of GRASP65 by MC and IC. RLG membranes were first incubated with MC (lanes 2–6) and then IC (lanes 3–6), in the absence or presence of microcystin (M.C., lane 4), Inhibitor-2 (In-2, lane 5) or okadaic acid (O.A., lane 6), followed by immunoblotting for GRASP65. Note that microcystin and okadaic acid inhibited dephosphorylation by IC, implicating PP2A in this process.
GRASP65 was phosphorylated by recombinant cdc2/B1 (Figure 1A and B, lane 1) and even more strongly by recombinant plk (Figure 1A and B, lane 2). Both kinases together labeled GRASP65 to a similar extent to that obtained using MC, which had the same kinase activity (Figure 1A and B, compare lanes 3 and 5). GM130 was also labeled by both kinases (Figure 1A and B, lanes 1–3), although the labeling was much weaker than for GRASP65, consistent with it having far fewer phosphorylation sites. GM130 has a single phosphorylation site for cdc2/B1 (Lowe et al., 1998) and a different one (or ones) for plk (not shown). Kinase-dead forms of cdc2 and plk showed no activity towards GRASP65 or GM130, and neither did active kinases pre-treated with kinase inhibitors such as olomoucine, roscovitine and staurosporine (not shown). Cyclin B2 in association with cdc2 labeled the same Golgi proteins as cyclin B1, with the same efficiency (not shown).
Phosphorylation of GRASP65 was also analyzed using a bandshift assay. Treatment of RLG membranes with MC led to a sharp increase in molecular weight on SDS–PAGE (Figure 1C, lane 2). This shift was caused by phosphorylation, since treatment of MGFs with calf intestine alkaline phosphatase (CIP), a non-specific protein phosphatase, restored the molecular weight almost back to its original value (lane 3). Restoration was abolished when β-glycerophosphate, a general phosphatase inhibitor, was included in the reaction (lane 4, β-GP). This result validates the bandshift assay as a means of studying the phosphorylation pattern of GRASP65.
We then tested the efficiency of GRASP65 phosphorylation by purified kinases and MC using the bandshift assay. Incubation of RLG membranes with purified cdc2/B1 had a modest effect (Figure 1D, lane 3) compared with plk (lane 4), consistent with the relative contributions of these kinases in phosphorylating GRASP65 (Lin et al., 2000). Neither kinase alone, however, could increase the molecular weight to the extent observed with MC (lanes 2 and 6) even when higher levels were used (not shown). Only when the two kinases were added together was the increase similar to that obtained using MC (Figure 1D, compare lanes 5 and 6). Together, these data argue that cdc2/B1 and plk kinases can largely mimic MC in its effect on the phosphorylation pattern of Golgi proteins and that endogenous GRASP65 is the major phosphorylation target.
Phosphorylated GRASP65 could be dephosphorylated by IC. The increase in molecular weight observed after treatment of RLG membranes with MC (Figure 1E, lane 2) could be reversed by subsequent treatment of the isolated membranes with IC (lane 3), suggesting the presence of a phosphatase or phosphatases that normally complete the mitotic Golgi cycle. Microcystin inhibited dephosphorylation by IC (lane 4), suggesting that PP1 and/or PP2A was the responsible phosphatase. Inhibitor-2 (In-2), a specific peptide inhibitor of PP1, showed no effect (lane 5), eliminating PP1 as the phosphatase. This was confirmed using okadaic acid, which inhibits PP2A at much lower concentrations than PP1. Low concentrations of okadaic acid inhibited dephosphorylation of GRASP65 (lane 6).
Microinjection of purified cdc2/B1 leads to fragmentation of the Golgi apparatus and phosphorylation of GM130
To test the effect in vivo of the recombinant cdc2/B1 and plk kinases we had prepared, they were microinjected into normal rat kidney (NRK) cells. The cells were fixed 30 min after injection and double labeled for phosphorylated GM130 (Figure 2, left panels, PS-25) and for total GM130 (Figure 2, right panels, GM130). Co-injected biotinylated bovine serum albumin (BSA) was detected by fluorescently labeled NeutrAvidin to identify the injected cells (indicated by asterisks).

Fig. 2. Microinjection of cdc2/B1 but not plk kinases leads to GM130 phosphorylation and Golgi fragmentation. NRK cells were injected with cdc2/B1 (A), plk (B) or cdc2/B1 and plk (C). Biotinylated BSA was used as an injection marker (asterisks). After 30 min at 37°C, cells were fixed and double labeled with polyclonal antibodies to phosphorylated GM130 (PS-25, left panels) and a monoclonal antibody to GM130 (GM130, right panels). Bar, 10 µm.
Microinjection of cdc2/B1 led to GM130 phosphorylation and Golgi disassembly. More than 98% of the injected cells showed phosphorylation of GM130, and 87% showed fragmentation of the Golgi manifested as breakdown of the juxta-nuclear ribbon into punctate structures (Figure 2A, cdc2/B1, Table I). Microinjection of cdc2 without cyclin B had no effect (Table I). These results are consistent with those obtained by Pines and colleagues using cells transfected with cDNAs encoding cdc2 and cyclin B (Draviam et al., 2001).
Table I. Quantitation of the kinase microinjected NRK cells.
| Protein injected | Cells counted | Cells with phosphorylated GM130 | Cells with fragmented Golgi |
|---|---|---|---|
| Cdc2 | 409 | 0% | 0% |
| Cdc2/B1 | 218 | 98% | 87% |
| plk | 330 | 0% | 0% |
| Cdc2/B1 + plk | 215 | 94% | 78% |
When cells were microinjected with plk, neither phosphorylation of GM130 at Ser25 nor breakdown of the Golgi ribbon was detected at the resolution afforded by light microscopy (Figure 2B, plk), consistent with work by Malhotra and colleagues using permeabilized cells (Sutterlin et al., 2001). Plk does phosphorylate GM130 in vitro (Figure 1A and B) but not at Ser25, the site phosphorylated by cdc2/B1, and the one recognized by the anti-phosphoserine antibodies, PS-25.
