DNA Array-Based Transcriptional Analysis of Asporogenous, Nonsolventogenic Clostridium acetobutylicum Strains SKO1 and M5 (original) (raw)
Abstract
The large-scale transcriptional program of two Clostridium acetobutylicum strains (SKO1 and M5) relative to that of the parent strain (wild type [WT]) was examined by using DNA microarrays. Glass DNA arrays containing a selected set of 1,019 genes (including all 178 pSOL1 genes) covering more than 25% of the whole genome were designed, constructed, and validated for data reliability. Strain SKO1, with an inactivated spo0A gene, displays an asporogenous, filamentous, and largely deficient solventogenic phenotype. SKO1 displays downregulation of all solvent formation genes, sigF, and carbohydrate metabolism genes (similar to genes expressed as part of the stationary-phase response in Bacillus subtilis) but also several electron transport genes. A major cluster of genes upregulated in SKO1 includes abrB, the genes from the major chemotaxis and motility operons, and glycosylation genes. Strain M5 displays an asporogenous and nonsolventogenic phenotype due to loss of the megaplasmid pSOL1, which contains all genes necessary for solvent formation. Therefore, M5 displays downregulation of all pSOL1 genes expressed in the WT. Notable among other genes expressed more highly in WT than in M5 were sigF, several two-component histidine kinases, spo0A, cheA, cheC, many stress response genes, fts family genes, DNA topoisomerase genes, and central-carbon metabolism genes. Genes expressed more highly in M5 include electron transport genes (but different from those downregulated in SKO1) and several motility and chemotaxis genes. Most of these expression patterns were consistent with phenotypic characteristics. Several of these expression patterns are new or different from what is known in B. subtilis and can be used to test a number of functional-genomic hypotheses.
The recent completion and tentative annotation of numerous genome sequences, and the many to follow, create both a wealth of new information for comparative genomic studies and enormous challenges. The latter include the need to develop fast if not high-throughput strategies to understand the major cellular programs of the newly sequenced organisms and to begin to assign functions to groups of genes or individual genes. These challenges are made especially difficult for the majority of cases where the genetics of the organism are not well developed, few genes have been transcriptionally examined or otherwise functionally assigned, mutants are scarce, and the closest related organisms are also minimally understood at the genomic level. The spore-forming strict anaerobe Clostridium acetobutylicum is typical of this situation. Its genome has been sequenced and computer annotated (18) and its physiology has been extensively studied, but only a small number of genes have been studied and functionally identified. The number of available mutants is small, and chromosomal integration for genetic studies remains a major hurdle. None of the other closely or distantly related clostridia is understood genetically (let alone genomically) any better than C. acetobutylicum, and thus, one has to rely on Bacillus subtilis as a prototypical organism for such studies. However, B. subtilis is not a strict anaerobe and is only distantly related to C. acetobutylicum. For example, although their sporulation and differentiation programs appear to be similar, many physiological and genetic differences exist (10, 18).
The objective of this study was to use DNA array-based large-scale transcriptional analysis in order to study two important mutants of C. acetobutylicum, SKO1 and M5, relative to the parent strain (wild type [WT]). SKO1 is the result of the chromosomal inactivation of the spo0A gene (10), which results in an asporogenous, filamentous, and solventogenesis-deficient phenotype. M5 is the result of the megaplasmid pSOL1 loss (3, 4) and is also asporogenous and nonsolventogenic. pSOL1 contains all essential genes for butanol and acetone formation. Although these two mutant strains have apparently similar phenotypes, they are genetically very different. Our aims include being able to relate gene-expression patterns to specific phenotypes and to discover gene expression differences between the two mutants but also to establish similarities to and differences from B. subtilis. An important aim is to assign functions to groups of or individual C. acetobutylicum genes and use this information to formulate specific hypotheses for further testing. DNA array-based analysis has been extensively used in human, yeast, and Escherichia coli systems but less so in B. subtilis (for example, see references 6 and 12) and not at all in clostridia or other anaerobes. We thus had first to develop and validate this high-throughput tool for the transcriptional analysis of C. acetobutylicum.
MATERIALS AND METHODS
Bacterial strains.
C. acetobutylicum ATCC 824 (American Type Culture Collection, Manassas, Va.) is the WT strain. Strain M5 is a degenerate strain lacking the pSOL1 megaplasmid (3). Mutant strain SKO1 (10) has a spo0A gene which has been disrupted with a macrolide-lincosamide-streptogramin B resistance marker.
Analytical methods.
Cell growth was determined by measuring the absorbance at 600 nm (_A_600) with a Thermo Spectronic (Rochester, N.Y.) BioMate3 spectrophotometer (26). Culture supernatants from the bioreactor samples were analyzed for acetate, butyrate, acetone, butanol, ethanol, acetoin, and glucose levels with a Waters (Milford, Mass.) high-pressure liquid chromatography system (2, 26).
Growth conditions and maintenance.
C. acetobutylicum strains were grown in an anaerobic chamber (Forma Scientific, Marietta, Ohio) at 37°C. Liquid cultures were grown in clostridium growth medium (CGM), and colonies were obtained from agar-solidified 2× YTG (28). WT liquid cultures were inoculated with a single colony, at least 4 days old, which had been heat shocked at 70°C for 7 min. Liquid cultures of the asporogenic M5 and SKO1 strains were inoculated with single colonies not older than 1 day, without heat shocking. The absence or presence of the pSOL1 megaplasmid in strains M5 and SKO1, respectively, was verified by monitoring amylase activity on 2× YTGMA plates (10, 20). Frozen stocks were prepared from cells at an _A_600 of 0.8 to 1.0 and were stored in CGM plus 20% glycerol at −85°C. SKO1 cultures were supplemented with 100 μg of erythromycin per ml unless otherwise noted.
