Increased ubiquitin-dependent degradation can replace the essential requirement for heat shock protein induction (original) (raw)
Abstract
Serine palmitoyltransferase, the first enzyme in ceramide biosynthesis, is required for resistance to heat shock. We show that increased heat shock sensitivity in the absence of serine palmitoyltransferase activity correlates with a lack of induction of the major heat shock proteins (Hsps) at high temperature. Normal heat shock resistance can be restored, without restoration of ceramide synthesis or induction of Hsps, by overexpression of ubiquitin. This function of ubiquitin requires the proteasome. These data imply that the essential function of Hsp induction is the removal of misfolded or aggregated proteins, not their refolding. This suggests that cells stressed by heat shock do not die because of the loss of protein activity due to their denaturation, but because of the inherent toxicity of the denatured and/or aggregated proteins.
Keywords: heat stress/Hsp/sphingoid base/ubiquitin–proteasome degradation
Introduction
All organisms respond to temperature increases by induction of a conserved set of proteins, the heat shock proteins (Hsps), which protect them from damage and facilitate recovery from such heat stresses. Most of these Hsps function as molecular chaperones that prevent the accumulation of aggregated proteins or promote refolding of misfolded proteins (Hendrick and Hartl, 1993; Parsell and Lindquist, 1993; Glover and Lindquist, 1998). In eukaryotic cells, ubiquitin and certain ubiquitin-conjugating enzymes are Hsps that function in the rapid turnover of denatured proteins. The major pathway for the selective degradation of abnormal proteins in the cytosol and nucleus is the ubiquitin–proteasome pathway (Ciechanover, 1994). In the budding yeast Saccharo myces cerevisiae, Hsp induction is caused by increased transcription of the corresponding genes (Lindquist, 1981). Two transcriptional control systems appear to be responsible for the gene expression changes upon heat stress, one involving the heat shock factor (Hsf1p) and the other one depending on Msn2p and Msn4p transcription factors. Hsf1p binds to the heat shock promoter element (HSE) found in the promoter region of many Hsp genes. In yeast, several genes have been identified that do not contain HSEs, but whose transcription is induced by heat and other stress signals, including osmotic shock, DNA damage and oxidative stress. Msn2/4p activates these genes through the stress response element (STRE), a cis regulatory sequence (Ruis and Schuller, 1995).
In addition to the induction of Hsps, heat shocked yeast cells display a number of characteristic phenotypes. Cells accumulate trehalose (a thermoprotectant), acquire thermotolerance, become transiently arrested in the G1 phase of the cell cycle and exhibit an increase in cellular levels of sphingoid bases and ceramides. Furthermore, de novo synthesis of sphingoid bases [phytosphingosine (PHS) and dihydrosphingosine (DHS)] is required for the yeast heat stress response (Patton et al., 1992; Jenkins et al., 1997). Sphingoid bases are potential mediators of the heat stress response, because treatment of cells with DHS activates transcription of the TPS2 gene encoding a subunit of trehalose synthase and causes trehalose to accumulate. DHS also induces expression of a STRE-LacZ reporter gene, showing that the global stress response pathway can be activated by sphingoid base signals (Dickson et al., 1997).
To understand the role of sphingoid bases in yeast heat stress response, we used the mutant strain lcb1-100, which has a thermosensitive defect in de novo sphingolipid synthesis and fails to grow at 37°C (Zanolari et al., 2000). The LCB1 gene encodes a subunit of serine palmitoyltransferase, an essential enzyme that catalyzes the first step in sphingoid base synthesis (Buede et al., 1991). Upon heat shock, lcb1-100 mutant cells show no increase in sphingoid base (PHS and DHS) synthesis, no transient cell cycle arrest and no resistance to heat stress, indicating a requirement for de novo synthesis of sphingoid bases for the heat shock response (Chung et al., 2000; Jenkins and Hannun, 2001). Here, we show that overexpression of the polyubiquitin gene UBI4 can abrogate the sphingoid base synthesis requirement for heat shock resistance and restore survival upon heat stress of the lcb1-100 mutant strain without induction of Hsps or ceramide synthesis. This suppressor effect of UBI4 is mediated via the ubiquitin–proteasome degradation pathway. These results suggest that the essential requirement for heat shock survival is the removal of misfolded or aggregated proteins, not their refolding and that cells stressed by heat shock do not die because of the loss of protein activity due to their denaturation, but because of the inherent toxicity of misfolded and/or aggregated proteins.
