Altered properties of volume-sensitive osmolyte and anion channels (VSOACs) and membrane protein expression in cardiac and smooth muscle myocytes from Clcn3−/− mice (original) (raw)
Abstract
ClC-3, a member of the large superfamily of ClC voltage-dependent Cl– channels, has been proposed as a molecular candidate responsible for volume-sensitive osmolyte and anion channels (VSOACs) in some cells, including heart and vascular smooth muscle. However, the reported presence of native VSOACs in at least two cell types from transgenic ClC-3 disrupted (_Clcn3_−/−) mice casts considerable doubt on this proposed role for ClC-3. We compared several properties of native VSOACs and examined mRNA transcripts and membrane protein expression profiles in cardiac and pulmonary arterial smooth muscle cells from Clcn3+/+ and _Clcn3_−/− mice to: (1) test the hypothesis that native VSOACs are unaltered in cells from _Clcn3_−/− mice, and (2) test the possibility that targeted inactivation of the Clcn3 gene using a conventional murine global knock-out approach may result in compensatory changes in expression of other membrane proteins. Our experiments demonstrate that VSOAC currents in myocytes from Clcn3+/+ and _Clcn3_−/− mice are remarkably similar in terms of activation and inactivation kinetics, steady-state current densities, rectification, anion selectivity (I− > Cl− ≫ Asp−) and sensitivity to block by glibenclamide, niflumic acid, DIDS and extracellular ATP. However, additional experiments revealed several significant differences in other fundamental properties of native VSOACs recorded from atrial and smooth muscle cells from _Clcn3_−/− mice, including: differences in regulation by endogenous protein kinase C, differential sensitivity to block by anti-ClC-3 antibodies, and differential sensitivities to [ATP]i and free [Mg2+]i. These results suggest that in response to Clcn3 gene deletion, there may be compensatory changes in expression of other proteins that alter VSOAC channel subunit composition or associated regulatory subunits that give rise to VSOACs with different properties. Consistent with this hypothesis, in atria from _Clcn3_−/− mice compared to Clcn3+/+ mice, quantitative analysis of ClC mRNA expression levels revealed significant increases in transcripts for ClC-1, ClC-2, and ClC-3, and protein expression profiles obtained using two-dimensional polyacrylamide gel electrophoresis revealed complex changes in at least 35 different unidentified membrane proteins in cells from _Clcn3_−/− mice. These findings emphasize that caution needs to be exercised in simple attempts to interpret the phenotypic consequences of conventional global Clcn3 gene inactivation.
The molecular identification of the protein responsible for volume-sensitive organic osmolyte and anion channels (VSOACs) in mammalian cells has been particularly difficult to resolve (Strange et al. 1996; Okada, 1997). One member of the ClC superfamily of voltage-dependent Cl− channels, ClC-3, has been under consideration as a molecular candidate for VSOACs in some mammalian cells (Duan et al. 1997). Stable or transient transfection of ClC-3 cDNA, cloned from guinea-pig heart (short isoform gpClC-3), into NIH/3T3 cells was reported to yield basally active outwardly rectifying chloride currents that were strongly modulated by cell volume and exhibited many properties similar to native VSOACs. Furthermore, site-directed mutagenesis altered rectification and anion selectivity of the expressed current, _I_gpClC-3, and altered its sensitivity to regulation by protein kinase C and cell volume (Duan et al. 1999_a_). More recently, native VSOACs in several different cell types and expressed _I_gpClC-3 have been found to be significantly inhibited by four different antibodies (Abs) targeting distinct ClC-3 epitopes (Duan et al. 2001; Wang et al. 2003; Jin et al. 2003) and by ClC-3 antisense oligonucleotides and/or cRNA (Wang et al. 2000; Hermoso et al. 2002).
However, several recent studies have provided conflicting and inconsistent data on the exact physiological role of ClC-3 Cl− channels (George et al. 2001; Jentsch et al. 2002; Nilius & Droogmans, 2003). Much of the current controversy surrounding the physiological role of ClC-3 Cl− channels can be attributed to the reported presence of native VSOACs in at least two cell types from transgenic ClC-3 disrupted (_Clcn3_−/−) mice (Stobrawa et al. 2001). In some studies, ClC-3 has been localized to intracellular membranes (Stobrawa et al. 2001; Li & Weinman, 2002) where it has been proposed to function primarily in vesicular acidification (Jentsch et al. 2002). However, other studies have clearly demonstrated plasma membrane localization of heterologously expressed ClC-3 (Huang et al. 2001; Weylandt et al. 2001; Schmieder et al. 2001; Ogura et al. 2002) and endogenous ClC-3 (Isnard-Bagnis et al. 2003; Olsen et al. 2003) in various cell types.
It is unknown whether the properties of native VSOACs recorded from cells of _Clcn3_−/− mice are similar, or different compared to native VSOACs in cells from Clcn3+/+ mice. The former possibility would be expected if ClC-3 is not responsible for native VSOACs, whereas the latter possibility would suggest that, in response to loss of expression of endogenous ClC-3, compensatory changes in expression of other proteins may alter VSOAC channel subunit composition or associated regulatory subunits and give rise to VSOACs with different properties. We tested these possibilities by comparing the properties of native VSOACs in cardiac and smooth muscle cells from Clcn3+/+ and _Clcn3_−/− mice, and cardiac expression levels of several different ClC Cl− channel mRNAs, and using two-dimensional gel electrophoresis, compared membrane protein expression profiles in atria from Clcn3+/+ and _Clcn3_−/− mice.
Methods
The Institutional Animal Use and Care Committee at the University of Nevada approved the use and treatment of all animals used in the experiments described here.
_Clcn3_−/− mice
Targeted inactivation of the Clcn3 gene was produced by replacement of part of exon 6 and all of exon 7 (Dickerson et al. 2002) with a replacement vector containing sequence for the neomycin resistance gene (NeoR). The excised allele contains the coding sequences for transmembrane domains B-D (Dutzler et al. 2002). Heterozygous 129/SvJ-C57BL/6 offspring were used to establish breeding colonies. Genotyping was performed using PCR as previously described (Dickerson et al. 2002). Northern blots confirmed expression of a smaller (∼0.26 kDa) ClC-3 transcript in heart and brain of _Clcn3_−/− mice (data not shown) and Western blot analysis has previously demonstrated the absence of ClC-3 protein in these _Clcn3_−/− mice (Arreola et al. 2002; Dickerson et al. 2002; Wang et al. 2003).
Total RNA extraction and RT-PCR
Total RNA was extracted from isolated heart and brain tissue with the use of a TRIZOL (Life Technology Inc., La Jolla, CA, USA) procedure and a SNAP total RNA isolation kit (Invitrogen, Carlsbad, CA, USA), respectively, as previously reported (Walker et al. 2001). The SUPERSCRIPT™. II RNase H− (Life Technology Inc., La Jolla, CA, USA) and 200 μg ml−1 of random hexamer (for tissues) were used to reverse transcribe the RNA sample. The PCR amplification profile was as follows: a 15 s denaturation step at 95°C and a 60 s primer extension step at 60°C using AmpliTag Gold(r) Taq DNA polymerase (PE Biosystems, Hayward, CA, USA). In the tissue RT-PCR, the amplification was performed for 30 cycles. The amplified products were separated by electrophoresis on a 2.0% agarose−1 × TAE (Tris, acetic acid, EDTA) gel, and the DNA bands were visualized by ethidium bromide staining. β-Actin primers that spanned two exons and an intron were used to confirm that the products generated were representative of RNA. Any cDNA preparation that amplified the β-actin intron was discarded. Each amplified product was sequenced by the chain termination method with an ABI PRIZM (model 310, PE Biosystems).
Primer sequences used for amplification.
ClC-1: Primers 5′CTGCATTTGGAAGGCTGGTAGGAG-3′ and 5′AATGACGGCTGTGGAGACTGTGTG-3′ Accession number XM_149848, amplicon = 161 bp, contains region of the molecule from 1557 to 1717.
ClC-2: Primers 5′-CGGGGAGTGGTGCTGAAAGAATA-3′ and 5′TCCGGGACTCATGCTCATAGATACC-3′ Accession number NM_009900, amplicon = 193 bp, contains region of the molecule from 657 to 849.
ClC-3: Primers 5′-CCCGAGGTGGAGAGAGACTGCT-3′ and 5′CCGGCTTTCAGAGAGGTTACG-3′ Accession number X78874, amplicon = 174 bp, contains region of the molecule from 41 to 214.
ClC-4: Primers 5′-TTATTGCTTGAGGACAGACGGGC-3′ and 5′-GGGGCAAGTGTTCAGCGTCAT-3′ Accession number Z49916, amplicon = 174 bp, contains region of the molecule from 36 to 193.
ClC-5: Primers 5′-CTCTTTAGGTGGCGTTTGTTGCTGT-3′ and 5′-CACCATTGTATGACTTGTTCCCTTCG-3′ Accession number NM_016691, amplicon = 189 bp, contains region of the molecule from 16 to 204.
Quantitative RT-PCR
Real time quantitative PCR was performed with the use of Syber Green chemistry on an ABI 5700 sequence detector (PE Biosystems) (Walker et al. 2001). Regression analysis of the mean values of six multiplex RT-PCRs for the log10 diluted cDNA was used to generate standard curves. Unknown quantities relative to the standard curve for a particular set of primers were calculated, yielding transcriptional quantification of ClC gene products relative to the endogenous standard (β-actin). The reproducibility of the assay was tested by analysis of variance (ANOVA) comparing repeat runs of samples, and mean values generated at individual time points were compared by Student's t test. These data were then analysed as a ratio of KO to WT mice to create the bars shown in Fig. 7B.
Figure 7. Qualitative and quantitative analysis of ClC-1–ClC-5 transcripts in atria and brain.