Fermentation experiments.
WT strain ATCC 824 and strain M5 were grown as static flask cultures in 400 ml of CGM at 37°C. The static flasks were inoculated with 8 ml (1/50) of preculture at an _A_600 of 0.6. Bioreactor fermentations with pH controlled at ≥5.0 were carried out as previously described (26). The reactor medium (CGM) was supplemented with 75 μg of clarithromycin (Abbott Labs, Abbott Park, Ill.) per ml and 0.15% antifoam.
RNA sampling, isolation, and purification.
Cell pellets from 5 to 15 ml of culture were collected by centrifugation at 4°C and 5,000 × g for 10 min. Pellets were resuspended in 200 μl of SET buffer (25% sucrose, 50 mM Tris [pH 8], 50 mM EDTA [pH 8]) with 20 mg of lysozyme per ml, and the samples were incubated at 37°C for 5 min (10). Cold TRIzol reagent (1 ml; Invitrogen, Carlsbad, Calif.) was added, and the samples were vortexed for 30 s. The TRIzol samples were immediately frozen at −85°C, and the RNA was purified within 1 month to avoid degradation. For isolation and purification, the TRIzol samples were thawed at room temperature and diluted fivefold in ice-cold TRIzol up to 1 ml. Chloroform (200 μl) was added to 1 ml of the diluted TRIzol-treated samples, vortexed, and allowed to stand for 2 min at room temperature. The samples were centrifuged at 12,000 × g for 15 min at 4°C, and the aqueous phase was transferred to a fresh tube. Isopropanol (0.5 ml) was added, the tubes were inverted several times, and the samples were allowed to stand for 10 min and then centrifuged at 12,000 × g for 10 min at 4°C. The resulting pellet was washed with 75% RNase-free ethanol and spun at 8,000 × g for 4 min at 4°C. After drying for 10 min, the RNA was finally resuspended in RNase-free water and quantitated with a UV spectrophotometer (_A_260 and _A_280). Each sample was run on a 1.2% agarose gel to check for lack of RNA degradation. Samples were stored at −85°C.
Northern analysis.
RNA samples (20 μg) were used for Northern analysis as described previously (10, 16), with the following modifications. RNA was transferred from the formaldehyde gel to a Nytran membrane (Schleicher and Schuell, Keene, N.H.) with a Bio-Rad vacuum blotter by using the supplied protocol. Probes for spo0A and phosphotransbutyrylase-butyrate kinase (_ptb_-buk) were prepared from PCR fragments produced with the following primers: spo0A, 5′-GCCTGACCTTGTTGTTCTCG-3′ and 5′-CGTGACCATGCAACTTCAATA-3′; ptb-buk, 5′-TGCAGATGCTATTCTTGTTGG-3′ and 5′-TCATTTTTGTTTCATGGCTGTC-3′. Probes for detection of the thiolase (thl) and aldehyde/alcohol dehydrogenase-acetoacetyl-coenzyme A (CoA):acetate-butyrate:CoA transferase (_aad_-_ctfA_-ctfB) mRNA transcripts were prepared as previously described (10). Double-stranded DNA probes were purified by using a GFX DNA purification column (Amersham Biosciences, Piscataway, N.J.).
cDNA microarrays.
cDNA microarrays with spots representing 1,019 open reading frames (ORFs), approximately one-fourth of the C. acetobutylicum genome, were printed by using the TIGR protocol (11). Genes in this generation of arrays include, among others, all 178 pSOL1 ORFs, 123 DNA replication and repair genes (90% of the total of such genes as identified by genome annotation [18]), 97 cell division- and sporulation-related genes (92%), 85 carbohydrate and/or primary metabolism genes (31%), 67 energy production genes (52%), 63 outer membrane and cell envelope genes (36%), 48 lipid metabolism genes (80%), and 42 motility and chemotaxis genes (39%). A complete list can be found at http://www.chem-eng.northwestern.edu/Faculty/papou.html. PCR primers (MWG Biotech, High Point, N.C.) were designed (Integrated Genomics, Chicago, Ill.) to amplify gene fragments with an average size of approximately 470 bp, such that nonspecific hybridization on the DNA arrays is minimized. PCRs (volume, 60 μl) with approximately 4 μg of chromosomal template DNA were performed with AmpliTaq DNA polymerase (Applied Biosystems, Foster City, Calif.) according to the manufacturer's suggested protocol. The resulting PCR products were run on an agarose gel to verify that fragments were of the proper size and that only a single DNA product was produced. Reactions with multiple bands were repeated under more stringent conditions. Additionally, 48 products were randomly chosen and sequenced on a 377 ABI sequencer (Applied Biosystems). The PCR products were purified with GFX columns, eluted with 60 μl of water, dried in a vacuum centrifuge, redissolved in a 35% dimethyl sulfoxide solution, and spotted at least in triplicate on Corning (New York, N.Y.) CMT-GAPS or TeleChem (Sunnyvale, Calif.) ArrayIt SuperAmine glass microarray slides with a BioRobotics (Woburn, Mass.) MicroGrid II DNA arrayer (125-μm spot size with 200-μm spacing). Many genes involved in solventogenesis and sporulation are represented by as many as 12 spots. In addition to the 1,019 ORFs, 22 control genes (3 from Clostridium pasteurianum, 9 from Saccharomyces cerevisiae, and 10 from Arabidopsis thaliana [SpotReport array validation system; Stratagene, La Jolla, Calif.]) with no known homologies to the C. acetobutylicum genome sequence were spotted as negative controls (see http://www.chem-eng.northwestern.edu/faculty/papou.html for a complete list). After spotting, the slides were UV cross-linked (Stratagene cross-linker) and baked in an oven at 80°C for 2 to 4 h.
cDNA labeling and hybridization.