Results and discussion
Overexpression of ubiquitin restores heat stress resistance to the lcb1-100 mutant
In this study we used a suppressor approach to identify proteins that are downstream effectors in the sphingoid base signaling pathway required for the heat shock response. The rationale of this study was based on the idea that overexpression of such proteins from a high copy number plasmid would result in an increased resistance of the lcb1-100 mutant to elevated temperature. The polyubiquitin gene UBI4 was isolated as a suppressor of the lcb1-100 mutation (Figure 1A), suggesting that ubiquitin overexpression can restore the heat stress defect due to the lack of sphingoid base synthesis. This effect was specific for the heat survival defect associated with the lcb1-100 mutation, because UBI4 overexpression did not suppress the endocytic defect of lcb1-100 mutant cells (data not shown) (Zanolari et al., 2000).

Fig. 1. (A) Increased ubiquitin expression suppresses the lcb1-100 temperature-sensitive phenotype. RH3809 (lcb1-100) cells carrying plasmids that overexpress either LCB1, UBI4, PKC1, TPS1, TPS3, HSC82 or SSA2 were streaked onto YPUAD plates and grown at the non-permissive temperature of 37°C (left and middle panels). The RH3809 (lcb1) strain bearing pYSGal104 (HSP104) or YCp50-GAL1-SSA1 (SSA1) plasmids was streaked onto an SGal/Raf-ura plate and tested for growth at 37°C (right panel). (B) Increased ubiquitin expression restores heat resistance to lcb1-100 cells. Mid log-phase cultures of WT, lcb1-100 (lcb1) cells or lcb1-100 mutant cells overexpressing UBI4 (+UBI4) were grown in YPUAD at 24°C and an aliquot was shifted to 44°C. Samples were taken in duplicate at the times indicated, diluted into ice-cold YPUAD, and immediately plated onto YPUAD agar at 24°C to assess cell viability. Survival at 44°C was plotted on a log scale as a percentage of colony forming units relative to that found at 24°C.
Survival at an elevated temperature was also examined. Log-phase cells of wild-type (WT), lcb1-100 and lcb1-100 mutant strains overexpressing the UBI4 gene, were heat shocked at 44°C and the percentage of cells able to form colonies was determined as a function of time (Figure 1B). The lcb1-100 mutant cells showed a clear defect in survival at high temperature when compared with WT cells. In contrast, lcb1-100 cells with UBI4 plasmid were 6- to 10-fold more resistant at 44°C than the parental lcb1-100 strain (Figure 1B). Consistent with this result, we found that increased expression of a single ubiquitin gene driven from the CUP1 promoter was also able to suppress the lcb1-100 mutation (data not shown). Thus, the suppression of the lcb1-100 heat shock defect results from increased ubiquitin expression.
The heat shock transcription factor Hsf1p and the stress-responsive transcription factors Msn2/4p are required for Hsp induction. To determine whether UBI4 overexpression could also suppress the temperature-sensitive growth defect displayed by the msn2 msn4, hsf1_-Δ_CTD, msn2 msn4 hsf1_-Δ_CTD, tetO-HSF1 and msn2 msn4 tetO-HSF1 mutant strains, these strains were transformed by the YEplac181-UBI4 plasmid bearing UBI4 and tested for growth at 37°C. None of these strains were suppressed by UBI4 overexpression, showing that this effect was specific for the temperature-sensitive defect associated with the lcb1-100 mutation.