A is a qualitative study. ClC-1 = 161 bp, ClC-2 = 193 bp, ClC-3 = 174 bp, ClC-4 = 174 bp, ClC-5 = 189 bp. NTC panel demonstrates that reactions lacking cDNA template did not amplify any products. Ladder bands indicate the molecular weight of amplicons and the sizes are indicated at the left side of panel A. B is a quantitative experiment that is measuring the ratio of expression of ClC channel transcripts of KO to that of WT mice. Therefore, if there is no expression (see, e.g. ClC-4) or no change in expression in KO compared to WT atria or brain tissues the ratio will be 1.
Preparation of heart extracts
Heart samples were initially prepared from WT C57BL/6 J mice and ClC-3 knock-out (_Clcn3_−/−) mice. Mice were anaesthetized with pentobarbital (i.p., 50 mg (kg body weight)−1) and hearts were quickly removed and washed twice with isolation solution (mm: 225 mannitol, 75 sucrose, 1 EDTA, 10 Hepes, pH 7.2). For each sample two hearts were pooled and minced with scissors in 3.5 ml isolation solution containing protease inhibitor cocktail (Sigma Chemical Co., Product P 8340). The mixtures were homogenized in a glass homogenizer fitted with a Teflon pestle. The intact cells, nuclei and debris were removed by centrifugation at 800 g for 10 min. The supernatants were centrifuged at 15 000 g for 15 min. The 15 000 g supernatants were further subjected to centrifugation at 100 000 g for 60 min to separate the membrane fraction (pellet) from the cytosolic fraction (supernatant). The membrane fraction was resuspended in 0.3 ml lysis buffer (150 mm NaCl, 1 mm EDTA, 50 mm dithiothreitol (DTT), 50 mm Tris-HCl, 20 μl protease inhibitor, 0.1% SDS, 1% Triton X-100, pH 7.4). All reagents and solutions for 2-D electrophoresis and protein assays, imobilized pH gradient (IPG) strips, and SDS polyacrylamide gels were obtained from Bio-Rad (Hercules, CA, USA) except as noted. All operations were carried out at 4°C.
Preparation of membrane proteins
Proteins from the membrane fraction were precipitated by the addition of 4 volumes of −20°C acetone. The solutions were kept at −20°C overnight and spun at 16 000 g for 10 min at 0°C. The supernatants were discarded and the pellets were washed twice with ice-cold acetone: water (4 : 1). The final pellets were air dried completely and then resuspended in 300 μl ReadyPrep Sequential Extraction Reagent 3 (5 m urea, 2 m thiourea, 2% CHAPS, 2% SB 3–10, 0.2% ampholytes). The protein content of the resultant supernatants was determined using the RC DC protein assay (Bio-Rad kit). After the addition of 50 mm dithiothreitol (DTT) and 1 μl 0.1% bromophenol blue, the extracts were spun at 16 000 g at room temperature for 5 min
Two-dimensional (2-D) gel electrophoresis
Each extract (185 μl) was loaded onto an IPG strip (linear pH 3–10, 11 cm) by overnight passive rehydration. Isoelectric focusing was carried out on a Bio-Rad Protean IEF cell using the following programme: 250 V, linear ramp for 20 min; 8000 V, linear ramp for 2 h 30 min; and 8000 V for a total of 20 000 V h (all steps with a maximum current of 50 μA per gel). Strips were stored at −80°C overnight, then thawed on the next day and incubated twice for 10 min each in 8 m urea, 2% SDS, 0.05 m Tris-HCl, pH 8.8, 20% glycerol. The first incubation contained 2% DTT and the second contained 2.5% iodoacetamide. The strips were then layered on 4–20% Criterion Tris-HCl gradient gels and embedded in place with 0.5% agarose, along with Invitrogen BenchMark Protein Ladder molecular weight markers. Electrophoresis was performed at a constant current of 200 mA until the dye front ran off the gel. Gels were washed with two changes of water (10 min each) and stained overnight with Bio-Safe Coomassie stain. Stained gels were imaged on a Bio-Rad VersaDoc imager and spot sets were created after images of gels from both wild-type (WT) and _Clcn3_−/− mouse heart were compared using Bio-Rad PDQuest version 7.1.1 software. The theoretical isoelectric point (pI) for ClC-3 was calculated using Compute pI/Mw (http://usexpasy.org).
Immunoblotting
The Coomassie-stained 2-D gel was equilibrated in transfer buffer (25 mm Tris, 192 mm glycine, 10% methanol) for 10 min. Proteins separated by 2-D electrophoresis were transferred to nitrocellulose membranes electrophoretically in transfer buffer at 100 V for 50 min using Criterion Blotter (Bio-Rad). The membrane was blocked in TNT buffer (100 mm Tris-HCl, pH 7.5, 150 mm NaCl, 0.1% Tween-20) containing 0.5% gelatin (Bio-Rad) for 1 h at room temperature. The blocking solution was removed. The membrane was incubated overnight at 4°C in anti-ClC-3 C670–687 primary antibody (Wang et al. 2003) diluted with TNT buffer containing 0.1% gelatin. The membrane was washed with TNT buffer three times for 10 min each. The membrane was then incubated in alkaline phosphatase-conjugated anti-rabbit IgG (Promega) diluted with TNT buffer containing 0.1% gelatin for 1 h at room temperature. The membrane was washed as previously described. The detection was performed with the Protoblot II AP system (Promega).
Cell preparation and electrophysiological techniques
Single atrial myocytes were enzymatically isolated (Levesque & Hume, 1995; Duan et al. 1999_b_, 2000) from 46 Clcn3+/+ and 17 _Clcn3_−/− adult mice. After the enzyme treatment, the cells were dissociated and stored at 4°C in a modified KB solution (Isenberg & Klockner, 1982) containing (mm): 70 potassium glutamate, 20 KCl, 1 MgCl2, 10 KH2PO4, 10 taurine, 10 EGTA, 10 glucose, 10 β-hydroxybutric acid, 10 Hepes, and 1 mg ml−1 bovine albumin; pH 7.2 with KOH; 300 mosmol kg−1 with mannitol. The myocytes were used for experiments within the same day of isolation. All cells studied were rod shaped, exhibited clear cross-striations, and lacked any visible blebs under isotonic conditions. Single pulmonary arterial smooth muscle cells (PASMCs) were obtained from eight Clcn3+/+ and six _Clcn3_−/− mice. The first to the third branches of pulmonary arteries were dissected and transferred to a low-Ca2+ physiological saline solution (PSS) containing (mm): 125 NaCl, 5.36 KCl, 0.336 Na2HPO4, 0.44 K2HPO4, 11 Hepes, 1.2 MgCl2, 0.05 CaCl2, 10 glucose, 2.9 sucrose; pH 7.4 with Tris; 300 mosmol kg−1 with sucrose. After connective tissues were removed, the arteries were transferred into a Ca2+-free PSS for 10 min, and then digested at 37°C for ∼20 min with a low-Ca2+ PSS supplemented with (in mg ml−1): 1.5 collagenase type XI, 0.5 papain, 0.15 dithiothreitol, 1 bovine albumin. The arteries were then washed with the low-Ca2+ PSS solution and triturated with a fire-polished Pasteur pipette. Freshly dispersed cells were kept at 4°C and used for experiments within the same day. Membrane currents were measured using whole-cell voltage-clamp technique. Patch pipettes (1.5 mm o.d. borosilicate glass electrodes) had a tip resistance of 1−3 MΩ when filled with pipette solutions. Voltage-clamp recordings were filtered at a frequency of 1 kHz and were obtained using an Axopatch200A patch-clamp amplifier (Axon Instruments, Foster City, CA, USA). Data acquisition and command potentials were controlled by pCLAMP 8.1 software (Axon Instruments). A 3 m KCl–agar bridge between the bath and the Ag–AgCl reference electrode was used to minimize changes in liquid junctional potential. All experiments were performed at room temperature and data are expressed as mean ± s.e.m.; n indicates the number of cells. Statistical analysis was performed using paired or unpaired Student's t test or one-way repeated measures analysis of variance (Student–Newman–Keuls method) where appropriate. Differences were considered to be significant at a two-tailed probability (P) of < 0.05. The blocking effects of Cl− channel inhibitors shown in Fig. 1_H_, were calculated from the equation:
Figure 1. Comparison of native VSOAC currents in cardiac atrial myocytes from Clcn3+/+ (WT) and _Clcn3_−/− (KO) mice.

A and B, time course of changes of VSOAC currents at +80 mV (•) and −80 mV (○) in atrial cells of WT (A) and KO (B) mice exposed to hypotonic (HYPO) and hypertonic (HYPER) solutions. The external solution initially was isotonic (ISO) solution. C and D, VSOAC currents in myocytes from WT mice (C) and from KO mice (D) in isotonic and hypotonic solutions, respectively. Currents were recorded at the time points (a and b) indicated in A and B. Here and in subsequent similar figures using atrial myocytes, the pulse protocol is the same as shown in the upper part of C; changes in whole-cell currents were monitored by applying 400 ms voltage-clamp steps to membrane potentials between −100 and +120 mV in +20 mV steps from a holding potential of −40 mV every 5 s. E, mean I_–_V relationships of VSOAC currents from WT (n = 18) and KO (n = 6) myocytes in isotonic and hypotonic solutions. F and G, anion permeability of VSOACs from WT and KO myocytes. H, comparison of inhibition by Cl− channel inhibitors (glibenclamide (0.2 mm), DIDS (0.1 mm), niflumic acid (0.1 mm) and extracellular ATP (1 mm)) of VSOACs in myocytes from WT and KO mice (n = 4 in each case); E_–_H, values represent mean ± s.e.m.
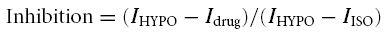
where _I_HYPO is the maximal amplitude of VSOAC currents in hypotonic solutions, _I_drug is the minimal VSOAC current amplitude after application of the inhibitors and _I_ISO is the current amplitude in isotonic solutions. The time for half-maximal VSOAC activation (_t_½) shown in Table 1 was calculated by fitting the activation time course with a Boltzmann function using Origin 7.0 (Microcal, Northampton, MA, USA).