When both cdc2/B1 and plk kinases were microinjected, the effect was very similar to injection of cdc2/B1 alone. GM130 was phosphorylated in 94% of the injected cells, and the Golgi ribbon broke down in 78% of them (Figure 2C, cdc2/B1 + plk, Table I). This slightly lower percentage was probably due to the lower concentration of the cdc2/B1 protein injected. Other mitotic phenomena were also detected, including condensation of the chromosomal DNA, breakdown of the nuclear envelope, rearrangement of the microtubule cytoskeleton and rounding up of the cells (not shown).
We also eliminated the possibility that any of these effects were the consequence of triggering apoptosis. Injected cells were not labeled by antibodies to cleaved PARP, an apoptotic marker (Lane et al., 2002) (Supplementary figure 2). Apoptotic cells also degrade GRASP65 so that this protein is no longer recognized by a monoclonal anti-GRASP65 antibody (Lane et al., 2002). In all cases, injected cells could be labeled with this antibody (Supplementary figure 2).
Regulation of cisternal stacking by protein phosphorylation
The consequence of phosphorylation by cdc2/B1 and plk kinases on Golgi structure was tested using isolated rat liver Golgi stacks. These were treated with purified cdc2/B1, plk or both kinases. Using the intersection method for quantitation (Rabouille et al., 1995b), 55 ± 7% of the cisternae were found in stacks in starting Golgi membranes (Figure 3A and F) and this was not significantly changed when they were treated with buffer alone (not shown). After treatment with cdc2/B1, many single cisternae were generated (Figure 3B) and the percentage of stacked cisternae dropped to 25 ± 3% (Figure 3F), suggesting that partial unstacking had occurred. When the cdc2/B1 inhibitor roscovitine was added, or when Golgi stacks were treated with the kinase-dead form of cdc2/B1, cyclin B1 or cdc2 alone, the Golgi cisternae remained stacked (not shown).

Fig. 3. Purified cdc2/B1 and plk kinases unstack Golgi cisternae. (A–E) RLG stacks were either left untreated (A), or treated with cdc2/B1 (B), plk (C) or both (D) at 37°C for 20 min. Membranes were re-isolated after kinase treatment (D) and further treated with IC for 60 min at 37°C (E). Membranes were fixed and processed for EM. Bar, 0.5 µm. (F) Quantitation of (A–E), by the intersection method, to estimate the percentage of single or stacked cisternal membranes. Results represent the mean of three independent experiments ±SEM. Note the increased number of single cisternae after kinase treatments.
A similar effect on unstacking was detected when Golgi membranes were treated with plk (Figure 3C), the percentage of stacked cisternae dropping from 55 ± 7 to 27 ± 3% (Figure 3F). The kinase-dead form of plk had no effect (not shown). When the Golgi stacks were treated with both cdc2/B1 and plk (Figure 3D), the percentage of stacked cisternae dropped even lower, to 11 ± 4% (Figure 3F), effectively converting most of the stacks into single cisternae. About 70% of total membranes were present in cisternae irrespective of the treatment, showing that the effect of these two kinases was restricted to unstacking.
We then tested whether single cisternae could re-stack, after dephosphorylation of GRASP65 using IC. When single cisternae generated by treatment with both recombinant kinases were incubated with IC, Golgi stacks were rebuilt (Figure 3E). Quantitation showed that the percentage of stacked cisternae rose from 11 ± 4 to 42 ± 3%, similar to the 55 ± 7% found in starting membranes (Figure 3F). These results suggest that the phosphorylation cycle of GRASP65 correlates with the cisternal stacking cycle.
Microinjection of mitotic cells with antibodies to GRASP65
To test the role of GRASP65 in vivo, affinity-purified antibodies, or non-specific IgG were microinjected into NRK cells in metaphase. After further incubation to allow the cells to complete cell division, the injected cells were processed for electron microscopy (EM) (Figure 4).

Fig. 4. Antibodies to GRASP65 prevent proper re-stacking of Golgi membranes in post-mitotic cells. NRK cells in metaphase were microinjected with affinity-purified, anti-GRASP65 antibodies (A and C–E) or non-specific rabbit IgGs as a control (B and F). After completion of cell division, the injected cells were processed for EM. Results of two individual experiments are shown in (A and B) and (C–F), respectively. Note the properly assembled stacks in cells injected with IgGs (B and F) and the disorganized Golgi structures in cells injected with antibodies to GRASP65 (A and C–E). Bar, 0.5 µm.
Microinjection of IgG had little, if any, discernible effect on the reassembly of Golgi stacks. Representative images in Figure 4B and F show closely apposed and flattened cisternal membranes, with 4–6 cisternae/stack, and ribbons of linked stacks (Figure 4F). In marked contrast, microinjection of anti-GRASP65 antibodies severely disrupted the reassembly of individual stacks. Structures ranged from tubulo-reticular networks (Figure 4A) to highly fenestrated, cisternal-like structures (Figure 4C). Stacked cisternae were observed, but they were often not properly aligned (Figure 4D and E) and the cross-sectional profiles were more reminiscent of swollen tubules, not flattened cisternae (Figure 4E). Quantitation showed that the percentage of stacked Golgi membranes was nearly 4-fold lower, 9.9 ± 2.7% (n = 26) in cells injected with anti-GRASP65 antibodies compared with 37 ± 7.0% (n = 25) in IgG-injected cells. This dramatic effect of anti-GRASP65 antibodies on the morphology of individual stacks did not extend to the overall organization of the Golgi ribbon, which appeared normal when visualized using antibodies to GM130, mannosidase II and TGN38 (data not shown). This implicates GRASP65 in the stacking of cisternae in individual stacks and not the overall ribbon-like structure.