Labeled cDNA was synthesized by random hexamer-primed reverse transcription reactions in the presence of Cy3-dUTP or Cy5-dUTP by using Moloney murine leukemia virus (Promega, Madison, Wis.) or SuperScript II (Invitrogen) reverse transcriptase. Twelve micrograms of RNA was mixed with 2.4 μg of random hexamer primers (Roche, Indianapolis, Ind.), heated to 70°C for 10 min, and cooled on ice for 1 min. Unlabeled deoxynucleoside triphosphates (0.60 mM dATP, 0.15 mM dTTP, and 0.40 mM dGTP and dCTP), either Cy3- or Cy5-labeled dUTP (Amersham), 400 U of reverse transcriptase, 5× reverse transcription buffer, and 0.50 μl of SUPERaseIn (Ambion, Austin, Tex.) were added to a final volume of 25 μl. The samples were incubated at 42°C for 2 h. The reaction was stopped by the addition of 20 mM EDTA, and the RNA was degraded by the addition of NaOH (30 mM final concentration) followed by incubation at 70°C for 10 min. The mixture was cooled on ice and neutralized by adding HCl (30 mM final concentration). The labeled probe was purified with a GFX purification kit, and the DNA was eluted with 50 μl of Tris-EDTA (pH 8). The purified probe was dried to completion in a rotary SpeedVac and stored at −20°C until use.
For hybridizations, the spotted arrays were incubated in prehybridization buffer (5× SSC [1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate], 0.1% sodium dodecyl sulfate, 1% bovine albumin) at 42°C for 45 min. The slides were then washed by dipping five times in Millipore water and twice in isopropanol and were then allowed to air dry. Oppositely labeled dried probes were resuspended in 5 μl of Tris-EDTA (pH 8) and mixed. One microliter of sonicated salmon sperm DNA (10 mg/ml; Stratagene) was added, and the mixture was denatured at 95°C for 3 min. An equal volume of 2× hybridization buffer (10× SSC, 50% formamide, 0.2% sodium dodecyl sulfate) was added, and the sample was loaded onto the array under a LifterSlip (Erie Scientific, Portsmouth, N.H.). The slides were hybridized 18 h at 42°C in Corning hybridization chambers with 100 μl of 10× SSC to maintain humidity. After hybridization, the slides were washed with TeleChem ArrayIt DNA microarray wash buffers A, B, and C for 5 min in each buffer. The slides were dried by centrifugation for 5 min at 500 × g. The hybridized arrays were analyzed with a GSI Lumonics scanner and ScanArray software (Perkin-Elmer Life Sciences, Boston, Mass.). Spot intensities were quantitated with QuantArray Microarray analysis software (Perkin-Elmer Life Sciences).
Microarray data analysis.
The data were normalized and genes which showed significant differences in expression levels were determined by a novel normalization and gene selection method (30). All array data were subjected to a filtering criterion based on the spot intensity (signal) for a given channel (which was background subtracted and corrected for nonspecific binding) and the standard deviation of the local background (noise). The criterion is described by the following:
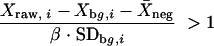
where _x_raw,i is the raw channel intensity, _x_bg,i is the local background for the given channel, _X_neg is the average nonspecific binding, β is a constant, and SDbg,i is the standard deviation for the local channel background. β was set to 1.96 so that the channel intensity (background and nonspecific binding corrected) was greater than the noise of the background at the 95% confidence level. This serves as a very conservative measure of the reliability of the intensity data, particularly at lower intensities. Spots which fail to meet the above criterion for both channels are ignored. Spots which fail to meet the criterion for one of the two channels are subjected to a stricter criterion for the second channel (β = 2.81 for 99.5% confidence). Those which fail to meet the more stringent criterion for the second channel are also ignored. For spots which pass the more stringent criterion, one can estimate the degree of overexpression (-fold) by dividing the intensity of the channel which exceeds the criterion by 1.96 times the standard deviation of the background for that channel. Data generated in this manner were utilized only to fill in any missing data and not for identification of differentially expressed genes.
Average linkage hierarchical clustering was performed with Cluster (5). Self-organizing-map (SOM) analysis (24) was performed with GeneCluster 2.0 (Whitehead Institute for Biomedical Research). Gene clusters were visualized in TreeView (5).
σ-factor and 0A box binding site and operon prediction.
The full set of C. acetobutylicum ATCC 824 intergenic regions was scanned for 0A boxes and sigma factor binding sites by using a dot plot-like strategy (9). Two mismatches were allowed for sigma factor binding sites, and only one was allowed for 0A boxes. Changes in the coding strand or the presence of a sigma factor consensus sequence was used to predict the start of transcriptional units. The following motifs (23) were utilized: σA, TTGACA(16-18)TATAAT; σD, TAAA(14-16)GCCGATAT; σE, ATA(16-18)CATACANT; σF, GYWTA(15)GGNRANANTW; σG, GNATR(15)CATNNTA, σH, RNAGGAWWW(11-12)RNNGAATWW; σL, TGGCA(5)CTTGCAT; spo0A, TGNCGAA; and reversed spo0A, TTCGNCA (the numbers in parentheses indicate spacing between nucleotide sequences; R = A or G; W = A or T; Y = C or T; N = A, C, G or T).
RESULTS
The complete set of DNA array expression data can be found at http://www.chem-eng.northwestern.edu/Faculty/papou.html.
Validation of the DNA array analysis protocol.