Table 1. Kinetics of VSOACs activation and deactivation.
| Half-times (min) | ||||
|---|---|---|---|---|
| Tissue | Mice | Condition | −80 mV | +80 mV |
| Atrial myocytes | WT (n = 18) | Hypotonic activation | 6.5 ± 0.5 | 5.8 ± 0.4 |
| Hypertonic deactivation | 5.8 ± 0.5 | 4.3 ± 0.4 | ||
| KO (n = 6) | Hypotonic activation | 6.2 ± 0.8 | 5.7 ± 0.4 | |
| Hypertonic deactivation | 5.0 ± 0.7 | 4.2 ± 0.6 | ||
| PASMCs | WT (n = 8) | Hypotonic activation | — | 5.0 ± 0.8 |
| KO (n = 5) | Hypotonic activation | — | 3.5 ± 0.3 |
Solutions and drugs
For VSOAC current recordings, extracellular and intracellular solutions were chosen to maximize recording of Cl− currents and reduce possible contamination by cation currents. Symmetrical chloride concentration was used for the bath and pipette solutions. For experiments with cardiac myocytes, the extracellular solutions contained (mm): 90 NaCl, 0.8 MgCl2, 1.0 CaCl2, 0.2 CdCl2, 10 CsCl, 2.0 BaCl2, 5.5 glucose, 10 Hepes; pH 7.4. The isotonic, hypotonic and hypertonic solutions were adjusted to 310, 210 and 360 mosmol kg−1 by adding mannitol, respectively. In some experiments, extracellular NaCl was replaced by an equimolar concentration (90 mm) of NaI or sodium aspartate. The standard intracellular pipette solution contained (mm): 108 _N_′-methyl-d-glucamine (NMDG), 108 HCl, 5 MgATP, 5 EGTA, 5 Hepes; pH 7.3; 290 mosmol kg−1 using mannitol. In experiments studying intracellular ATP depletion, MgATP was omitted and the amount of free Mg2+ in the pipette solution was maintained at the same concentration (0.652 mm) as in the standard pipette solution. The high free Mg2+ plus ATP-depleted pipette solution was prepared by omitting MgATP and adding 3.737 mm MgCl2 (to replace NMDG-Cl) to increase the free Mg2+ concentration (2.0 mm). Free Mg2+ was calculated by using MaxChelator software (WINMAXC 2.40; provided by Dr Chris Patton of Stanford University, Pacific Grove, CA, USA). For cystic fibrosis transmembrane conductance regulator (CFTR) Cl− current recordings, the extracellular solutions contained (mm): 122 NaCl, 0.8 MgCl2, 1.0 CaCl2, 0.2 CdCl2, 10 CsCl, 2.0 BaCl2, 5.5 glucose, 10 Hepes; pH 7.4; 320 mosmol kg−1 by adding mannitol. The intracellular pipette solution (mm): 140 NMDG, 140 HCl, 5 MgATP, 0.1 NaGTP, 5 EGTA, 5 Hepes; pH 7.3; 300 mosmol kg−1 using mannitol. For experiments with PASMCs, the hypotonic (230 mosmol kg−1) bath solution contained (mm): 107 NMDG-Cl, 1.5 MgCl2, 2.5 MnCl2, 0.5 CdCl2, 0.05 GdCl3, 10 glucose, and 10 Hepes; pH 7.4 with NMDG. The isotonic (310 mosmol kg−1) and hypertonic (350 mosmol kg−1) solutions were prepared by adding mannitol. The pipette solution contained (mm): 96 CsCl, 20 TEA-Cl, 5 ATP-Mg, 5 EGTA, 80 mannitol and 5 Hepes; pH 7.2 with CsOH; 300 mosmol kg−1 with mannitol.
Preparation and intracellular dialysis with the newly developed anti-ClC-3 antibody (A1–14 Ab) have been previously described (Wang et al. 2003). A1–14 Ab was added to the pipette solution at a final concentration of 1.5 μg ml−1. The osmolarity of the pipette dialysis solution was not altered by inclusion of the Ab. Usually, 15 min was allowed for adequate intracellular dialysis of the Ab before cells were challenged with hypotonic solutions. Glibenclamide (Sigma; St Louis, MO, USA), niflumic acid (Sigma), 4,4′-diisothiocyanostilbene-2,2′-disulphonate (DIDS; Sigma), phorbol 12,13-dibutyrate (PDBu; Sigma), bisindolylmaleimide I-HCl (BIM; Calbiochem, San Diego, CA, USA) and 3-isobutyl-1-methyxanthine (IBMX; Calbiochem), were prepared as stock solutions in dimethyl sulphoxide (DMSO). The final concentration of DMSO in the bath solutions was < 0.1%, which, by itself, did not affect Cl− currents. 8-Br-cAMP (Sigma) was freshly dissolved in the bath solution. All other chemicals were purchased from Sigma.
Results
Comparison of native VSOACs in atrial myocytes from Clcn3+/+ and _Clcn3_−/− mice
Figure 1 illustrates the activation of native VSOAC currents and compares their properties in single atrial cells isolated from Clcn3+/+ wild-type (WT) and _Clcn3_−/− knockout (KO) mice. Figure 1_A_ (WT) and B (KO) shows the time course of change in the amplitudes of whole-cell currents at +80 mV (•) and −80 mV (○) in cells initially exposed to isotonic, hypotonic and hypertonic solutions. Small whole-cell currents were observed in both WT and KO atrial cells in the presence of isotonic bath solutions. Current amplitudes in both WT and KO cells gradually increased after changing to the hypotonic solution, and were almost completely reversed by subsequent exposure to hypertonic solutions. There were no significant differences between WT (n = 18) and KO cells (n = 6) with regard to the mean time to start current activation upon hypotonic challenge (3.3 ± 0.4 min in WT versus 2.9 ± 0.5 min in KO), the mean time for maximal current activation (8.8 ± 0.3 min in WT versus 8.2 ± 0.3 min in KO), and the mean time for current deactivation upon exposure to hypertonic solution (13.8 ± 1.3 min in WT versus 12.3 ± 1.2 min in KO). Table 1 compares the half-times for maximal current activation or deactivation between WT and KO cells, which were not significantly different. Figure 1_C_ (WT) and D (KO) shows representative currents elicited by the voltage protocol illustrated under isotonic and hypotonic conditions, respectively. These currents were obtained at the corresponding time points (a) and (b) in Fig. 1_A_ and B. The hypotonic-induced VSOAC currents in both WT and KO cells displayed moderate outward rectification and time-dependent inactivation at positive potentials. Comparison of current–voltage (I_–_V) relationships shown in Fig. 1_E_ demonstrates that VSOAC current densities in WT cells were not significantly different from that in KO cells at all test potentials, nor were the reversal potentials (_V_rev) between WT (−2.9 ± 1.7 mV) and KO cells (−4.5 ± 3.3 mV), both of which were close to the estimated Cl− equilibrium potential (_E_Cl = 0 mV with symmetrical Cl− solutions). These properties are consistent with previous reports for native VSOACs in cardiac cells (Hagiwara et al. 1992; Sorota, 1992; Vandenberg et al. 1994; Sakaguchi et al. 1997; Yamazaki & Hume, 1997; Duan et al. 1999_a_; Nagasaki et al. 2000). When the external NaCl was replaced with an equimolar concentration of NaI or sodium aspartate (Fig. 1_F_; WT, n = 5; Fig. 1_G_; KO, n = 6), I− substitution of [Cl−]o shifted _V_rev of the currents from −2.4 ± 1.4 mV to −8.8 ± 1.8 mV (WT) and from −3.3 ± 2.9 mV to −10.4 ± 2.6 mV (KO), respectively, and Asp− substitution shifted _V_rev to 26.4 ± 3.8 mV (WT) and 27.6 ± 4.3 mV (KO), respectively. Permeability ratios (_P_x/_P_Cl) were calculated from the shift of _V_rev using the modified Goldman–Hodgkin–Katz equation. For myocytes from WT and KO mice, _P_I/_P_Cl ratios were 1.56 ± 0.13 and 1.61 ± 0.13, respectively, and _P_Asp/_P_Cl ratios were 0.41 ± 0.07 and 0.37 ± 0.04, respectively. These results indicate that VSOAC currents recorded from atrial myocytes from WT and KO mice are similar in terms of current densities, kinetics, rectification, and anion permeability (I− > Cl−≫ Asp−).
To compare the pharmacological properties of VSOACs in atrial myocytes from WT and KO mice, we examined the effects of the Cl− channel inhibitors glibenclamide (0.2 mm) (Sakaguchi et al. 1997; Yamazaki & Hume, 1997), 4,4′-diisothiocyanostilbene-2,2′-disulphonate (DIDS; 0.1 mm) (Hagiwara et al. 1992; Sorota, 1992; Vandenberg et al. 1994), niflumic acid (0.1 mm) (Sorota, 1994), and extracellular ATP (1 mm) (Duan et al. 1997, 1999_a_). Figure 1_H_ shows the mean blocking effects of these compounds at +80 and −80 mV (see Methods). Glibenclamide and niflumic acid inhibited VSOACs in cells from WT by 25.2 ± 9.2% (n = 4) and 65.0 ± 12.1% (n = 4), respectively, at +80 mV and by 20.4 ± 8.8 and 57.3 ± 11.2%, respectively, at −80 mV, and inhibited VSOACs in cells from KO by 7.8 ± 12.1% (n = 4) and 63.5 ± 18.8% (n = 4), respectively, at +80 mV and 15.3 ± 7.9 and 53.1 ± 10.4%, respectively, at −80 mV. At +80 mV, DIDS significantly inhibited VSOACs from WT (n = 4) and KO (n = 4) by 51.4 ± 14.9 and 32.8 ± 22.9%, respectively, whereas less potent effects of these compounds were observed at −80 mV (2.3 ± 5.1 and 3.4 ± 2.0%, respectively). Extracellular ATP inhibited VSOACs from WT and KO myocytes by 89.6 ± 12.6 and 95.1 ± 27.3%, respectively, at +80 mV, and by 29.4 ± 12.9 and 15.9 ± 14.0%, respectively, at −80 mV. These pharmacological results illustrate voltage-independent inhibition by glibenclamide and niflumic acid, and voltage-dependent inhibition by DIDS and extracellular ATP of VSOAC currents in myocytes from both WT and KO mice that were virtually indistinguishable in the two cell types.