GRASP65 forms homodimers
One possible mechanism for cisternal stacking is for GRASP65 in adjacent membranes to interact, forming oligomers that hold the cisternae together (Barr et al., 1998). Earlier work using gel filtration suggested that GRASP65 can form dimers or trimers, but the precise number of molecules in the complex was unclear (Barr et al., 1998). To determine the oligomeric state, GRASP65 tagged with maltose-binding protein (MBP–GRASP65) and His-tagged GRASP65 (His-GRASP65) were co-expressed in bacteria and the proteins purified using affinity columns.
As shown in Figure 5A, the ratio of MBP–GRASP65 to His-GRASP65 in the lysate was ∼1:3 (lane 1). After purification utilizing the His tag and nickel affinity chromatography, the ratio of the two proteins changed to ∼1:8 (Figure 5A, lane 2). When this sample was purified further on an amylose column utilizing the MBP tag, this ratio of the two tagged proteins in the complex was ∼1:1 (Figure 5A, lane 3). Quantitation of the blots from three independent experiments gave a mean ratio ± SEM of 1.06 ± 0.15.

Fig. 5. Recombinant GRASP65 forms stable dimers. (A and B) MBP-tagged GRASP65 and His-tagged GRASP65 were co-expressed in E.coli and purified sequentially on nickel followed by amylose columns (A), or the reverse (B). Samples were fractionated by SDS–PAGE. Note that the ratio of the two tagged proteins in the complex was ∼1:1, irrespective of the order of purification. (C) Purified heterodimers of MBP–GRASP65/His-GRASP65 bound to amylose beads were treated with buffer alone (lane 1), MC ( lane 2), MC and the general kinase inhibitor staurosporine (lane 3), IC (lane 4), cdc2/B1 (lane 5), plk (lane 6) or both (lane 7). Beads (bound, upper panel) or supernatant (unbound, lower panel) were analyzed by immunoblotting for GRASP65. The extra bands in lanes 6 and 7 are cGST–plk (arrowhead) and its fragments (asterisk). Note that none of the treatments changed the ratio of the two proteins in the bound or unbound fractions.
The purification was then carried out using the same two columns in the reverse order. Once again, the final ratio was ∼1:1 (Figure 5B, lane 3). This ratio was also unaffected by differences in the expression level of the two proteins, which ranged from 1:1 to 1:5 for MBP–GRASP65:His-GRASP65. This eliminates the possibility that the complex is a tetramer or higher, even-numbered oligomer. Such oligomers would contain the tagged GRASP65 proteins at a ratio reflecting their relative expression levels. The oligomeric state was also not the consequence of interactions between the tags since co-expressed MBP and His-GRASP65 did not co-purify. Taken together, these results strongly suggest that recombinant GRASP65 is a homodimer.
GRASP65 homodimers are resistant to treatment with mitotic kinases
If GRASP65 homodimers link adjacent cisternae, then we might expect them to be sensitive to treatment with mitotic kinases. To test this possibility, purified heterodimers containing MBP–GRASP65 and His-GRASP65 were bound to amylose beads and treated with either MC or IC from HeLa cells, or purified cdc2/B1 and/or plk kinases (Figure 5C). Both proteins shifted in molecular weight when treated with MC (Figure 5C, lane2), or purified cdc2/B1 and/or plk kinases (lane 5–7), showing that both proteins were phosphorylated. No shift was detected when the proteins were treated with IC (lane 4) or with MC treated with the general kinase inhibitor staurosporine (lane 3). Incubation of the beads under these conditions released some protein from the amylose beads, but the ratio of the two proteins, either bound to the beads or in the supernatant, remained unchanged irrespective of their phosphorylation state (Figure 5C). Further binding of the supernatant proteins to nickel or amylose beads showed that the released proteins were still heterodimers of the two tagged GRASP65s (not shown). These results show that mitotic phosphorylation of recombinant GRASP65 dimers does not break them down into monomeric units.
GRASP65 homodimers form higher order oligomers under interphase but not mitotic conditions
We then tested whether GRASP65 homodimers can oligomerize further to form complexes that might help stack cisternae. Purified His-GRASP65 was incubated with or without cdc2/B1 kinase and loaded onto glycerol gradients. After centrifugation and fractionation, GRASP65 was visualized by western blotting. In the absence of kinase, GRASP65 distributed throughout the gradient, with a peak near the top and smaller peaks in the long tail (Figure 6A, upper panel). After treatment with cdc2/B1 kinase, the profile was much simpler, with a single peak near the top of the gradient (fraction 3, Figure 6A, lower panel). These data show that GRASP65 forms a mixture of differently sized oligomers that apparently can be reduced to a single oligomeric complex in the presence of mitotic kinase.

Fig. 6. GRASP65 dimers form higher order oligomers in a cell cycle-dependent manner. (A) His-GRASP65 was incubated in the absence or presence of cdc2/B1, sedimented in glycerol gradients and fractions analyzed by western blotting. Note that the mixture of GRASP65 oligomers is reduced to a single oligomeric complex after treatment with mitotic kinase. (B) Separately expressed and purified MBP–GRASP65 and His-GRASP65 proteins were mixed and incubated in the presence of buffer (control, lanes 1–3), MC (lanes 4–6) or IC (lanes 7–9). The protein complex was then isolated using nickel beads. Equal portions of the input (I), unbound (U) or bound (B) fraction were analyzed by immunoblotting for GRASP65. Note the absence of MBP–GRASP65 in the bound complexes treated with MC (lane 6). (C) As in (B) except that purified dimers were incubated with cdc2/B1 and plk alone or in combination. Complexes were isolated on nickel beads (upper panel) or amylose beads (lower panel) and processed as in (B). Note that bound complexes under control conditions contained both tagged GRASP65 proteins (lane 3) whereas kinase treatment separated them (lanes 6, 9 and 12).