The heat shock response pathway activates several genes that are under the control of the HSE and/or the STRE regulons. The UBI4, HSP12, HSP26 and HSP104 genes contain both stress inducible regulons (Boy-Marcotte et al., 1999; Simon et al., 1999; Amoros and Estruch, 2001). Other genes contain only the STRE regulon, including genes for trehalose biosynthetic enzymes TPS1 and TPS2. Most of the classical heat shock protein genes are heat inducible only via Hsf1p, including the Hsp70s encoded by the SSA1-4 genes. To determine if overexpression of other heat inducible genes could also suppress the lcb1-100 temperature-sensitive defect, the lcb1-100 strain was transformed with high copy number plasmids bearing HSC82, TPS1, TPS2, TPS3, TSL1, SSA2 or SSA4 genes (Figure 1A; data not shown for TPS1, TSL1 and SSA4 overexpression) and with a centromeric plasmid bearing the SSA1 or HPS104 gene under the control of the inducible GAL1 promoter (Figure 1A). The transformants were tested for survival at 37°C. None of the genes tested was able to suppress the lcb1-100 temperature-sensitive growth defect, even the HSP104 gene, which like the UBI4 gene contains both the HSE and STRE regulons in its promoter region. This result shows that in contrast to ubiquitin, overexpression of different chaperones or subunits of the trehalose synthase are not sufficient to restore survival upon heat stress in the lcb1-100 strain. The UBI4 gene is stress inducible and important for the survival under diverse stresses (Finley et al., 1987; Fraser et al., 1991; Jungmann et al., 1993; Cheng et al., 1994). This probably reflects the need for adequate ubiquitin levels to enable the ubiquitylation system to control the turnover of damaged proteins.
The protein kinase C–mitogen-activated protein (Pkc1–MAP) kinase pathway is inducible by elevated temperature and this activation is required for acquired thermotolerance. Indeed, activation of the MAP kinase branch of the pathway is sufficient to confer acquired thermotolerance (Kamada et al., 1995). It is therefore possible that activation of the _PKC1_–MAP kinase pathway by overexpressing effectors of this pathway, could rescue the heat survival of the lcb1-100 mutant cells. This MAP kinase signaling pathway is composed of four downstream effectors, Bck1p, Mkk1p/Mkk2p and Mpk1p (Lee and Levin, 1992; Irie et al., 1993; Lee et al., 1993), which are homologs of the MAP kinase cascade effectors in mammalian cells. The lcb1-100 mutant was transformed with high copy number plasmids bearing PKC1, BCK1, MKK1 or MPK1 genes and survival at 37°C was tested. We also overproduced Pkc1p activity by transformation with a low copy number plasmid bearing a dominant, activated allele of PKC1 (PKC1-R398P) (Nonaka et al., 1995). None of these kinases was able to suppress the heat sensitivity associated with the lcb1-100 mutation (Figure 1A, panel lcb1+PKC1; and data not shown) despite the fact that Pkc1p overexpression can suppress the endocytic defect of lcb1-100 cells (Friant et al., 2000). In yeast, the Pkh1/2p kinases that phosphorylate and activate Pkc1p, are stimulated by sphingoid bases. This sphingoid base-mediated signaling pathway is required for endocytosis (Friant et al., 2001). These results suggest that the lack of sphingoid base synthesis observed in the lcb1-100 strain upon heat shock, may result in a decrease of Pkh1/2p and Pkc1p kinase activity. Therefore, we tested whether PKH1 or PKH2 overexpression could suppress the temperature-sensitive phenotype displayed by the lcb1-100 mutant. Neither of the two kinases tested was able to restore growth of lcb1-100 at 37°C, even though they are also able to suppress the endocytic defect of this strain (Friant et al., 2001). Taken together, these results indicate that the suppressor effect of UBI4 is specific and is not mediated via the Pkc1p–MAP kinase pathway. These results suggest that elevated levels of ubiquitin expression can bypass the need for de novo sphingoid base synthesis for survival upon heat stress.