Differential regulation of native VSOACs by protein kinase C in atrial myocytes from Clcn3+/+ and _Clcn3_−/− mice
We next examined the regulation of native VSOACs in atrial myocytes from WT and KO mice by endogenous protein kinase C (PKC). As shown in Fig. 2_A_, native VSOACs in atrial myocytes from WT mice were strongly inhibited by activation of endogenous PKC using phorbol 12,13-dibutyrate (PDBu; 100 nm). From I_–_V relationships (n = 6) shown in Fig. 2_C_, the mean densities of VSOAC currents preactivated by exposure to hypotonic solutions were significantly decreased by PDBu from 5.29 ± 0.81 to 1.59 ± 0.41 pA pF−1 at +80 mV (P < 0.01), and from −2.96 ± 0.82 to −1.24 ± 0.69 pA pF−1 at −80 mV (P < 0.01). In contrast, native VSOACs in atrial myocytes from KO mice were surprisingly unaffected by PDBu, as shown in Fig. 2_B_ and the mean I_–_V relationships (n = 7) in Fig. 2_D_. The mean densities of VSOAC currents in these cells preactivated by exposure to hypotonic solutions, and after exposure to PDBu were 6.13 ± 1.05 and 7.20 ± 1.70 pA pF−1 at +80 mV, respectively, and −2.62 ± 0.53 and −3.04 ± 0.67 pA pF−1 at −80 mV, respectively.
Figure 2. Comparison of endogenous PKC regulation of VSOAC currents in atrial myocytes from Clcn3+/+ (WT) and _Clcn3_−/− (KO) mice.

A and B, time course of inhibition of VSOAC currents at +80 mV (•) and −80 mV (○) in atrial cells of WT (A) and KO (B) mice exposed to hypotonic solution and the subsequent application of 100 nm PDBu. C and D, mean I_–_V relationships of VSOAC currents from WT (n = 6) mice and KO (n = 7) mice at the time points (a and b) indicated in A and B. E and F, time course of activation of VSOAC currents at +80 mV (•) and −80 mV (○) in atrial myocytes of WT (E) and KO (F) mice in isotonic or hypotonic solution in the presence of 100 nm BIM. G and H, mean I_–_V relationships of VSOAC currents from WT (n = 6) mice and KO (n = 7) mice, respectively, at the time points (a, b and c) indicated in E and F. I, mean I_–_V relationships of BIM-induced VSOAC currents in myocytes from WT (•) mice and KO (○) mice in isotonic solution.
We also compared the sensitivity of native VSOACs in atrial myocytes from WT and KO mice to inhibition of endogenous PKC activity, using bisindolylmaleimide I-HCl (BIM; 100 nm), which at this concentration is a more selective inhibitor of conventional (c)PKC isozymes, compared to novel (n)PKC isozymes (Martiny-Baron et al. 1993). As shown in Fig. 2E, exposure of atrial cells from WT mice to BIM (100 nm) under isotonic conditions caused activation of native VSOACs, which were further activated by subsequent exposure of cells to hypotonic solutions. Figure 2_G_ summarizes the mean I_–_V relationships (n = 6) under these conditions. Mean current densities at +80 and −80 mV were, respectively, 1.56 ± 0.07 and −1.02 ± 0.50 pA pF−1 (isotonic), 2.52 ± 0.20 and −1.18 ± 0.42 pA pF−1 (isotonic + BIM) and 5.51 ± 1.15 and −2.31 ± 0.98 pA pF−1 (hypotonic + BIM). In marked contrast, VSOACs in atrial myocytes from KO mice were insensitive to inhibition of endogenous PKC activity by BIM (Fig. 2_F_ and H). Mean current densities at +80 and −80 mV were, respectively, 1.66 ± 0.43 and −0.93 ± 0.40 pA pF−1 (isotonic), 1.83 ± 0.46 and −1.02 ± 0.39 pA pF−1 (isotonic + BIM) and 5.95 ± 0.47 and −2.91 ± 0.44 pA pF−1 (hypotonic + BIM).
It is possible that the lack of PKC modulation of VSOACs observed in atrial myocytes from KO mice compared to WT might be due to down-regulation of an important effector in the endogenous PKC pathway. In order to test this possibility, we examined the integrity of the PKC pathway in these cells from KO mice by testing whether or not PKC activation leads to stimulation of CFTR (cystic fibrosis transmembrane conductance regulator) Cl− channels in these cells (Levesque et al. 1992; Nagel et al. 1992). Figure 3_A_ shows the time course of activation of whole-cell currents at +80 and −80 mV in an atrial myocyte under symmetrical Cl− (140 mm) conditions. Application of 100 nm PDBu caused an increase in membrane currents, which was further enhanced by addition of a cAMP cocktail containing 0.5 mm 8-Br-cAMP and 2 mm IBMX. The original current traces at the indicated time points are displayed in Fig. 3_B_, and the mean I_–_V relations are shown in Fig. 3_C_. The difference current traces depicted in Fig. 3_B_ represent the PDBu-sensitive (b – a) and cAMP cocktail-sensitive (c – b) currents, respectively. The mean difference currents (n = 6) shown in Fig. 3_D_ clearly demonstrate a linear I_–_V relation with reversal potentials close to 0 mV (i.e. the theoretical Cl− equilibrium potential). These characteristics are identical to those of CFTR Cl− currents observed in cardiac myocytes from WT mice (Duan et al. 1999_b_), and are distinct from VSOACs which exhibit outward rectification under these conditions. These data demonstrate that atrial myocytes from KO mice retain a functional PKC pathway.
Figure 3. PKC-activated CFTR currents in atrial myocytes from _Clcn3_−/− (KO) mice.

A, time course of changes of CFTR currents at +80 mV (•) and −80 mV (○) in atrial cells of KO mice before (arrow at time a), after exposure to 100 nm PDBu (arrow at time b) and after exposure to cAMP cocktail (0.5 mm 8-Br-cAMP +2.0 mm IBMX; arrow at time c). B, CFTR currents were activated by PDBu and further enhanced by cAMP cocktail. Currents were recorded at the time points (a, b and c) indicated in A; traces labelled b ∓ a and c ∓ b are PDBu- and cAMP cocktail-induced difference currents, respectively. C, mean I_–_V relationships at the same time points (a, b and c) in A. PDBu activated CFTR current in 4 out of 15 cells, and cAMP cocktail enhancement was observed in all 4 cells. D, mean I_–_V relationships of PDBu- (b – a) and cAMP cocktail- (c – b) induced difference currents.
VSOACs in atrial myocytes from Clcn3+/+ and _Clcn3_−/− mice exhibit differential sensitivity to block by anti-ClC-3 antibody
It has previously been shown that intracellular dialysis of a commercially available carboxy terminus anti-ClC-3 antibody (Ab) (Duan et al. 2001) and two newly developed Abs against either an amino terminus epitope (A1–14 Ab) or a carboxy terminus epitope (C670–687 Ab) strongly inhibit native VSOACs in guinea-pig cardiac atrial and canine pulmonary arterial smooth muscle cells (Wang et al. 2003). We examined the effects of intracellular dialysis of the new A1–14 Ab on native VSOACs in mouse atrial myocytes from Clcn3+/+ (WT) and _Clcn3_−/− (KO) mice. Figure 4_A_ and B illustrates the effects of intracellular dialysis of A1–14 Ab on activation of VSOACs by exposure to hypotonic solutions in WT and KO atrial cells. There was a marked suppression of VSOAC currents in WT atrial cells dialysed with A1–14 Ab, which was not observed in cells dialysed with the antigen-preabsorbed A1–14 Ab (data not shown). In contrast, the activation of VSOACs in KO atrial cells was unaffected by intracellular dialysis with A1–14 Ab (Fig. 4_B_ and D). The mean I_–_V relationships of currents recorded in isotonic and hypotonic solutions for WT and KO atrial cells dialysed with A1–14 Ab is shown in Fig. 4_E_ and F. The hypotonic-sensitive difference currents (b – a in Fig. 4_E_ and F) show that the VSOAC current densities for WT (n = 10) and KO (n = 7) cells were −0.92 ± 1.0 and 6.12 ± 0.94 pA pF−1 (P < 0.01) at +120 mV, respectively (Fig. 4_G_). These results demonstrate that native VSOACs in myocytes from WT mice were completely inhibited by intracellular dialysis with the ClC-3 A1–14 Ab, while VSOACs in KO mice appeared to be completely insensitive to ClC-3 A1–14 Ab.
Figure 4. Comparison of effects of intracellular dialysis of anti-ClC-3 Ab (A1–14 Ab) on VSOAC currents in atrial myocytes from Clcn3+/+ (WT) and _Clcn3_−/− (KO) mice.

A and B, time course of changes of VSOAC currents at +80 mV (•) and −80 mV (○) in atrial cells from WT (A) and KO (B) mice with intracellular dialysis of 1.5 μg ml−1 anti-ClC-3 antibody (A1–14 Ab). C and D, VSOAC currents in myocytes from WT mice (Ca and b) and from KO mice (Da and b) in isotonic and hypotonic solutions, respectively. Currents were recorded at the time points (a and b) indicated in A and B. E and F, mean I_–_V relationships of WT (n = 10) and KO (n = 7) in isotonic and hypotonic solutions, respectively. G, mean I_–_V relationships of hypotonic-activated Cl− currents in myocytes from WT (•) and KO (○) mice.