We also tested the oligomerization of GRASP65 using differently tagged dimers. MBP–GRASP65 and His-GRASP65 were expressed and purified separately in bacteria, so that each should form homodimers. The two proteins were then mixed and incubated with either buffer alone, MC or IC. Protein complexes were isolated using nickel beads, and bound proteins were analyzed by immunoblotting for GRASP65.
Under control conditions, almost all MBP–GRASP65 was brought down by nickel beads, showing that it had formed a complex with His-GRASP65 (Figure 6B, compare lanes 2 and 3). Similar results were obtained in the presence of IC, though some of the MBP–GRASP65 was present in the unbound fraction (Figure 6B, compare lanes 8 and 9). Treatment with MC shifted the molecular weight of both proteins, showing that phosphorylation had occurred (Figure 6B, compare lanes 1 and 4). Furthermore, almost all of the MBP–GRASP65 was present in the unbound fraction, leaving only His-GRASP65 bound to the nickel beads (Figure 6B, compare lanes 5 and 6). These data argue that mitotic phosphorylation of GRASP65 dimers prevents the formation of higher order oligomers.
These data were confirmed using the recombinant kinases. Separately purified MBP–GRASP65 and His-GRASP65 proteins were mixed and incubated together with purified cdc2/B1 and/or plk kinases. Protein complexes were isolated using either nickel (Figure 6C, upper panel) or amylose (lower panel) beads. Nickel beads brought down His-GRASP65 and associated MBP–GRASP65 under control conditions (lanes 1–3), but MBP–GRASP65 was almost all in the unbound fraction after treatment with any of the kinases, alone or in combination (lanes 4–12). The efficacy of the kinases was judged by the shift in molecular weight compared with control conditions. Similar results were obtained using amylose beads, though the efficiency of binding MBP–GRASP65 was lower. Nevertheless, His-GRASP65 only bound to the MBP–GRASP65 on the amylose beads under control conditions. After kinase treatment, His-GRASP65 was found almost exclusively in the unbound fraction. Together, these data argue that phosphorylation of GRASP65 dimers by cdc2/B1 and/or plk prevents the association of dimers into higher order oligomers.
Higher order oligomers are sufficient to cause aggregation of GRASP65-coated beads in a mitotically regulated manner
The next step was to test whether the higher order oligomers could act to link adjacent cisternal membranes. As a model system, we chose to coat beads with GRASP65 proteins and ask whether these would then aggregate in a manner dependent on GRASP65 oligomerization. We chose Dynal magnetic beads since coupled proteins are located exclusively on the surface. Purified His-GRASP65 or BSA (as a control) was coupled by direct cross-linking.
The coated beads were incubated with either IC or MC, or purified cdc2/B1 and plk kinases, or BSA (as a control). The beads were then transferred to glass slides and observed under bright-field illumination. As shown in Figure 7A, in the presence of BSA, beads coated with His-GRASP65 formed aggregates, whereas control beads formed none. Quantitation confirmed these observations, showing that 24% of His-GRASP65 beads were found in aggregates compared with 0.7% of control beads (Figure 7C). Aggregates were defined as clusters of five or more beads.

Fig. 7. GRASP65 can form trans oligomers. (A) Purified His-GRASP65 (upper panel), or BSA (as control, lower panel) was covalently coupled to Dynal beads and incubated with BSA, IC, MC or cdc2/B1 and plk (kinases). After incubation, the beads were placed on glass slides and random fields photographed. A representative image of each condition is shown. Bar, 500 µm. (B) As in (A) except that the GRASP65 beads were first aggregated using IC and then treated with either BSA, MC or a combination of cdc2/B1 and plk (kinases). Kinase-treated beads were treated further with IC (IC→kinases→IC). Bar, 500 µm. (C and D) Quantitation of (A) and (B) and MBP–GRASP65 beads. Note that aggregates only formed in the presence of buffer and IC and that these aggregates were disassembled reversibly by MC and recombinant kinases.
Aggregation was even more dramatic when the beads were incubated with IC. More than 91% of His-GRASP65-coated beads were found in the aggregates compared with 1% of the control beads (Figure 7C). When the beads were incubated in the presence of MC (Figure 7A, MC), the percentage of beads in aggregates was found to be very low, <3% for the His-GRASP65 beads compared with 0.9% for the control beads (Figure 7C, column 3). Similar results were also obtained using the purified kinases (Figure 7A, kinases). About 8% of the beads were found in the aggregates for His-GRASP65 beads, whereas no aggregates were detected for the control beads (Figure 7C, column 4).
Other controls were also carried out, treating His-GRASP65 beads with reagents known to inhibit cisternal stacking. NEM alkylates GRASP65 and prevents cisternal stacking (Barr et al., 1997). It also prevented the aggregation of the beads in the presence of IC. Only 28 ± 3% of NEM-treated beads aggregated in the presence of IC compared with 84 ± 5% in the absence of such treatment.
Anti-GRASP65 antibodies and soluble GRASP65 also inhibit cisternal stacking (Barr et al., 1997), and they inhibited the aggregation of His-GRASP65 beads. Aggregation in the presence of anti-GRASP65 antibodies was 28 ± 5% compared with 77 ± 4% for control antibodies. Only 9 ± 2% aggregated in the presence of soluble His-GRASP65 compared with 43 ± 6% in the presence of a control His-tagged protein, the tail and acidic domain of the tethering protein p115 (Dirac-Svejstrup et al., 2000).