We used two independent strategies for validation of the DNA array analysis protocol. The first was based on the genes of the megaplasmid pSOL1, which is absent in strain M5 (4). In a comparative analysis of WT and M5, any gene which resides on the pSOL1 megaplasmid should fall into one of two categories: significantly upregulated in the WT or nondifferentially expressed. An Eisen plot (5) of the 56 pSOL1 genes which are differentially expressed at the 95% confidence level for at least two time points is shown in Fig. 1. The predominance of green in the Eisen plot, indicating higher expression in the WT, demonstrates the validity of the DNA arrays and associated protocols. pSOL1 genes with no expression in the WT at a particular time point will have expression ratios that are randomly distributed about zero. It is therefore not entirely unexpected to see a modicum of light red (higher M5 expression) on the Eisen plot. In 2,244 classifications (132 pSOL1 genes [not including hypothetical proteins] analyzed on 17 arrays), 2,231 were proper identifications (99.4%). Analysis of pSOL1 hypothetical proteins revealed significant regions of homology with chromosomal genes, despite the effort to minimize such homologies during designing of the cDNA probes for array spotting. Of the 13 misidentified genes, only one was present on more than one slide. Nine of the misidentifications had expression ratios less than 1.9, while only one was greater than 2.5.
FIG. 1.

Expression profile of differentially expressed pSOL1 genes for the WT and M5. The average expression for the entire cluster is shown across the top of the cluster. Genes are identified by their assigned annotation number (18).
A second validation method is based on direct comparison (Fig. 2) of array data to Northern analysis data for the strain pair WT/SKO1 (spo0A mutant). mRNA levels generated for this study by Northern analysis were normalized against levels of the thiolase gene (thl), which shows a relatively constant expression in these experiments (10, 25). Because spo0A is disrupted in SKO1, Northern blots show up to a 38-fold decrease in transcript levels where the array analysis shows up to a 3.5-fold decrease (Fig. 2a). The sol operon (aad-ctfAB) and the adc gene have been shown to be directly regulated by Spo0A (10). For the aad-ctfAB, adc, and spo0A transcripts, the normalized ratios follow the same pattern of downregulation but underestimate the overall ratio, a well-known characteristic of cDNA microarrays. This suggests that our DNA array technology and computational tools (30) are conservatively reliable for transcriptional analysis.
FIG. 2.

Comparison of expression ratios between SKO1 and 824 for spo0A (a), aad-ctfAB (b), and adc (c) (no Northern probe was used in this study). Open bars show ratios calculated from normalized microarray results; for polycistronic transcripts (aad-ctfAB and ptb-buk), the average ratio for all genes is shown. Solid bars show ratios from Northern blots made from the same RNA samples as the microarrays; gray bars show values calculated from previously published Northern blots (10) and are paired with microarray data based on growth kinetic similarities. The Northern blots were normalized against thl expression at the same time point. ∗, transcript differentially expressed to a 95% confidence interval on at least one array; # and †, ratios of 1 calculated from Northern and microarray analyses, respectively.
Comparison of SKO1 against the WT strain.
Batch fermentations of pH-controlled SKO1 produced negligible amounts of solvents due to inactivation of the spo0A gene (Fig. 3). The WT strain produced an average (n = 2) of 280 mM total solvents (166 mM butanol, 90 mM acetone, and 24 mM ethanol) compared to 21 mM for SKO1 (13 mM butanol, 4 mM acetone, and 4 mM ethanol; n = 2). In contrast, SKO1 had a greater average peak acid concentration than the WT (256 and 112 mM, respectively). SKO1 samples from the bioreactors were plated on 2× YTGMA (10, 20) and shown by iodine staining to degrade starch, indicating that the strain still contained pSOL1. Growth kinetics was also inhibited: the doubling times for the WT and SKO1 were 1.07 and 1.45 h, respectively. These results agree with previously reported characterizations of the strains (10). Samples from nine time points (Fig. 3, panel IA) were analyzed on 17 DNA arrays. All duplicate arrays (n = 3 for sample C) were hybridized with reverse-labeled samples (e.g., WT-Cy3/M5-Cy5 and M5-Cy3/WT-Cy5) except points H and I, for which only one labeled sample was successfully analyzed. Array data were normalized, and genes that were identified as differentially expressed for at least two time points at the 95% significance level were further analyzed by average-linkage hierarchical clustering (5) and SOM analysis (24). A total of 211 genes were identified and were organized into six clusters by using SOMs (Fig. 4). The two most distinct clusters are S2 and S0 (Fig. 4). Cluster S2 consists of genes differentially upregulated in SKO1, most of which are directly and indirectly inhibited by Spo0A. Particularly noticeable are the >25 genes related to motility and chemotaxis. These include the genes of an operon homologous to the B. subtilis flaA operon (which includes fliFGHIJK and likely is made up of genes CAC2165 to CAC2156 [18] or CAC2165 to CAC2154 based on our sigma factor binding site computational analysis) but also several genes from four more predicted (by our computational analysis) motility and chemotaxis operons (CAC2152 to CAC2139, CAC2214 to CAC2204, the monocistronic CAC2203, and CAC2225 to CAC2218). Five chemotaxis genes (two cheA genes, cheC, CAC0250, and CAP0048) are also included in this cluster, some in the aforementioned operons. Two similar genes (flgD [CAC2156] of the flaA operon discussed above and motA [CAC1846]) show similar expression kinetics but are in cluster S5. This category of motility and chemotaxis genes was previously designated category II (directly or indirectly inhibited by Spo0A) in B. subtilis (6). Motility and chemotaxis genes are affected by Spo0A indirectly through σD-mediated transcription and regulation (6). The flagellin of C. acetobutylicum is known to be posttranslationally glycosylated (15), and we accordingly noted upregulation of two glycosyltransferase genes (CAC2186 and CAC2522). CAC3647, annotated as the transitional state gene regulator abrB, is included in cluster S2 and in B. subtilis in category II. PCR primers were designed to amplify a 242-bp fragment from this gene. However, there are two other genes in the C. acetobutylicum genome annotated as abrB, each having a high degree of homology with the CAC3647 PCR product as evaluated by BLASTN (CAC0310, 173 of 202 bp; CAC1941, 155 of 199 bp), and it is impossible to discriminate among transcripts from these three genes. A notable gene of cluster S2 is ftsZ (CAC1693), which is part of a predicted bicistronic operon (CAC1692 and CAC1693). The cell division GTPase FtsZ and its partner FtsA play an essential role in cell division and sporulation in B. subtilis and all eubacteria (7, 13). Of the genes in cluster S2, 72% belong to transcriptional units (operons) with promoter regions predicted to have a 0A box, while 44% have a predicted σF binding site. This is statistically significant compared to the entire genome (52 and 31%, respectively), further suggesting that these genes are regulated by Spo0A.