VSOACs in atrial myocytes from Clcn3+/+ and _Clcn3_−/− mice exhibit differential sensitivity to [ATP]i and free [Mg2+]i
A well known property of VSOACs in many mammalian cells is the dependence for activation on intracellular ATP and Mg2+ (Strange et al. 1996; Okada, 1997; Nilius & Droogmans, 2003). We examined the dependence of native VSOACs in atrial myocytes from Clcn3+/+ and _Clcn3_−/− mice on [ATP]i and free [Mg2+]i. To deplete intracellular ATP, cells were initially exposed to glucose-free external solutions containing NaCN (2 mm), and were dialysed with an ATP-free pipette solution, containing 0.652 nm free [Mg2+]i. In atrial cells from WT animals (Fig. 5_A_), intracellular ATP depletion significantly decreased the rate of activation of VSOACs during exposure to hypotonic solutions (control, _t_½ = 6.9 ± 0.8 min (n = 18); ATP depletion, _t_½ = 12.1 ± 1.6 min (n = 4); P < 0.005) but not in atrial cells from KO animals (control, _t_½ = 5.7 ± 0.4 min (n = 6); ATP depletion, _t_½ = 5.2 ± 1.0 min (n = 4); n.s.) (Fig. 5_B_). The maximum densities of VSOAC currents activated in the two cell types were similar in the steady state. VSOAC current densities after 30 min exposure to hypotonic solutions were 5.62 ± 0.67 pA pF−1 for WT cells (Fig. 5_C_) and 6.36 ± 0.60 pA pF−1 for KO cells (Fig. 5_D_) at +80 mV.
Figure 5. Comparison of sensitivity to [ATP]i and free [Mg2+]i of VSOAC currents in atrial myocytes from Clcn3+/+ (WT) and _Clcn3_−/− (KO) mice.

A and B, time course of activation of VSOAC currents at +80 mV (•) and −80 mV (○) in atrial cells from WT and KO mice with ATP depletion (0 mm ATP in pipette solution (0 mm[ATP]i) and 0 mm glucose +2 mm NaCN in bathing solution). C and D, mean I_–_V relationships of WT (n = 4) and KO (n = 4) cells at the time points (a, b and c) of indicated in A and B, respectively. E and F, time course of inhibition of VSOAC currents at +80 mV (•) and −80 mV (○) in myocytes from WT (E) and KO (F) with ATP depletion plus 2 mm intracellular free Mg2+ (high free [Mg2+]i). G and H, mean I_–_V relationships of VSOAC currents from WT (n = 4) and KO (n = 4) mice at the time points indicated in E and F. I, mean I_–_V relationships of hypotonic-activated difference currents in myocytes from WT (•) and KO (○) mice.
We then compared the effects of high free [Mg2+]i (2.0 mm) on native VSOACs in [ATP]i-depleted atrial myocytes from Clcn3+/+ and _Clcn3_−/− mice. As shown in Fig. 5_E_, the activation of VSOACs in cells from WT mice was completely suppressed by the increase in free [Mg2+]i, whereas some residual VSOAC current activation was still observed under identical conditions in cells from KO mice (Fig. 5_F_). The mean I_–_V relationships of currents recorded in isotonic and hypotonic solutions for WT and KO atrial cells are shown in Fig. 5_G_ and H. The hypotonic-sensitive difference currents (b – a in Fig. 5_G_ and H) show that the VSOAC current densities for WT (n = 10) and KO (n = 7) cells were 0.49 ± 0.27 and 2.34 ± 0.66 pA pF−1(P < 0.05) at +120 mV, respectively (Fig. 5_I_). These results demonstrate that native VSOACs in atrial myocytes from WT and KO mice exhibit significant differences in their sensitivity to [ATP]i depletion and high free [Mg2+]i.
VSOACs in pulmonary arterial smooth muscle cells from Clcn3+/+ and _Clcn3_−/− mice exhibit differential sensitivity to PKC and anti-ClC-3 A1–14 Ab
To test whether or not the observed differences (described above) in the properties of VSOACs in cardiac myocytes from _Clcn3_−/− mice, compared to VSOACs from myocytes of Clcn3+/+ mice, may be more general and characteristic of other cells as well, we also compared the sensitivity of VSOACs in pulmonary arterial smooth muscle cells (PASMCs) (Yamazaki et al. 1998; Zhong et al. 2002) from WT and KO mice, to PDBu and to the anti-ClC-3 A1–14 Ab. As shown in Fig. 6, mean VSOAC current densities from WT cells preactivated by exposure to hypotonic solutions were significantly decreased by exposure to PDBu from 2.77 ± 0.35 to 2.0 ± 0.16 pA pF−1 at +80 mV (P < 0.05), and from −0.75 ± 0.17 to −0.54 ± 0.10 pA pF−1 at −80 mV (P < 0.05). However, mean VSOAC current densities from KO myocytes were unaffected by PDBu (2.65 ± 0.21 and 2.85 ± 0.48 pA pF−1 at +80 mV and −0.66 ± 0.12 and −0.68 ± 0.16 pA pF−1 at −80 mV, for hypotonic and hypotonic + PDBu solutions, respectively).
Figure 6. Comparison of the effects of PDBu and anti-ClC-3 (A1–14) Ab on native VSOACs in pulmonary artery smooth muscle cells (PASMCs) from Clcn3+/+ (WT) and _Clcn3_−/− mice (KO).

A, representative VSOAC currents recorded in isotonic, hypotonic and hypotonic +100 nm PDBu solutions. Membrane currents were elicited by 400 ms voltage steps ranging from −80 to +80 mV in 20 mV increments from a holding potential of −40 mV. B, mean current densities measured at −80 mV (downward bars) and +80 mV (upward bars) in isotonic, hypotonic, and hypotonic + PDBu solutions. C, mean current densities measured at +80 mV (upward bars) and −80 mV (downward bars) in isotonic and hypotonic solutions for PASMCs from WT and KO mice intracellularly dialysed with 1.5 μg ml−1 A1–14 Ab.
In PASMCs intracellularly dialysed with 1.5 μg l−1 A1–14 Ab, mean current densities in isotonic and hypotonic solutions for WT cells were 2.08 ± 0.54 and 2.69 ± 0.78 pA pF−1 at +80 mV (n.s.) and −0.49 ± 0.10 and −0.59 ± 0.15 pA pF−1 at −80 mV (n.s.), respectively; mean current densities in isotonic and hypotonic solutions for KO cells were 1.56 ± 0.47 and 3.25 ± 0.73 pA pF−1 at +80 mV (P < 0.05) and −0.52 ± 0.09 and −0.79 ± 0.08 pA pF−1 at −80 mV (P < 0.05), respectively. These results confirm that similar to atrial myocytes, VSOACs in PASMCs from WT mice were inhibited by bath application of PDBu or intracellular dialysis of A1–14 Ab, whereas VSOACs in PASMCs from KO mice were insensitive to both PDBu and the anti-ClC-3 A1–14 Ab. No significant differences were observed for VSOAC current activation kinetics between WT and KO mice (Table 1).
Altered ClC Cl− channel mRNA expression in atria and brain from _Clcn3_−/− mice
We next compared mRNA transcript levels for ClC-1–ClC-5 in atria and brain from Clcn3+/+ and _Clcn3_−/− mice to determine if there may be compensatory changes in expression of message of any of these Cl− channels in response to Clcn3 gene inactivation. The rationale for these experiments was that ClC-3, ClC-4 and ClC-5 belong to the same subfamily of ClC Cl− channels and therefore have the potential of combining to form heterodimeric channels (Jentsch et al. 2002). As illustrated in Fig. 7, quantitative RT-PCR revealed an approximate 100-fold increase in mRNA transcript levels for ClC-1 and ClC-2, and an approximate 4- to 5-fold increase in mRNA transcript levels for ClC-3 in atria from _Clcn3_−/− compared to Clcn3+/+ mice. In brain, mRNA transcript levels for ClC-2 and ClC-3 were also significantly higher in _Clcn3_−/−, compared to Clcn3+/+ mice. In contrast to atria, little change in ClC-1 transcript levels were observed in brain. mRNA transcripts for ClC-4 were close to undetectable in atria and brain, and although low transcript levels for ClC-5 were detectable in brain, they were undetectable in atria. No significant changes in ClC-4 or ClC-5 transcripts were observed in atria or brain from _Clcn3_−/− and Clcn3+/+ mice.
Altered membrane protein expression in atrial myocytes from _Clcn3_−/− mice
To test the possibility that targeted inactivation of the Clcn3 gene using a conventional murine global knock-out approach might result in compensatory changes in expression of other membrane proteins in cells from _Clcn3_−/− mice, cardiac membrane proteins were isolated from Clcn3+/+ and _Clcn3_−/− mice and analysed using two-dimensional (2-D) polyacrylamide gel electrophoresis. 753 distinct membrane proteins were initially identified in atrial cell membrane extracts, and up to 104 proteins appeared to exhibit altered expression levels in cells from _Clcn3_−/− mice, compared to atrial cells from WT mice. In order to minimize spurious results that might arise due to expected small variations in protein spot densities that normally occur from gel to gel, we established a minimal detection criterion of a two-fold change in spot density in order to more reliably quantify actual changes in protein expression. Using this criterion, comparisons of 2-D gels from membrane extracts isolated from Clcn3+/+ and _Clcn3_−/− mice (Fig. 8) consistently revealed significant changes in the expression of at least 35 distinct membrane proteins (6 missing proteins, 2 new proteins, 9 up-regulated proteins, 15 down-regulated proteins, and 2 translocated proteins) from hearts of _Clcn3_−/− mice (n = 6) compared to Clcn3+/+ mice (n = 6). One of the six missing proteins was identified as ClC-3 by Western blot analysis of the 2-D gels (data not shown). The location (molecular mass = 85 kDa and pI = 6.9) of the ClC-3 protein spot (No. 3812) in the 2-D gels from WT mice (□) is consistent with the predicted molecular mass of 84 kDa (Borsani et al. 1995) and pI (6.91) for ClC-3.
Figure 8. Two-dimensional electrophoresis analysis of protein expression patterns in membranes of cardiac cells from Clcn3+/+ and _Clcn3_−/− mice.