The pattern of results was not affected by the nature of the tag attached to the N-terminus of GRASP65. MBP–GRASP65 beads gave results very similar to those obtained using His-GRASP65 beads (Figure 7C and D). The manner in which the proteins were coupled to the beads also had no effect. Antibodies to the tags gave the same pattern of results as cross-linking (not shown). The size of the beads was also unimportant, and differently sized beads gave rise to mixed aggregates (not shown). Taken all together, these data show that immobilized GRASP65 dimers can link structures by forming trans oligomers and that this is prevented by all treatments known to affect cisternal stacking in vitro.
Aggregates of GRASP65-coated beads can be disaggregated by mitotic kinases and thenre-aggregated
Since mitotic kinases act on pre-existing Golgi stacks to unstack them (Figure 3), we asked whether the GRASP65 bead aggregates could be disaggregated by mitotic kinases. To test this possibility, His-GRASP65 or control (BSA-coated) beads were first incubated with IC (Figure 7B, IC) then washed and incubated with buffer either containing BSA (IC→BSA), MC (IC→MC) or purified cdc2/B1 and plk (IC→kinases).
When treated with BSA, most of the aggregates remained, the percentage of His-GRASP65 aggregates falling from 91 to 73% (Figure 7B and D). In marked contrast, treatment with MC lowered the percentage of His-GRASP65 aggregates to only 12%. The recombinant kinases were even more effective, lowering the percentage of His-GRASP65 aggregates to a mere 1%. Less than 5% of the control beads were found in aggregates irrespective of the treatment (Figure 7B and D, BSA beads). A similar pattern of results was obtained using MBP–GRASP65 beads (Figure 7C and D).
Finally, we tested the possibility that the beads disaggregated by the two recombinant kinases could be re-aggregated after dephosphorylation. As shown in Figure 7B, treatment of beads with IC to dephosphorylate GRASP65 (Figure 1E) caused the beads to aggregate. The percentage aggregation rose from 1 ± 1% (after treatment with kinases) to 93 ± 5% (after treatment with IC). These data provide further corroboration that the system is mimicking the mitotic stacking cycle of Golgi cisternae mediated by the phosphorylation state of GRASP65.
Discussion
GRASP65 was first identified as a Golgi protein, whose function was sensitive to the alkylating agent, NEM, but only after disassembly of Golgi stacks in the presence of MC (Barr et al., 1997). Since NEM treatment of Golgi membranes after disassembly (but not before) was known to inhibit subsequent reassembly of stacked membranes (Rabouille et al., 1995a), this suggested that GRASP65 might be involved in the stacking process. This was confirmed by subsequent experiments using soluble, recombinant GRASP65 or GRASP65 antibodies, which inhibited cisternal stacking but not cisternal regrowth (Barr et al., 1997). Further study also showed that cleavage of GRASP65 during apoptosis facilitates Golgi fragmentation (Lane et al., 2002). These experiments did not, however, determine whether GRASP65 played a direct role in cisternal stacking, or aided the process indirectly through as yet unidentified stacking factors. The possibility of an indirect role was strengthened recently by work suggesting that GRASP proteins might act as a checkpoint control in budding yeast (Norman et al., 1999), which in animal cells might help prevent entry of cells into mitosis until after the Golgi ribbon is fragmented (Sutterlin et al., 2002).
We first studied the effect of cdc2/B1 and plk on RLG membranes. These two kinases are responsible for most of the mitotic phosphorylation of GRASP65 in cells transfected with cDNA encoding this protein (Lin et al., 2000). Using 32P labeling and bandshift assays, we showed that endogenous GRASP65 in Golgi membranes is phosphorylated by these two kinases almost as well as by using MC. Furthermore, GRASP65 is the major target of these two kinases in isolated RLG membranes. Most of the shift in molecular weight of endogenous GRASP65 was caused by plk, not cdc2/B1 (Figure 1D), in agreement with the experiments by Erikson and colleagues, mapping the different phosphorylation site (Lin et al., 2000). In contrast, recombinant GRASP65 was shifted more by cdc2/B1, not plk (Figures 5C and 6C). We do not yet understand the reason for this, though it might have to do with the absence of GM130, which is tightly bound to endogenous GRASP65, via a PDZ-like motif at the C-terminus (Barr et al., 1998).
We then studied the effect of these kinases on cisternal stacking. Purified RLG stacks treated with recombinant kinases released single cisternae. Either cdc2/B1 or plk were competent, but the two together had the greatest effect, lowering the percentage of stacked cisternae from 55 to 11%, a 5-fold decrease. Since these conditions of incubation did not lead to vesiculation or tubulation of cisternae (the percentage of membrane in cisternae remained unchanged at ∼70% irrespective of the treatment), these data show that treatment with cdc2/B1 and plk suffices for separation of stacked cisternae. The key role played by phosphorylation was demonstrated by the fact that kinase-dead forms of cdc2 and a range of kinase inhibitors prevented cisternal unstacking. Dephosphorylation by IC also led to re-stacking of single cisternae. The fact that GRASP65 is the major phosphorylation target in RLG membranes further argues that this protein mediates the effect triggered by kinases and phosphatases.