FIG. 3.

Fermentation kinetics from WT/SKO1 (I) and WT/M5 (II) fermentations. (A) Growth curves for WT (□) and SKO1 or M5 (▪), with sampling points used for array analysis labeled A through I. Arrows indicate time points at which Northern blotting was performed: open arrows correspond to this study, and closed arrows show previously published results (10). (B and C) Product formation for the WT (B), SKO1 (I, C), and M5 (II, C). Symbols: ○, acetate; •, acetone; ▴, butanol; ▵, butyrate; ◊, ethanol.
FIG. 4.

Expression profile of differentially expressed genes for WT and SKO1. SOM analysis of the 211 differentially expressed genes for WT and SKO1 arranged in six gene clusters as the logarithm of expression ratios (SKO1/WT). Detailed gene annotation is shown only for clusters S0 and S2. Annotation details for the remaining clusters can be found at http://www.chemeng.northwestern.edu/Faculty/papou.html.
Cluster S0 (Fig. 4) includes 59 genes identified as differentially downregulated in SKO1, i.e., as positively controlled directly or indirectly by Spo0A. This is confirmed by the presence in this cluster of the major sol locus genes (aad, ctfAB, and adc, all on pSOL1), which are known to be positively regulated by Spo0A (10), but also of the butanol dehydrogenase (bdhAB) genes. Several sporulation genes were also included in cluster S0 (downregulated in SKO1), including sigF and spoIIAB (anti-sigF), spoVS, and spoVAD. The sigF tricistronic operon is known to be positively regulated by Spo0A. spoVAD was included in cluster S0, although its expression profile seems to make it an outlier. The spoVAE and spoVAD genes are part of the spoVA operon in B. subtilis, and spoVAE was found to belong to category II (inhibited by Spo0A [6]), which is opposite to the classification of spoVAD shown here for C. acetobutylicum. The product of CAC3319 is a signal transduction kinase that bears much similarity to the B. subtilis KinA (the phosphorelay protein that helps initiate sporulation by phosphorylating Spo0F) and SpoIIJ. Also included in cluster S0 were several genes related to electron transport, including those encoding flavodoxin (CAC0587), ferredoxin (CAC0303), thioredoxin (CAC1547), and three oxidoreductases (CAC2018, CAC2459, and CAP0135). Several sugar metabolism genes were also downregulated in strain SKO1 during stationary phase, including fruB (CAC0232), a gene encoding a regulator of sugar metabolism (CAC0231), a phosphomannomutase gene (CAC2337), amyP (CAP0168), a glucoamylase gene (CAC2810), and an endoglucanase gene (CAC2556). Production of amylases and glucoamylases in B. subtilis and many other organisms is also known to be associated with stationary-phase metabolism. In summary, cluster S0 contains genes which are positively regulated by Spo0A and expressed during the stationary phase of the culture.
Transcriptional analysis of strain M5.
Fermentations of WT and M5 cultures in static flasks were used for DNA array analysis. The strains grew at nearly the same rate (Fig. 3, panel IIA), allowing RNA samples (A to I) used for DNA array analysis to be closely paired. The WT culture (Fig. 3, panel IIB) produced typical amounts of solvents (69 mM butanol, 38 mM acetone, and 10 mM ethanol) and acids (44 mM butyrate and 48 mM acetate) for static culture. Strain M5 produced no acetone or butanol (Fig. 3, panel IIC), less ethanol (7 mM) and acetate (25 mM), but significantly more butyrate (79 mM). Samples from nine time points (Fig. 3, panel IIA) were analyzed on 17 DNA arrays. All duplicate arrays were hybridized with reverse-labeled samples (e.g., WT-Cy3/M5-Cy5 and M5-Cy3/WT-Cy5) except for point D, for which only one labeled sample was successfully analyzed. Of the 1,019 ORFs represented on the arrays, 253 genes were determined to be differentially expressed (same criteria as SKO1 analysis) and were clustered by SOM analysis (Fig. 5).
FIG. 5.

Expression profile of differentially expressed genes for the WT versus M5. Results of SOM analysis of the 253 differentially expressed genes for the WT versus M5 arranged in eight gene clusters are shown as the logarithm of expression ratios (M5/WT). Detailed gene annotation is shown only for clusters D2 and D7. Annotation details for the remaining clusters can be found at http://www.chem-eng.northwestern.edu/Faculty/papou.html.