A, representative 2-D gel depicts Coomassie-stained proteins from wild-type (Clcn3+/+) mouse heart. B, representative 2-D gel depicts Coomassie-stained proteins from _Clcn3_−/− mouse heart. C, spot sets created from images of 2-D gels of both wild-type and _Clcn3_−/− mouse heart run under the same conditions as the gels in A and B and compared using Bio-Rad PDQuest version 7.1.1 software. Three gels were run for each mouse heart type; two hearts were pooled to provide proteins for each gel. The filled symbols indicate changes in protein patterns in _Clcn3_−/− compared to wild-type. A total of 35 proteins consistently changed (minimum criteria: > 2-fold change) in membranes from _Clcn3_−/− mouse heart in all 3 experiments (6 missing proteins, 2 new proteins, 9 up-regulated proteins, 15 down-regulated proteins, and 2 translocated proteins). The open squares (□) in A, B, and C indicate the location (molecular mass 85 kDa and pI 6.9) of the ClC-3 protein spot (No. 3812) in the 2-D gels, which was independently confirmed by Western blotting using a specific anti-ClC-3 C670–687 antibody.
Discussion
The original demonstration of the presence of native VSOACs in hepatocytes and pancreatic acinar cells from _Clcn3_−/− mice (Stobrawa et al. 2001) has been considered strong evidence arguing against a significant role of ClC-3 as a molecular candidate responsible for native VSOACs (Wills & Fong, 2001; Jentsch et al. 2002; Li & Weinman, 2002; Nilius & Droogmans, 2003). Yet, an alternative explanation for this observation, that in response to loss of expression of endogenous ClC-3 another protein may be up-regulated that alters VSOAC channel subunit composition or an associated regulatory subunit that may give rise to VSOACs with different properties, has yet to be tested. There are numerous examples of activation of compensatory mechanisms in response to conventional gene inactivation, which can complicate the accurate assessment of the phenotypic impact of the gene in question. One example in heart is the significant electrical remodelling that has been shown to occur in response to targeted deletion of several K+ channel subunit genes (Nerbonne et al. 2001; Warth & Barhanin, 2002). In the present study, we compared the properties of native VSOACs in atrial myocytes from Clcn3+/+ and _Clcn3_−/− mice to determine whether the presence of VSOACs in myocytes from _Clcn3_−/− mice can be attributed to lack of involvement of ClC-3 as an endogenous protein responsible for native VSOACs, as previously suggested (Stobrawa et al. 2001), or whether the properties of VSOACs in these cells may differ, possibly due to compensatory up-regulation of a different protein in response to Clcn3 gene deletion.
Our experiments showed that VSOAC currents activated by hypotonic cell swelling in atrial myocytes from WT and KO mice were remarkably similar in appearance, including activation kinetics, steady-state current densities and slight outward rectification. They also exhibited similarities in anion selectivity (I− > Cl− ≫ Asp−) and sensitivity to block by glibenclamide, niflumic acid, DIDS and extracellular ATP (Fig. 1). Based on these criteria alone, VSOACs in the two cell types appeared indistinguishable. However, further examination revealed significant differences in the sensitivity of VSOACs from WT and KO mice to modulation by endogenous PKC, inhibition by intracellular dialysis with a new anti-ClC-3 A1–14 Ab, and differences in sensitivity to [ATP]i depletion and high free [Mg2+]i. These observations suggest that the properties of VSOACs in atrial myocytes are significantly altered by targeted deletion of the Clcn3 gene.
Previous reports provide data suggesting an essential role of endogenous PKC phosphorylation/dephosphorylation in the cell volume regulatory mechanisms controlling native VSOAC activity in guinea-pig cardiac myocytes (Duan et al. 1999_a_), canine smooth muscle cells (Zhong et al. 2002), Xenopus oocytes (Souktani et al. 2000), rat brain endothelial cells (von Weikersthal et al. 1999), human ciliary epithelial cells (Coca-Prados et al. 1996), and recombinant ClC-3 Cl− channels expressed in NIH/3T3 cells (Duan et al. 1999_a_). In these cells, VSOACs are strongly inhibited by activation of endogenous PKC, and can be activated under isotonic conditions by inhibition of endogenous PKC. These results are consistent with a proposed model (Horowitz et al. 1999) in which cell swelling promotes a shift in equilibrium to favour channel dephosphorylation and channel opening, and cell shrinkage promotes channel phosphorylation and channel closure; similar to models recently proposed to account for cell volume regulation of ClC-2 Cl− channels as well (Rutledge et al. 2002; Furukawa et al. 2002). In contrast, in many cells native VSOACs have been found to be rather insensitive to regulation by endogenous PKC (Nilius & Droogmans, 2003), or have been reported to be regulated in the opposite direction by endogenous PKC (Du & Sorota, 1999; Ellershaw et al. 2002).
We previously demonstrated that native VSOACs in cardiac and smooth muscle cells, and Xenopus oocytes, as well as expressed _I_gpClC-3, were found to be significantly inhibited by intracellular dialysis of a commercially available anti-ClC-3 Ab (Duan et al. 2001), although the specificity of this Ab has been questioned (Stobrawa et al. 2001). More recently, VSOACs elicited by hypotonic cell swelling in canine PASMCs and guinea-pig atrial myocytes were shown to be nearly completely abolished by intracellular dialysis with two new anti-ClC-3 Abs specifically targeting the ClC-3 carboxy (C670–687 Ab) and amino terminus (A1–14 Ab) (Wang et al. 2003). These new Abs produced a common prominent immunoreactive band with an apparent molecular mass of 90−92 kDa in guinea-pig heart, canine PASMCs, and brain from homozgygous Clcn3+/+ mice, but not from homozygous _Clcn3_−/− mice. The inhibitory effects of these Abs on native VSOACs appear to be attributable to a specific interaction with endogenous ClC-3 since preabsorption of the Abs with corresponding antigens prevents the inhibitory effects, the Abs have no effect on swelling-induced augmentation of the slow component of the delayed rectifying potassium current (_I_Ks) in guinea-pig ventricular myocytes, and intracellular dialysis with an Ab targeting Kv1.1 potassium channels failed to inhibit native VSOACs. It also has been recently shown that ClC-3 antisense oligonucleotides or antisense cRNA treatment significantly reduces endogenous ClC-3 immunoreactivity, assessed using these new anti-ClC-3 Abs, and significantly reduces the density of native VSOACs and regulatory volume decreases in Hela cells and Xenopus oocytes (Hermoso et al. 2002). The lack of inhibitory effect of these anti-ClC-3 Abs on native VSOACs in atrial myocytes and PASMCs from _Clcn3_−/− mice observed here, is further evidence strongly supporting a specific interaction of these Abs with endogenous ClC-3 in cells from Clcn3+/+ mice. These data also support the conclusion that native VSOACs in cells from _Clcn3_−/− mice are mediated or regulated by a yet to be identified protein, distinct from ClC-3.
It is interesting that VSOACs in atria and PASMCs from _Clcn3_−/− mice exhibit both a lack of sensitivity to the inhibitory effects of anti-ClC-3 Abs and phorbol esters, suggesting that these two properties are somehow related. Differential sensitivity of native VSOACs to anti-ClC-3 Abs and phorbol esters in different cell types in normal animals is consistent with the possible expression of distinct VSOAC subtypes. The properties of VSOACs in atria and PASMCs from _Clcn3_−/− mice may therefore represent a VSOAC subtype normally expressed in some cells from normal animals. For example, activation of PKC potentiates native VSOACs in cells from rabbit portal vein (Ellershaw et al. 2002) and these currents are also unaffected by dialysis with a commercially available ClC-3 Ab (Zhong et al. 2001). This result has also recently been confirmed using the new A1–14 ClC-3 Ab (G.-X. Wang & J. R. Hume, unpublished observation).
Low [ATP]i and high [Mg2+]i are known to inhibit VSOACs in a large variety of cells (Strange et al. 1996; Okada, 1997; Nilius & Droogmans, 2003), including cardiac myocytes (Sakaguchi et al. 1997), by a mechanism independent of ATP hydrolysis or phosphorylation. The dependence on [ATP]i may indicate non-hydrolytic binding of ATP to the channel or to an accessory protein. In our experiments, intracellular ATP depletion in atrial cells from Clcn3+/+ mice significantly decreased the rate of activation of VSOACs during exposure to hypotonic solutions. This effect may indicate that VSOACs in these cells may normally be activated by both an ATP-dependent pathway and a slower ATP-independent pathway, as previously demonstrated in neuroblastoma cells (Bond et al. 1999). In marked contrast, VSOACs recorded from atrial cells from _Clcn3_−/− mice were relatively insensitive to depletion of [ATP]i and exhibited lower sensitivity to block by high [Mg2+]i. This finding, along with the relative insensitivity of these channels to regulation by PKC, demonstrates that additional experiments are required to determine the regulatory pathways responsible for linking changes in cell volume to activation of VSOACs in cells from _Clcn3_−/− mice.
The finding that mRNA transcripts for ClC-1, ClC-2, and ClC-3 were significantly elevated in atria from _Clcn3_−/− mice, compared to Clcn3+/+ mice, may be physiologically relevant to the appearance of VSOACs with different properties in _Clcn3_−/− mice. Our finding that mRNA transcript levels for ClC-4 and ClC-5 were unchanged in atria and brain of _Clcn3_−/− mice confirms earlier findings reported by Stobrawa and colleagues (Stobrawa et al. 2001). In the case of elevated transcript levels of ClC-3 mRNA, this may not be physiologically significant, since expression of the truncated transcript in _Clcn3_−/− mice fails to be transcribed into protein (Stobrawa et al. 2001; Dickerson et al. 2002; Wang et al. 2003). In contrast, up-regulation of mRNA transcripts for ClC-1 and ClC-2, may be physiologically relevant to the appearance of a novel VSOACs in _Clcn3_−/− mice. While it is unlikely this can be directly attributed to either ClC-1 or ClC-2 alone, since the properties of these channels are clearly distinct from typical VSOACs, the possibility exists that these proteins may combine with each other or with other related proteins to form heterodimeric channels with unique properties (Jentsch et al. 2002).