We next tested the role of GRASP65 in vivo, since its role as a stacking factor has relied solely on experiments using our cell-free reassembly assay (Barr et al., 1997). Antibodies were microinjected into NRK cells in the early phases of mitosis and the cells allowed to complete mitosis and cell division before analysis by electron microscopy. Microinjection of non-specific IgGs had no discernible effect on reassembly, whereas antibodies to GRASP65 severely disrupted this process. Qualitatively, the Golgi membranes were more swollen, fenestrated and improperly aligned. Quantitatively, the percentage of stacked membrane was nearly 4-fold lower than control cells. The fact that some stacking does occur might reflect the action of GRASP55, which stacks medial rather than cis cisternae (Shorter et al., 1999). The absence of any discernible effect on the Golgi ribbon at the light level suggests that the role of GRASP65 is restricted to the reconstruction of individual stacks.
We next tried to mimic stacking using purified GRASP65 so as to determine whether this protein alone could mediate this process. We first determined the oligomeric state of GRASP65 using differently tagged proteins. Co-expression showed that recombinant GRASP65 is dimeric, but these dimers could not be reduced to the monomeric state using either MC or recombinant kinases, suggesting that dimerization alone is unlikely to be the mechanism of Golgi stacking as previously speculated (Barr et al., 1998). We further showed, however, that these dimers could form higher order oligomers whose formation was sensitive to phosphorylation by MC and kinases. This suggested that GRASP65 dimers on one membrane might be able to interact with GRASP65 dimers on another and so stack cisternae.
We tested this idea using a model system comprising Dynal beads coated with recombinant GRASP65. This was attached to the surface of the beads either by cross-linking or by using antibodies to the tags. We used both MBP- and His-tagged GRASP65 to eliminate effects that might be due to the nature of the tags. In all cases, the results were the same. Beads coated with tagged GRASP65 formed extensive aggregates. Beads coated with BSA did not. Interestingly, the size of the aggregates formed with GRASP65 beads was much larger in the presence of IC than BSA. This suggests that there are components in IC that enhance the formation of aggregates, and this assay might provide the means of identifying them.
GRASP65 beads did not aggregate after treatments that were known to prevent cisternal stacking in vitro. Aggregation was inhibited after treatment with NEM or antibodies to GRASP65. Aggregation was also inhibited in the presence of soluble, recombinant GRASP65. Most importantly, aggregation of GRASP65 beads did not occur in the presence of MC or the two mitotic kinases. Aggregates formed in the presence of IC could even be disaggregated by treatment with either MC or the two kinases. It also proved possible to disaggregate the beads with the two kinases and then re-aggregate them using IC (Figure 7B). This latter experiment exactly follows the conditions we used to unstack and re-stack RLG membranes (Figure 3), arguing that the aggregation/disaggregation cycle of the beads is faithfully recapitulating the stacking/unstacking cycle of Golgi membranes.
In conclusion, these data provide the best evidence to date that GRASP65 plays a direct role in cisternal stacking. We envisage dimers on adjacent cisternal membranes interacting to form higher order, trans oligomers. Direct phosphorylation by cdc2/B1 and plk kinases would break up the trans oligomers, leading to cisternal unstacking. Subsequent dephosphorylation would re-stack the cisternae. GRASP65 is restricted to cis cisternae, so this does not explain why unstacking is nearly complete (Barr et al., 1997). GRASP55, however, is located more towards the middle of the stack (Shorter et al., 1999) and can also be mitotically phosphorylated by several kinases including cdc2/B1 (Y.Wang and G.Warren, unpublished results). Unstacking might therefore be the consequence of phosphorylation of both GRASP family members. Further work will determine whether they are sufficient to stack Golgi cisternae, or just necessary.
Materials and methods
Reagents
All reagents were from Sigma Co., Roche or Calbiochem, unless otherwise stated. The following antibodies were used: polyclonal antibodies against GM130, phosphorylated GM130 (PS-25, M.Lowe) and MBP (NEB); monoclonal antibodies against GM130 (Transduction Laboratories), GRASP65 (F.Barr), α-tubulin (K.Gull) and RGS-His (Qiagen). Secondary antibodies were from Molecular Probes. Antibodies against GST–GRASP65 (amino acids 202–451) were raised in rabbits and purified using His-GRASP65.
Purification of kinases
Baculoviruses encoding human cyclin B1, cyclin B2, cyclin A, cdc2 wild-type (cdc2WT), cdc2 kinase-dead form (cdc2KD, K33R) and cdc2 constitutively active form (cdc2AF, T14A/Y15F) were kindly provided by M.Jackman (Cambridge, UK). His-tagged human cdc2–cyclin complexes, and single cyclins or cdks were expressed in Hi-5 cells using a baculoviral system (Krude et al., 1997) and purified on nickel (Ni-NTA) columns. The constitutively active form of cdc2–cyclin B1complex (cdc2/B1) was used in most experiments. Histone H1 kinase activity was measured as described (Lowe et al., 1998).
Baculoviruses and plasmids encoding GST-tagged human plkWT (wild-type), plkKD (kinase-dead form, K82M) or plkΔC (amino acids 1–356) were kindly provided by Q.Y.Lin (Harvard). Proteins were expressed in Hi-5 insect cells or in BL21(DE3)CodonPlus-RIL bacteria (Stratagene) and purified using glutathione–Sepharose (Amersham-Phamacia) (Lin et al., 2000).
The specific activity of plk was measured using His-GRASP65 as the substrate. Plk in TBMD buffer (Lin et al., 2000) was mixed with 0.5 µCi/µl [γ-32P]ATP, 0.2 µg/µl His-GRASP65 and 25 µM ATP. After 30 min at 37°C, His-GRASP65 was pulled down by nickel beads and analyzed by SDS–PAGE and phosphoimaging.