Sphingolipid synthesis is defective in the lcb1-100 cells overexpressing UBI4
The suppressor effect of the UBI4 gene could be due to restoration of normal sphingoid base synthesis in the lcb1-100 mutant strain. To investigate this possibility, sphingolipid synthesis was measured in cells overexpressing UBI4. WT and lcb1-100 cells were grown at 24°C and metabolically labeled with [3H]_myo_inositol at 24°C or upon heat shock at 37°C. Lipids were extracted, treated with mild base to identify sphingolipids which are base-resistant, separated by thin layer chromatography (TLC) and visualized using a PhosphorImager (Figure 2). At 24°C, both WT and mutant cells overexpressing UBI4 or not, synthesize sphingolipids [inositolphosphocera mide (IPC), mannosylated inositolphosphoceramide (MIPC) and mannosylated di-inositolphosphoceramide (M(IP)2C)], although the lcb1-100 strains showed less sphingolipid synthesis than the WT cells. After mild heat shock (37°C), WT cells showed normal synthesis of sphingolipids. In contrast, the lcb1-100 mutant cells showed a reduction in sphingolipid synthesis. The reduction was the same in lcb1-100 mutant cells overexpressing UBI4 (Figure 2). This result shows that overexpression of UBI4 does not restore synthesis of sphingolipids in lcb1-100 mutant cells and suggests that the requirement for heat-induced increase in sphingolipid synthesis can be overcome by UBI4 overexpression.

Fig. 2. Increased ubiquitin expression does not restore sphingolipid synthesis in lcb1-100 cells. WT, lcb1-100 mutant (lcb1) or lcb1-100 mutant cells overexpressing UBI4 (+UBI4) were grown in SDYE at 24°C, preshifted to 24 or 37°C and labeled with [3H]_myo_inositol. Incorporation of [3H]_myo_inositol into the total lipid fraction was quantified and equal c.p.m. were directly applied to TLC plates, or treated with mild base to identify sphingolipids [IPC, MIPC and M(IP)2C].
Accumulation of a novel intermediate in the sphingolipid synthesis pathway upon UBI4 overexpression could explain the restoration of the viability of the lcb1-100 cells upon heat stress. To test this hypothesis, WT and lcb1-100 mutant cells were labeled with [3H]DHS. Addition of DHS to the lcb1-100 mutant restores synthesis of sphingolipids at 37°C (Zanolari et al., 2000). Exogenously added DHS can be incorporated into phosphorylated DHS, ceramides and sphingolipids, allowing an analysis of the sphingolipid biosynthetic pathway in the lcb1-100 strain. WT and lcb1-100 strains overexpressing UBI4 or not, were grown at 24°C, preincubated for 15 min at 37°C to induce the heat shock response, then [3H]DHS was added and incubation was continued for 15 min. Lipids were extracted, separated by TLC and visualized using a PhosphorImager (data not shown). There was no difference in the lipid pattern between the strains bearing no plasmid and the ones overexpressing UBI4, showing that the suppressor effect is not due to a difference in sphingolipid synthesis.
The lcb1-100 mutant shows a defect in heat induction of Hsps
Sphingoid bases are potential mediators of the heat stress response, because treatment of cells with DHS mimics heat-induced activation of several reporter genes (Dickson et al., 1997). The lcb1-100 mutant cells show very low synthesis of sphingoid bases (DHS or PHS) upon heat treatment. To determine whether the lack of sphingoid base synthesis results in loss of induction of heat shock activated genes, we tested heat induction of a reporter gene having the HSP26 or SSA1 gene fused in frame to the Escherichia coli lacZ gene in WT and lcb1-100 mutant strains with or without a UBI4 plasmid (Figure 3A). The WT cells showed an induction of β-galactosidase activity upon heat treatment, whereas this induction was defective in the lcb1-100 mutant cells (lcb1) and in the lcb1-100 cells overexpressing UBI4 (lcb1+UBI4) for both reporter genes (Figure 3A). The HSP26 gene promoter region contains both regulons controlling stress response whereas the SSA1 gene is heat inducible only via Hsf1p. This result shows that activation of two different reporter genes that are under the control of the STRE and/or the HSE regulons is defective in the absence of sphingolipid synthesis and this activation is not restored upon UBI4 overexpression.