Clusters D2 and D3 (Fig. 5), which include 60% of the pSOL1 genes, represent genes with higher expression in the WT throughout the culture. The major solvent formation genes aad (_adhe_1, CAP0162), cftA (CAP0163), ctfB (CAP0164), adc (CAP0165), and adh (CAP0059) are present, while _adhe_2 (CAP0035; we note that _adhe_2 and aad/_adhe_1 were incorrectly identified in the original genome annotation) has an expression pattern similar to that of cluster D2 but was placed in cluster D7 (Fig. 5). Strong expression of these genes correlates well with the onset of solvent production. Since M5 sporulation is abolished due to pSOL1 loss, one or more of the pSOL1 genes expressed in WT are absolutely necessary for the sporulation process. The most likely candidates for such genes are sporulation-like and transcriptional-regulator genes. Among those is a spo0J regulator of the soj/parA family (CAP0177). The spo0J gene located on the chromosome (CAC3731) was differentially expressed at the 90% confidence level at four of the nine time points, with an expression pattern very similar to that of cluster D1, suggesting that spo0J is utilized at the later stages in the WT but not M5. spo0J is required for chromosome partitioning during sporulation, and spo0J mutants produce anucleate cells during vegetative growth (23). Cluster D2 also contains a special sigma factor belonging to the sigF/sigE/sigG family (CAP0157). Sigma factors of the sigF/sigE/sigG family are responsible for controlling a number of early sporulation processes. Interestingly, the chromosomal copy of the sporulation specific sigma factor σF also falls within this cluster. CAP0009 is a two-component response regulator possibly involved in autolysis. There are also two transcriptional regulators with unknown functions: CAP0087 (highly expressed in the WT) and CAP0178.
Chromosomal genes which fall into one of these two clusters (D2 and D3), and to a great extent clusters D6 and D7, are likely to have higher expression in the WT as a result of pSOL1 loss. These genes may therefore play a role in generating the asporogenous phenotype of M5. As previously mentioned, sigF (cluster D2) has persistently higher expression in the WT. σF is the first compartment-specific sigma factor activated by spo0A in B. subtilis. spo0A (CAC2071) is in cluster D7 and is generally expressed to a greater extent in the WT, with the exception of the very first time points. Cluster D7 (Fig. 5) also contains a gene for a two-component signal transduction histidine kinase (CAC3319) with high homology to kinA of B. subtilis, which as discussed above may be involved in initiation of sporulation. Cluster D2 (Fig. 5) contains genes for two additional two-component histidine kinases: cheA (CAC2220), involved in chemotaxis, and CAC1701, involved in general phosphate regulation. Cluster D6 contains cheC, an inhibitor of chemotaxis protein methylation through CheD binding (19).
An important family of genes that show differential expression is that related to stress response. Nine stress-related genes are included in cluster D7 (Fig. 5), the majority having higher expression in M5 during early exponential and late stationary phases, with substantially higher expression in the WT during late exponential and transition phases. It has been reported that the onset of solvent production (not seen in M5) during late exponential and transition phases is preceded or accompanied by induction of the stress response genes (1, 21). Stress genes in cluster D7 include the major stress response genes from the groE operon, groES and groEL (CAC2704 and CAC2703, respectively), and the dnaK operon, hrcA, dnaK, and dnaJ (CAC 1280, CAC1282, and CAC1283). Both operons are negatively regulated by HrcA (1). Also included are the molecular chaperones of the small- and large-heat-shock-protein families, _hsp_18 (CAC3714) and _hsp_90 (CAC3315), respectively. Cluster D7 also includes a member of the class III _hsp_100 heat shock family, clpC (CAC3189), and an associated transcriptional regulator of class III heat shock proteins, ctsR (CAC3192). Additional members of the clp family which are differentially expressed include clpA (CAC1824, cluster D0), clpP (CAC2640, cluster D6), a clpP family serine protease gene (CAC1893), and clpX (CAC2639, cluster D1). Two additional stress-related genes from the lon protease family, lonA (CAC0456, cluster D6) and CAC2637 (cluster D3), show slightly lower expression in M5 during the late stationary and transition phases. Members of both the clp and lon families have been indirectly implicated in both positive and negative regulation of several sporulation-specific sigma factors, including sigF and sigH (14, 22). clpP has been shown to indirectly affect intracellular Spo0A levels by regulating the activity of enzymes responsible for its phosphorylation (17).
Four genes from the fts family, involved in cell division and chromosome segregation through the establishment of cellular asymmetry during sporulation, are differentially regulated. ftsA and ftsZ (CAC1692 and CAC1693, cluster D1) form a predicted operon whose expression appears to be elevated during the late transition phase in M5 but shifts to higher expression in the WT during the stationary phase (differential expression of ftsA is at the 90% confidence level). ftsX (CAC0498, cluster D2) has persistently lower expression levels in M5, while ftsK (CAC3709, cluster D7) expression is lower in M5 through the transition phase of growth and higher in M5 during the late transition phase. The significantly different expression patterns of fts family genes suggest altered cell division and chromosome segregation programs.
Several genes involved in control of DNA replication and topology are also dramatically altered in M5. Genes with DNA gyrase (CAC0006 and CAC0007) and DNA helicase (CAC2262) functions are expressed at a lower level in M5 during the exponential and transition phases with increased expression into the stationary phase (clusters D5 and D6). Transcription of most E. coli operons is sensitive to DNA supercoiling (8). In C. acetobutylicum, DNA topology has been shown to affect the rate of transcription of several acid and solvent formation genes and has been implicated as a possible signal for the onset of solventogenesis (27, 29). In fact, clusters D5 and D6 also contain the acid formation genes pta (CAC1742), ack (CAC1743), buk (CAC3075), and ptb (CAC3076), along with 12 other genes involved in carbon metabolism. Genes involved in glycolysis (except for hexokinase [CAC2613]) are downregulated during exponential growth and subsequently upregulated during stationary phase. Genes responsible for conversion of acetoacetyl-CoA to butyryl-CoA also follow this pattern. The chromosomal thiolase gene (CAC2873), however, is upregulated throughout. Expression of the acetate formation genes remains lower in M5; however, expression of butyrate formation genes is greater in M5 during the stationary phase of growth. Production of butyrate was observed to continue through the entire course of the M5 culture, while acetate production was lower at each stage of the fermentation (Fig. 3, panel II). As expected, all genes involved in solvent formation, including those located on the chromosome, were expressed at a higher level in the WT.