Phosphorylation and dephosphorylation of Golgi proteins
A 5 µg aliquot of RLG was mixed with 500 µg of HeLa cytosol, or equivalent kinase activity as described (Lowe et al., 1998). For [γ-32P]ATP labeling, 0.5 µCi/µl of [γ-32P]ATP and 5 µM ATP were used. After 60 min at 37°C, the membranes were pelleted through 0.4 M sucrose at 55 000 r.p.m. for 30 min in a TLA55 rotor (Beckman). Samples were analyzed by immunoprecipitation, SDS–PAGE, autoradiography and/or phosphoimaging.
Phosphorylation of GRASP65 was also analyzed by a bandshift assay. A 5 µg aliquot of MGFs was treated with 20 U of CIP or 100 µg of IC at 37°C for 60 min and analyzed by immunoblotting. In some reactions, phosphatase inhibitor β-glycerophosphate (50 mM), microcystin LR (0.5 µM), In-2 (0.5 µM) or okadaic acid (0.1 µM) was included.
Microinjection experiments
NRK cells were injected with: (i) 4.8 mg/ml plk; (ii) 0.7 mg/ml cdc2 (without cyclin); (iii) 2.5 mg/ml cdc2/B1; and (iv) 1.2 mg/ml cdc2/B1 + 2.4 mg/ml plk. Biotinylated BSA followed by Alexa 350 NeutrAvidin was used as the co-injection marker. After 30 min at 37°C, cells were processed for immunofluorescence (Seemann et al., 2000).
Cells in metaphase were injected with affinity–purified GRASP65 antibodies (4 mg/ml) or non-specific rabbit IgGs, and incubation continued for 3 h. Fluorescein isothiocyanate (FITC)–dextran was co-injected so that the divided cells could be identified under the fluorescence microscope and the uninjected cells were removed by scraping with a microinjection needle. These divided cells were then processed for EM (Seemann et al., 2000).
Stacked Golgi membranes were quantitated using the intersection method (Rabouille et al., 1995b). Profiles of Golgi cisternae, vesicles and tubules were identified using morphological criteria (Souter et al., 1993) and were readily distinguishable from ER and organelles on the endocytic pathway. Stacked regions were defined as Golgi membranes ≤15 nm apart and ≥150 nm in length. This length was chosen to exclude tethered vesicles from the calculation.
Cell-free Golgi disassembly and reassembly assay
Reactions were performed with cytosol (Rabouille et al., 1995b) or equivalent kinase activities at 37°C for 20 min. For reassembly, MGFs were collected through 0.4 M sucrose in KHM buffer (Shorter and Warren, 1999), treated with IC at 37°C for 60 min and processed for EM. Membrane profiles were quantitated by the intersection method (Rabouille et al., 1995b).
GRASP65 oligomerization
GRASP65 cDNA in pMAL-2CX (NEB) and pET30a (Novagen) were co-transformed into BL21(DE3)Gold bacteria. The proteins were purified on nickel (Qiagen) or amylose (NEB) agarose. This was followed by a second purification on the alternative resin. Purified MBP–GRASP65 and His-GRASP65 dimers on nickel or amylose beads were incubated with MC, IC or kinases for 60 min at 37°C.
To test oligomerization of GRASP65, separately expressed and purified MBP–GRASP65 and His-GRASP65 were treated with IC, MC or kinases. The protein complex was isolated using amylose beads and analyzed by immunoblotting.
For sedimentation analysis, His-GRASP65 purified on a nickel column followed by gel filtration (Superose-6 HR10/30, Amersham-Phamacia) was incubated at 37°C for 2 h in gradient buffer [25 mM HEPES-KOH pH 7.4, 150 mM KCl, 5 mM MgCl2, 1 mM dithiothreitol (DTT)] with or without cdc2/B1. Glycerol gradients [10–35% (w/v)] were centrifuged (2 h, 65 000 r.p.m., VTI65.1 rotor) and fractions analyzed by western blotting.
Aggregation of GRASP65-coated beads
His- and MBP–GRASP65, or BSA were clarified by ultracentrifugation and cross-linked to Dynalbeads M500. The beads were incubated for 60 min at 37°C with IC (+staurosporine), MC or kinases (+microcystine LR) in KHM with nocodazole and observed under a microscope. NEM treatments were carried out as described (Barr et al., 1997). In some experiments, bead aggregates were washed with KHM buffer, incubated with IC at 37°C for 60 min, washed again and treated further with MC or kinases at 37°C for 60 min.
For quantitation, single beads or aggregated beads from 12 random pictures of each sample were counted. Aggregates were defined as those with ≥5 beads since all controls had ≤4 beads. For large aggregates, only visible, surface beads were counted; therefore, the number of beads in these aggregates was underestimated.
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We thank M.Jackman for the baculoviruses, cyclins and cdc2, Q.Y.Lin for the baculoviruses and cDNAs for plk, M.Lowe, K.Gull and T.Hunt for antibodies, J.Lee for constructing the MBP-GRASP65 plasmid, M.Beard and H.Meyer for critical reading of the manuscript, C.Horensavitz for help with the EM, and the entire Warren/Mellman group for discussions and support. This work was funded by the NIH.