Alternatively, the altered properties of VSOACs in _Clcn3_−/− mice may be due to compensatory expression of a protein unrelated to the ClC Cl− channel family, which may act to maintain the fundamental role of VSOACs in cell volume regulatory mechanisms in _Clcn3_−/− mice (Arreola et al. 2002). Indeed, the results of two-dimensional gel electrophoresis analysis comparing membrane protein expression patterns revealed alterations in at least 35 distinct proteins in atrial myocytes from _Clcn3_−/− mice, compared to myocytes from Clcn3+/+ mice.
In summary, our results reveal several significant differences in properties of VSOACs recorded from atrial and smooth muscle cells from Clcn3+/+ and _Clcn3_−/− mice, including differences in regulation by endogenous PKC, sensitivity to block by anti-ClC-3 Abs, and [ATP]i and free [Mg2+]i sensitivity. These data suggest that in response to loss of expression of endogenous ClC-3, another protein may be up-regulated that may interact with VSOACs, alter VSOAC channel subunit composition, or may represent a channel-associated regulatory subunit in cells from _Clcn3_−/− mice. The fact that sensitivity to inhibition by anti-ClC-3 Abs, and sensitivity to PKC and changes in volume are conferred by specific amino acid residues in the ClC-3 protein (Wang et al. 2003) argues that it is part of the channel protein itself that is altered in _Clcn3_−/− mice, rather than alteration of an associated regulatory protein. This is also supported by data in which mutations of specific amino acids in recombinant ClC-3 alter PKC and volume sensitivity (Duan et al. 1999_a_) of the expressed currents. Yet, similarities in current kinetics, steady-state current densities, rectification, anion selectivity (I−> Cl− ≫ Asp−) and sensitivity to block by Cl− channel inhibitors in cells from _Clcn3_−/− mice might be taken as evidence that ClC-3 itself plays no role in the pore forming part of VSOACs, and that the altered properties observed are due to changes in a channel-associated regulatory subunit. However, an earlier N579K mutation of gpClC-3 produced marked alterations in rectification and anion selectivity (Duan et al. 1997) of the expressed currents. Clearly, in comparing VSOACs in cells from Clcn3+/+ and _Clcn3_−/− mice, more definitive information on single channel properties and both fast and slow voltage-dependent gating mechanisms (Dutzler et al. 2003; Estevez & Jentsch, 2002) is required to conclusively decide whether or not ClC-3 contributes to the pore forming part of the channel. The use of other, simpler strategies to eliminate endogenous ClC-3 expression, including antisense oligonucleotides and/or cRNA, or siRNA, which may not produce such dramatic compensatory changes in other proteins, may be fruitful approaches to examine this question. Future studies carefully identifying the specific compensatory membrane proteins that are altered in cells from _Clcn3_−/− mice will be useful in providing a unique opportunity to gain new insights into the molecular diversity of the proteins responsible for native VSOAC subtypes. Finally, these data show that conventional methods to produce targeted inactivation of the murine Clcn3 gene result in complex compensatory changes in the expression of a variety of membrane proteins. These changes must be carefully considered when interpreting the physiological consequences of loss of ClC-3.
Acknowledgments
The authors would like to thank Lisa Miller, Linda Ye, Keith Murray, Xiaomin Shen, Yanping Dai, Phillip Keller, Susan Tamowski and Paul Scowen for excellent technical assistance. This study was supported by NIH grants HL49254, and P20RR15581, P20RR16464 from the National Center for Research Resources (NCRR), and is dedicated to the memory of our colleague and friend Burton Horowitz.
References
- Arreola J, Begenisich T, Nehrke K, Nguyen HV, Park K, Richardson L, Yang B, Schutte BC, Lamb FS, Melvin JE. Secretion and cell volume regulation by salivary acinar cells from mice lacking expression of the Clcn3 Cl (-) channel gene. J Physiol. 2002;545:207–216. doi: 10.1113/jphysiol.2002.021980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond T, Basavappa S, Christensen M, Strange K. ATP dependence of the ICl,swell channel varies with rate of cell swelling Evidence for two modes of channel activation. J General Physiol. 1999;113:441–456. doi: 10.1085/jgp.113.3.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borsani G, Ruggarli EI, Taglialatela M, Wong C, Ballabio A. Characterization of a human and murine gene (CLCN3) sharing similarities to voltage-gated chloride channels and to a yeast integral membrane protein. Genomics. 1995;27:131–141. doi: 10.1006/geno.1995.1015. [DOI] [PubMed] [Google Scholar]
- Coca-Prados M, Sanchez-Torres J, Peterson-Yantorno K, Civan MM. Association of ClC-3 channel with Cl− transport by human nonpigmented ciliary epithelial cells. J Memb Biol. 1996;150:197–208. doi: 10.1007/s002329900044. [DOI] [PubMed] [Google Scholar]
- Dickerson LW, Bonthius DJ, Schutte BC, Yang B, Barna TJ, Bailey MC, Nehrke K, Williamson RA, Lamb FS. Altered GABAergic function accompanies hippocampal degeneration in mice lacking ClC-3 voltage-gated chloride channels. Brain Res. 2002;958:227–250. doi: 10.1016/s0006-8993(02)03519-9. [DOI] [PubMed] [Google Scholar]
- Du XY, Sorota S. Protein kinase C stimulates swelling-induced chloride current in canine atrial cells. Pflugers Arch Eur J Physiol. 1999;437:227–234. doi: 10.1007/s004240050773. [DOI] [PubMed] [Google Scholar]
- Duan D, Cowley S, Horowitz B, Hume JR. A serine residue in clc-3 links phosphorylation-dephosphorylation to chloride channel regulation by cell, volume. J General Physiol. 1999a;113:57–70. doi: 10.1085/jgp.113.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan D, Winter C, Cowley S, Hume JR, Horowitz B. Molecular identification of a volume-regulated chloride channel. Nature. 1997;390:417–421. doi: 10.1038/37151. [DOI] [PubMed] [Google Scholar]
- Duan D, Ye L, Britton F, Horowitz B, Hume JR. A novel anionic inward rectifier in native cardiac myocytes. Circ Res. 2000;86:e63–e71. [PubMed] [Google Scholar]
- Duan D, Ye L, Britton F, Miller L, Yamazaki J, Horowitz B, Hume JR. Purinergic-coupled Cl− channels in mouse heart: a novel, alternative pathway for CFTR regulation. J Physiol. 1999b;523:705–717. doi: 10.1111/j.1469-7793.1999.00043.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan D, Zhong J, Hermoso M, Satterwhite CM, Rossow CF, Hatton WJ, Yamboliev I, Horowitz B, Hume JR. Functional inhibition of native volume-sensitive outwardly rectifying anion channels in muscle cells and Xenopus oocytes by anti-ClC-3 antibody. J Physiol. 2001;531:437–444. doi: 10.1111/j.1469-7793.2001.0437i.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutzler R, Campbell EB, Cadene M, Chait BT, MacKinnon R. X-ray structure of a ClC chloride channel at 3.0 A reveals the molecular basis of anion selectivity. Nature. 2002;415:287–294. doi: 10.1038/415287a. [DOI] [PubMed] [Google Scholar]
- Dutzler R, Campbell EB, MacKinnon R. Gating the selectivity filter in ClC chloride channels. Science. 2003;300:108–112. doi: 10.1126/science.1082708. [DOI] [PubMed] [Google Scholar]
- Ellershaw DC, Greenwood IA, Large WA. Modulation of volume-sensitive chloride current by noradrenaline in rabbit portal vein myocytes. J Physiol. 2002;542:537–547. doi: 10.1113/jphysiol.2002.018770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estevez R, Jentsch TJ. CLC chloride channels: correlating structure with function. Curr Op Struct Biol. 2002;12:531–539. doi: 10.1016/s0959-440x(02)00358-5. [DOI] [PubMed] [Google Scholar]
- Furukawa T, Takehiko O, Zheng Y-J, Tsuchiya H, Nakaya H, Katayama Y, Inagaki N. Phosphorylation and functional regulation of ClC-2 chloride channels expressed in Xenopus oocytes by M cyclin-dependent protein kinase. J Physiol. 2002;540:883–893. doi: 10.1113/jphysiol.2001.016188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George AL, Jr, Bianchi L, Link EM, Vanoye CG. From stones to bones: the biology of ClC chloride channels. Curr Biol. 2001;11:R620–R628. doi: 10.1016/s0960-9822(01)00368-2. [DOI] [PubMed] [Google Scholar]
- Hagiwara N, Masuda H, Shoda M, Irisawa H. Stretch-activated anion currents of rabbit cardiac myocytes. J Physiol. 1992;456:285–302. doi: 10.1113/jphysiol.1992.sp019337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermoso M, Satterwhite CM, Andrade YN, Hidalgo J, Wilson SM, Horowitz B, Hume JR. ClC-3 is a fundamental molecular component of volume-sensitive outwardly rectifying Cl− channels and volume regulation in HeLa cells and Xenopus laevis oocytes. J Biol Chem. 2002;277:40066–40074. doi: 10.1074/jbc.M205132200. [DOI] [PubMed] [Google Scholar]
- Horowitz B, Duan D, Dick GM, Yamazaki J, Hume JR. Volume-regulated chloride channels in heart and smooth muscle. In: Kozlowski RZ, editor. Chloride Channels. Oxford: Isis Medical Media, Ltd; 1999. pp. 109–120. [Google Scholar]