Clusters D5 and D6 have numerous motility and chemotaxis genes, including fliFGHL (flaA operon), fliP, fliY, flgD, and flgK. Cluster D4 has two additional motility genes, a flagellin gene (CAC1555) and fliS (CAC2206). All these genes exhibit lower expression in M5 during early exponential growth with increasing expression through the stationary phase. This is the opposite of the expression pattern of spo0A (Fig. 5) and is in direct agreement with observations for SKO1 and B. subtilis (6).
DISCUSSION
The validity of the cDNA array analysis protocol used in this study was clearly demonstrated by two independent methods. In addition to Northern analysis, DNA array results were previously compared to results of quantitative RT-PCR (30), and agreement between the two methods was observed. Further, DNA array results from a duplicate WT-M5 experiment were consistent with the data reported here. Nevertheless, in SKO1, the level of spo0A downregulation measured by DNA arrays is much lower than that observed with Northern analysis (3.5- versus 38-fold). This is likely due to production of a short truncated spo0A transcript in SKO1 (10), which may result in artificially lower ratios. This assessment is supported by the fact that overexpression of spo0A [strain ATCC 824(pMSPOA)] (10) produces DNA array ratios up to 17-fold higher (data not shown).
This is the first study in which DNA arrays were used for large-scale transcriptional analysis in C. acetobutylicum. A number of both expected and unexpected patterns of gene expression were captured. Many genes known to be directly or indirectly influenced by Spo0A in B. subtilis are similarly regulated in SKO1, including increased expression of the chemotaxis and motility genes, sigF, and spoIIAB. An enrichment of genes with predicted 0A boxes and σF binding sites in clusters containing genes known to be controlled by Spo0A was also noted. While the majority of gene expression patterns in SKO1 are similar to known patterns in B. subtilis, several differences exist. For example, classification of spoVAD as positively controlled by Spo0A is the opposite of that seen in B. subtilis. Upregulation of the glycosyltransferases was unique to C. acetobutylicum. Finally, several genes related to electron transport were discovered to be differentially regulated (different groups of genes in opposite directions).
Several similarities were observed between SKO1 and M5, which have very similar phenotypes (asporogenous and deficient in solventogenesis). As expected, both strains had significantly lower expression of the solvent formation genes. Both strains also had decreased expression of spo0A, sigF, and CAC3319 (kinA). However, significant differences were also noted. For example, in contrast to SKO1, the chemotaxis genes cheA and cheC have significantly lower expression in M5 than in the WT. The fts genes also show significantly different expression patterns. Several unique classes of genes were noted as being differentially regulated in M5 but not in SKO1. The gyrases and DNA helicases were differentially expressed, and their expression pattern appears to be closely linked to expression of genes involved in acid formation and glycolysis; this lends support to their role in regulating C. acetobutylicum metabolism. Finally, expression of the stress response genes in M5 is greatly altered, including regulation of clpP and lonA with known roles in regulation of Spo0A and sporulation specific sigma factors, respectively.
Acknowledgments
This work was supported by a National Science Foundation grant (BES-9905669).
We acknowledge use of the Keck Biophysics Facility at Northwestern University, and we thank Nadereh Jafari from the Microarray Core Facility Center for Genetic Medicine at Northwestern University, Abbott Laboratories for donation of clarithromycin, Marija Tesic for S. cerevisiae DNA, and J. W. Peters and B. Lemon for WT C. pasteurianum.
REFERENCES
- 1.Bahl, H., H. Muller, S. Behrens, H. Joseph, and F. Narberhaus. 1995. Expression of heat shock genes in Clostridium acetobutylicum. FEMS Microbiol. Rev. 17**:**341-348. [DOI] [PubMed] [Google Scholar]
- 2.Buday, Z., J. C. Linden, and M. N. Karim. 1990. Improved acetone butanol fermentation analysis using subambient HPLC column temperature. Enzyme Microb. Technol. 12**:**24-27. [Google Scholar]
- 3.Clark, S. W., G. N. Bennett, and F. B. Rudolph. 1989. Isolation and characterization of mutants of _Clostridium acetobutylicu_m ATCC 824 deficient in acetoacetyl-coenzyme A:acetate/butyrate:coenzyme A transferase and in other solvent pathway enzymes. Appl. Environ. Microbiol. 55**:**970-976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cornillot, E., R. V. Nair, E. T. Papoutsakis, and P. Soucaille. 1997. The genes for butanol and acetone formation in Clostridium acetobutylicum ATCC 824 reside on a large plasmid whose loss leads to degeneration of the strain. J. Bacteriol. 179**:**5442-5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eisen, M. B., P. T. Spellman, P. O. Brown, and D. Botstein. 1998. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. USA 95**:**14863-14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fawcett, P., P. Eichenberger, R. Losick, and P. Youngman. 2000. The transcriptional profile of early to middle sporulation in Bacillus subtilis. Proc. Natl. Acad. Sci. USA 97**:**8063-8068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feucht, A., I. Lucet, M. D. Yudkin, and J. Errington. 2001. Cytological and biochemical characterization of the FtsA cell division protein of Bacillus subtilis. Mol. Microbiol. 40**:**115-125. [DOI] [PubMed] [Google Scholar]
- 8.Gellert, M. 1981. DNA topoisomerases. Annu. Rev. Biochem. 50**:**879-910. [DOI] [PubMed] [Google Scholar]
- 9.Gribskov, M. 1999. Sequence comparison, p. 105-120. In M. Bishop (ed.), Genetics databases. Academic Press, New York, N.Y.