References
- Barr F.A., Puype,M., Vandekerckhove,J. and Warren,G. (1997) GRASP65, a protein involved in the stacking of Golgi cisternae. Cell, 91, 253–262. [DOI] [PubMed] [Google Scholar]
- Barr F.A., Nakamura,N. and Warren,G. (1998) Mapping the interaction between GRASP65 and GM130, components of a protein complex involved in the stacking of Golgi cisternae. EMBO J., 17, 3258–3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr F.A., Preisinger,C., Kopajtich,R. and Korner,R. (2001) Golgi matrix proteins interact with p24 cargo receptors and aid their efficient retention in the Golgi apparatus. J. Cell Biol., 155, 885–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirac-Svejstrup A.B., Shorter,J., Waters,M.G. and Warren,G. (2000) Phosphorylation of the vesicle-tethering protein p115 by a casein kinase II-like enzyme is required for Golgi reassembly from isolated mitotic fragments. J. Cell Biol., 150, 475–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draviam V.M., Orrechia,S., Lowe,M., Pardi,R. and Pines,J. (2001) The localization of human cyclins B1 and B2 determines CDK1 substrate specificity and neither enzyme requires MEK to disassemble the Golgi apparatus. J. Cell Biol., 152, 945–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farquhar M.G. and Palade,G.E. (1981) The Golgi apparatus (complex)—(1954–1981)—from artifact to center stage. J. Cell Biol., 91, 77s–103s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jesch S.A., Lewis,T.S., Ahn,N.G. and Linstedt,A.D. (2001) Mitotic phosphorylation of Golgi reassembly stacking protein 55 by mitogen-activated protein kinase ERK2. Mol. Biol. Cell, 12, 1811–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornfeld R. and Kornfeld,S. (1985) Assembly of asparagine-linked oligosaccharides. Annu. Rev. Biochem., 54, 631–664. [DOI] [PubMed] [Google Scholar]
- Krude T., Jackman,M., Pines,J. and Laskey,R.A. (1997) Cyclin/Cdk-dependent initiation of DNA replication in a human cell-free system. Cell, 88, 109–119. [DOI] [PubMed] [Google Scholar]
- Kuo A., Zhong,C., Lane,W.S. and Derynck,R. (2000) Transmembrane transforming growth factor-α tethers to the PDZ domain-containing, Golgi membrane-associated protein p59/GRASP55. EMBO J., 19, 6427–6439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane J.D., Lucocq,J., Pryde,J., Barr,F.A., Woodman,P.G., Allan,V.J. and Lowe,M. (2002) Caspase-mediated cleavage of the stacking protein GRASP65 is required for Golgi fragmentation during apoptosis. J. Cell Biol., 156, 495–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C.Y., Madsen,M.L., Yarm,F.R., Jang,Y.J., Liu,X. and Erikson,R.L. (2000) Peripheral Golgi protein GRASP65 is a target of mitotic polo-like kinase (Plk) and Cdc2. Proc. Natl Acad. Sci. USA, 97, 12589–12594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe M., Rabouille,C., Nakamura,N., Watson,R., Jackman,M., Jamsa,E., Rahman,D., Pappin,D.J. and Warren,G. (1998) Cdc2 kinase directly phosphorylates the _cis_-Golgi matrix protein GM130 and is required for Golgi fragmentation in mitosis. Cell, 94, 783–793. [DOI] [PubMed] [Google Scholar]
- Misteli T. and Warren,G. (1995) A role for tubular networks and a COP I-independent pathway in the mitotic fragmentation of Golgi stacks in a cell-free system. J. Cell Biol., 130, 1027–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman T.C. et al. (1999) Genetic selection of peptide inhibitors of biological pathways. Science, 285, 591–595. [DOI] [PubMed] [Google Scholar]
- Pfeffer S.R. (2001) Constructing a Golgi complex. J. Cell Biol., 155, 873–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabouille C., Levine,T.P., Peters,J.M. and Warren,G. (1995a) An NSF-like ATPase, p97 and NSF mediate cisternal regrowth from mitotic Golgi fragments. Cell, 82, 905–914. [DOI] [PubMed] [Google Scholar]
- Rabouille C., Misteli,T., Watson,R. and Warren,G. (1995b) Reassembly of Golgi stacks from mitotic Golgi fragments in a cell-free system. J. Cell Biol., 129, 605–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambourg A. and Clermont,Y. (1997) Three-dimentional structure of the Golgi apparatus in mammalian cells. In Berger,E.G. and Roth,J. (eds), The Golgi Apparatus. Birkhauser Verlag, Basel, Switzerland, pp. 37–61. [Google Scholar]
- Seemann J., Jokitalo,E.J. and Warren,G. (2000) The role of the tethering proteins p115 and GM130 in transport through the Golgi apparatus in vivo. Mol. Biol. Cell, 11, 635–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short B., Preisinger,C., Korner,R., Kopajtich,R., Byron,O. and Barr,F.A. (2001) A GRASP55–rab2 effector complex linking Golgi structure to membrane traffic. J. Cell Biol., 155, 877–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorter J. and Warren,G. (1999) A role for the vesicle tethering protein, p115, in the post-mitotic stacking of reassembling Golgi cisternae in a cell-free system. J. Cell Biol., 146, 57–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorter J., Watson,R., Giannakou,M.E., Clarke,M., Warren,G. and Barr,F.A. (1999) GRASP55, a second mammalian GRASP protein involved in the stacking of Golgi cisternae in a cell-free system. EMBO J., 18, 4949–4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnichsen B., Lowe,M., Levine,T., Jamsa,E., Dirac-Svejstrup,B. and Warren,G. (1998) A role for giantin in docking COPI vesicles to Golgi membranes. J. Cell Biol., 140, 1013–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souter E., Pypaert,M. and Warren,G. (1993) The Golgi stack reassembles during telophase before arrival of proteins transported from the endoplasmic reticulum. J. Cell Biol., 122, 533–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutterlin C., Lin,C.Y., Feng,Y., Ferris,D.K., Erikson,R.L. and Malhotra,V. (2001) Polo-like kinase is required for the fragmentation of pericentriolar Golgi stacks during mitosis. Proc. Natl Acad. Sci. USA, 98, 9128–9132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutterlin C., Hsu,P., Mallabiabarrena,A. and Malhotra,V. (2002) Fragmentation and dispersal of the pericentriolar Golgi complex is required for entry into mitosis in mammalian cells. Cell, 109, 359–369. [DOI] [PubMed] [Google Scholar]