- Huang P, Di Lui JA, Robinson NC, Musch MW, Kaetzel MA, Nelson DJ. Regulation of human CLC-3 channels by multifunctional Ca2+/calmodulin-dependent protein kinase. J Biol Chem. 2001;276:20093–20100. doi: 10.1074/jbc.M009376200. [DOI] [PubMed] [Google Scholar]
- Isenberg G, Klockner U. Calcium tolerant ventricular myocytes prepared by preincubation in a ‘KB medium’. Pflugers Arch Eur J Physiol. 1982;395:6–18. doi: 10.1007/BF00584963. [DOI] [PubMed] [Google Scholar]
- Isnard-Bagnis C, DaSilva N, Beaulieu VYuASL, Brown D, Breton S. Detection of ClC-3 and ClC-5 in epidedymal epithelium: immunofluorescence and RT-PCR after LCM. Am J Physiol. 2003;284:C220–C232. doi: 10.1152/ajpcell.00374.2001. [DOI] [PubMed] [Google Scholar]
- Jentsch TJ, Stein V, Weinreich F, Zdebik AA. Molecular structure and physiological function of chloride channels. Physiol Rev. 2002;82:503–568. doi: 10.1152/physrev.00029.2001. [DOI] [PubMed] [Google Scholar]
- Jin NG, Kim JK, Yang DK, Cho SJ, Kim JM, Koh EJ, Jung HC, So I, Kim KW. A fundamental role of ClC-3 in volume-sensitive Cl- channel function and cell volume regulation in AGS cells. Am J Physiol. 2003;285:G938–G948. doi: 10.1152/ajpgi.00470.2002. [DOI] [PubMed] [Google Scholar]
- Levesque PC, Hart PJ, Hume JR, Kenyon JL, Horowitz B. Expression of cystic fibrosis transmembrane regulator Cl− channels in heart. Circ Res. 1992;71:1002–1007. doi: 10.1161/01.res.71.4.1002. [DOI] [PubMed] [Google Scholar]
- Levesque PC, Hume JR. ATPo but not cAMPi activates a chloride conductance in mouse ventricular myocytes. Cardiovas Res. 1995;29:336–343. [PubMed] [Google Scholar]
- Li X, Weinman SA. Chloride channels and hepatocellular function: prospects for molecular identification. Ann Rev Physiol. 2002;64:609–633. doi: 10.1146/annurev.physiol.64.090501.145429. [DOI] [PubMed] [Google Scholar]
- Martiny-Baron G, Kazanietz MG, Mischak H, Blumberg PM, Kochs G, Hug H, Marme D, Schachtele C. Selective inhibition of protein kinase C isozymes by the indolocarbazole Go 6976. J Biol Chem. 1993;268:9194–9197. [PubMed] [Google Scholar]
- Nagasaki MYeL, Duan D, Horowitz B, Hume JR. Intracellular cAMP inhibits native and recombinant volume-regulated chloride channels from heart. J Physiol. 2000;523:705–717. doi: 10.1111/j.1469-7793.2000.00705.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel G, Hwang TC, Nastiuk KL, Nairn AC, Gadsby DC. The protein kinase A-regulated cardiac Cl− channel resembles the cystic fibrosis transmembrane conductance regulator. Nature. 1992;360:81–84. doi: 10.1038/360081a0. [DOI] [PubMed] [Google Scholar]
- Nerbonne JM, Nichols CG, Schwarz TL, Escande D. Genetic manipulation of cardiac K+ channel function in mice: what have we learned, and where do we go from here? Circ Res. 2001;89:944–956. doi: 10.1161/hh2301.100349. [DOI] [PubMed] [Google Scholar]
- Nilius B, Droogmans G. Amazing chloride channels: an overview. Acta Physiol Scand. 2003;177:119–147. doi: 10.1046/j.1365-201X.2003.01060.x. [DOI] [PubMed] [Google Scholar]
- Ogura T, Furakawa T, Toyozaki T, Yamada K, Zheng Y-J, Katayama Y, Nakaya H, Inagaki N. ClC-3B, a novel ClC-3 splicing variant that interacts with EBP50 and facilitates expression of CFTR-regulated ORCC. FASEB J. 2002;16:863–865. doi: 10.1096/fj.01-0845fje. [DOI] [PubMed] [Google Scholar]
- Okada Y. Volume expansion-sensing outward-rectifier Cl− channel: fresh start to the molecular identity and volume sensor. Am J Physiol. 1997;273:C755–C789. doi: 10.1152/ajpcell.1997.273.3.C755. [DOI] [PubMed] [Google Scholar]
- Olsen ML, Schade S, Lyons SA, Amaral MD, Sontheimer H. Expression of voltage-gated chloride channels in human glioma cells. J Neurosci. 2003;23:5572–5582. doi: 10.1523/JNEUROSCI.23-13-05572.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutledge E, Denton J, Strange K. Cell cycle- and swelling-induced activation of a Caenorhabditis elegans ClC channel is mediated by CeGLC-7α/β phosphatases. J Cell Biol. 2002;158:435–444. doi: 10.1083/jcb.200204142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi M, Matsuura H, Ehara T. Swelling-induced Cl− current in guinea-pig atrial myocytes: inhibition by glibenclamide. J Physiol. 1997;505:41–52. doi: 10.1111/j.1469-7793.1997.041bc.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmieder S, Lindenthal S, Ehrenfeld J. Tissue-specific N-glycosylation of the ClC-3 chloride channel. Biochem Biophys Res Comm. 2001;286:635–640. doi: 10.1006/bbrc.2001.5407. [DOI] [PubMed] [Google Scholar]
- Sorota S. Swelling-induced chloride-sensitive current in canine atrial cells revealed by whole-cell patch-clamp method. Circ Res. 1992;70:679–687. doi: 10.1161/01.res.70.4.679. [DOI] [PubMed] [Google Scholar]
- Sorota S. Pharmacologic properties of the swelling-induced chloride current of dog atrial myocytes. J Cardiovas Electrophysiol. 1994;5:1006–1016. doi: 10.1111/j.1540-8167.1994.tb01143.x. [DOI] [PubMed] [Google Scholar]
- Souktani R, Berdeaux A, Ghaleh B, Giudicelli JF, Guize L, Le Heuzey JY, Henry P. Induction of apoptosis using sphingolipids activates a chloride current in Xenopus laevis oocytes. Am J Physiol. 2000;279:C158–C165. doi: 10.1152/ajpcell.2000.279.1.C158. [DOI] [PubMed] [Google Scholar]
- Stobrawa SM, Breiderhoff T, Takamori S, Engel D, Schweizer M, Zdebik AA, Bosl MR, Ruether K, Jahn H, Draguhn A, Jahn R, Jentsch TJ. Disruption of ClC-3, a chloride channel expressed on synaptic vesicles, leads to a loss of the hippocampus. Neuron. 2001;29:185–196. doi: 10.1016/s0896-6273(01)00189-1. [DOI] [PubMed] [Google Scholar]
- Strange K, Emma F, Jackson PS. Cellular and molecular physiology of volume-sensitive anion channels. Am J Physiol. 1996;270:C711–C730. doi: 10.1152/ajpcell.1996.270.3.C711. [DOI] [PubMed] [Google Scholar]
- Vandenberg JI, Yoshida A, Kirk K, Powell T. Swelling-activated and isoprenaline-activated chloride currents in guinea pig cardiac myocytes have distinct electrophysiology and pharmacology. J General Physiol. 1994;104:997–1017. doi: 10.1085/jgp.104.6.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Weikersthal SF, Barrand MA, Hladky SB. Functional and molecular characterization of a volume-sensitive chloride current in rat brain endothelial cells. J Physiol. 1999;516:75–84. doi: 10.1111/j.1469-7793.1999.075aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker RL, Hume JR, Horowitz B. Differential expression and alternative splicing of TRP channel genes in smooth muscles. Am J Physiol. 2001;280:C1184–C1192. doi: 10.1152/ajpcell.2001.280.5.C1184. [DOI] [PubMed] [Google Scholar]
- Wang G-X, Hatton WJ, Wang GL, Zhong J, Yamboliev I, Duan D, Hume JR. Functional effects of novel anti-ClC-3 antibodies on native volume-sensitive osmolyte and anion channels (VSOACs) in cardiac and smooth muscle cells. Am J Physiol. 2003;285:H1453–H1463. doi: 10.1152/ajpheart.00244.2003. [DOI] [PubMed] [Google Scholar]
- Wang L, Chen L, Jacob TJ. The role of ClC-3 in volume-activated chloride currents and volume regulation in bovine epithelial cells demonstrated by antisense inhibition. J Physiol. 2000;524:63–75. doi: 10.1111/j.1469-7793.2000.t01-1-00063.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warth R, Barhanin J. The multifaceted phenotype of the knockout mouse for the KCNE1 potassium channel gene. Am J Physiol. 2002;282:R639–R648. doi: 10.1152/ajpregu.00649.2001. [DOI] [PubMed] [Google Scholar]
- Weylandt KH, Valverde MA, Nobles M, Raguz S, Amey JS, Diaz M, Nastrucci C, Higgins CF, Sardini A. Human ClC-3 is not the swelling-activated chloride channel involved in cell volume regulation. J Biol Chem. 2001;276:17461–17467. doi: 10.1074/jbc.M011667200. [DOI] [PubMed] [Google Scholar]
- Wills NK, Fong P. ClC chloride channels in epithelia: recent progress and remaining puzzles. News Physiol Sci. 2001;16:161–166. doi: 10.1152/physiologyonline.2001.16.4.161. [DOI] [PubMed] [Google Scholar]
- Yamazaki J, Duan D, Janiak R, Kuenzli K, Horowitz B, Hume JR. Functional and molecular expression of volume-regulated chloride channels in canine vascular smooth muscle cells. J Physiol. 1998;507:729–736. doi: 10.1111/j.1469-7793.1998.729bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki J, Hume JR. Inhibitory effects of glibenclamide on cystic fibrosis transmembrane regulator, swelling-activated, and Ca2+-activated Cl− channels in mammalian cardiac myocytes. Circ Res. 1997;81:101–109. doi: 10.1161/01.res.81.1.101. [DOI] [PubMed] [Google Scholar]
- Zhong J, Walsh MP, Hume JR. Regulation of volume-sensitive outwardly rectifying anion channels in vascular smooth muscle cells by PKC. Proceedings of the XXXIV Congress IUPS (Christchurch) 2001:A303. [Google Scholar]
- Zhong J, Wang G-X, Hatton WJ, Yamboliev I, Walsh MP, Hume JR. Regulation of volume-sensitive anion channels in pulmonary arterial smooth muscel cells by PKC. Am J Physiol. 2002;283:C1627–C1636. doi: 10.1152/ajpcell.00152.2001. [DOI] [PubMed] [Google Scholar]