- 10.Harris, L. M., N. E. Welker, and E. T. Papoutsakis. 2002. Northern, morphological, and fermentation analysis of spo0A inactivation and overexpression in Clostridium acetobutylicum ATCC 824. J. Bacteriol. 184**:**3586-3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hegde, P., R. Qi, K. Abernathy, C. Gay, S. Dharap, R. Gaspard, J. E. Hughes, E. Snesrud, N. Lee, and J. Quackenbush. 2000. A concise guide to cDNA microarray analysis. BioTechniques 29**:**548-562. [DOI] [PubMed] [Google Scholar]
- 12.Helmann, J. D., M. F. W. Wu, P. A. Kobel, F. J. Gamo, M. Wilson, M. M. Morshedi, M. Navre, and C. Paddon. 2001. Global transcriptional response of Bacillus subtilis to heat shock. J. Bacteriol. 183**:**7318-7328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kemp, J. T., A. Driks, and R. Losick. 2002. FtsA mutants of Bacillus subtilis impaired in sporulation. J. Bacteriol. 184**:**3856-3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu, J. J., W. M. Cosby, and P. Zuber. 1999. Role of Lon and ClpX in the post-translational regulation of a sigma subunit of RNA polymerase required for cellular differentiation in Bacillus subtilis. Mol. Microbiol. 33**:**415-428. [DOI] [PubMed] [Google Scholar]
- 15.Lyristis, M., Z. L. Boynton, D. Petersen, Z. Kan, G. N. Bennett, and F. B. Rudolph. 2000. Cloning, sequencing, and characterization of the gene encoding flagellin, flaC, and the post-translational modification of flagellin, FlaC, from Clostridium acetobutylicum ATCC824. Anaerobe 6**:**69-79. [Google Scholar]
- 16.Nair, R. V., E. M. Green, D. E. Watson, G. N. Bennett, and E. T. Papoutsakis. 1999. Regulation of the sol locus genes for butanol and acetone formation in Clostridium acetobutylicum ATCC 824 by a putative transcriptional repressor. J. Bacteriol. 181**:**319-330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nanamiya, H., K. Takahashi, M. Fujita, and F. Kawamura. 2000. Deficiency of the initiation events of sporulation in Bacillus subtilis clpP mutant can be suppressed by a lack of the Spo0E protein phosphatase. Biochem. Biophys. Res. Commun. 279**:**229-233. [DOI] [PubMed] [Google Scholar]
- 18.Nolling, J., G. Breton, M. V. Omelchenko, K. S. Makarova, Q. D. Zeng, R. Gibson, H. M. Lee, J. Dubois, D. Y. Qiu, J. Hitti, Y. I. Wolf, R. L. Tatusov, F. Sabathe, L. Doucette-Stamm, P. Soucaille, M. J. Daly, G. N. Bennett, E. V. Koonin, and D. R. Smith. 2001. Genome sequence and comparative analysis of the solvent-producing bacterium Clostridium acetobutylicum. J. Bacteriol. 183**:**4823-4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rosario, M. M. L., and G. W. Ordal. 1996. CheC and CheD interact to regulate methylation of Bacillus subtilis methyl-accepting chemotaxis proteins. Mol. Microbiol. 21**:**511-518. [DOI] [PubMed] [Google Scholar]
- 20.Sabathe, F., C. Croux, E. Cornillot, and P. Soucaille. 2002. amyP, a reporter gene to study strain degeneration in Clostridium acetobutylicum ATCC 824. FEMS Microbiol. Lett. 210**:**93-98. [DOI] [PubMed] [Google Scholar]
- 21.Sauer, U., and P. Durre. 1995. Differential induction of genes related to solvent formation during the shift from acidogenesis to solventogenesis in continuous culture of Clostridium acetobutylicum. FEMS Microbiol. Lett. 125**:**115-120. [Google Scholar]
- 22.Serrano, M., S. Hovel, C. P. Moran, A. O. Henriques, and U. Volker. 2001. Forespore-specific transcription of the lonB gene during sporulation in Bacillus subtilis. J. Bacteriol. 183**:**2995-3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sonenshein, A., J. Hoch, and R. Losick (ed.). 2002. Bacillus subtilis and its closest relatives: from genes to cells. ASM Press, Washington, D.C.
- 24.Tamayo, P., D. Slonim, J. Mesirov, Q. Zhu, S. Kitareewan, E. Dmitrovsky, E. S. Lander, and T. R. Golub. 1999. Interpreting patterns of gene expression with self-organizing maps: methods and application to hematopoietic differentiation. Proc. Natl. Acad. Sci. USA 96**:**2907-2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tummala, S., and E. T. Papoutsakis. 1999. Development and characterization of a gene-expression reporter system for Clostridium acetobutylicum ATCC 824. Appl. Environ. Microbiol. 65**:**3793-3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tummala, S. B., S. G. Junne, and E. T. Papoutsakis. 2003. Design of antisense RNA constructs for downregulation of the acetone formation pathway of Clostridium acetobutylicum. J. Bacteriol. 185**:**1923-1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ullmann, S., A. Kuhn, and P. Durre. 1996. DNA topology and gene expression in Clostridium acetobutylicum: implications for the regulation of solventogenesis. Biotechnol. Lett. 18**:**1413-1418. [Google Scholar]
- 28.Wiesenborn, D. P., F. B. Rudolph, and E. T. Papoutsakis. 1988. Thiolase from Clostridium acetobutylicum ATCC 824 and its role in the synthesis of acids and solvents. Appl. Environ. Microbiol. 54**:**2717-2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wong, J., and G. N. Bennett. 1996. The effect of novobiocin on solvent production by Clostridium acetobutylicum. J. Ind. Microbiol. 16**:**354-359. [DOI] [PubMed] [Google Scholar]
- 30.Yang, H., H. Haddad, C. Tomas, K. Alsaker, and E. T. Papoutsakis. 2003. A segmental nearest neighbor normalization and gene identification method gives superior results for DNA-array analysis. Proc. Natl. Acad. Sci. USA 100**:**1122-1127. [DOI] [PMC free article] [PubMed] [Google Scholar]