Fig. 3. (A) lcb1-100 mutant cells are defective in Hsp induction. WT, lcb1-100 mutant (lcb1) or lcb1-100 mutant cells overexpressing UBI4 (+UBI4) were transformed with plasmids carrying SSA1-LacZ or HSP26-LacZ reporter constructs. After growth at 24°C, transformants were shifted to 37°C for the indicated time to induce the heat shock response and β-galactosidase expression driven from these promoters was quantified. (B) Normal heat induction of UBI4 in the lcb1-100 mutant cells. WT or lcb1-100 mutant (lcb1) cells were transformed with a plasmid carrying the UBI4-LacZ reporter gene and treated as described for (A).
The UBI4 gene promoter region also contains both regulons controlling stress induction, HSE and STRE that contribute independently to heat shock regulation of the UBI4 gene (Simon et al., 1999). Overexpression of the UBI4 gene restored viability of the lcb1-100 mutant strain upon heat stress. To determine whether the UBI4 gene was heat inducible in the lcb1-100 mutant, we tested heat induction of a reporter gene having the UBI4 gene fused in frame to the E.coli lacZ gene in WT and lcb1-100 mutant strains (Figure 3B). Both WT and lcb1-100 mutant cells (lcb1) showed an induction in β-galactosidase activity upon heat treatment. This result shows that the UBI4 gene is heat inducible in lcb1-100 mutant cells, in contrast to HSP26 or SSA1 genes. Therefore, heat shock activation of UBI4 expression is preserved in the absence of sphingoid base synthesis suggesting a difference in the mechanism of activation from the one used to induce HSP26 or SSA1. UBI4 oxerexpression could therefore restore normal heat shock resistance to the lcb1-100 strain, because this heat shock protein is still heat inducible in the absence of sphingoid base synthesis in contrast to the other Hsps. In several genes with an essential role in stress protection, such as HSP26, HSP104 or UBI4, Hsf1p and Msn2/4p act redundantly, assuring the expression of these genes even when one of the regulatory pathways is inactive (Amoros and Estruch, 2001). Consistent with our results, the expression of UBI4 was not completely abolished in cells deficient for both stress pathways, suggesting the involvement of additional transcription factor(s) (Simon et al., 1999). Activation of this additional transcription factor(s) upon heat shock could be independent of sphingoid base synthesis, explaining the normal heat induction of UBI4 in the lcb1-100 mutant cells.
The lcb1-100 mutant is defective in Hsp synthesis and sustained trehalose accumulation upon heat shock
Activation of the heat shock response in yeast results in increased synthesis of Hsps of 100, 90 and 70 kDa, as monitored by pulse-labeling and one-dimensional SDS–PAGE (Miller et al., 1979). To determine whether the lcb1-100 mutant cells exhibit a defect in Hsp synthesis upon heat stress, the general heat shock response was analyzed in the lcb1-100 mutant cells with or without the UBI4 plasmid and compared to WT cells. Hsp synthesis was induced by a temperature shift from 24 to 44°C. The proteins were labeled with a mix of [35S]methionine and [35S]cysteine 15 min after the temperature shift, because the induction of Hsps is transient, with a maximum expression at 15–20 min (Smith and Yaffe, 1991; Martinez-Pastor et al., 1996). Proteins were separated by SDS–PAGE and visualized using a PhosphorImager in WT, lcb1-100 and lcb1-100 overexpressing UBI4 strains (Figure 4A). This analysis allows the detection of heat shock proteins Hsp104, Hsp90, Hsp82 and Hsp70s (Ssa1-4), identified according to their molecular weight. Following heat shock, the WT strain showed the expected production of Hsps, whereas the lcb1-100 strain overexpressing UBI4 or not, showed a strong reduction in all Hsp synthesis (Figure 4A). Hsp104p and Hsp82p were barely detected in the lcb1 mutant strain overexpressing UBI4 or not, whereas these two proteins were expressed in the WT cells. Hsp90p and Hsp70p proteins synthesis was greatly reduced in the lcb1 mutant strain when compared with WT cells. Mutant lcb1+UBI4 cells, which retain viability under these conditions similarly to WT cells (Figure 1B), were also defective in Hsp expression showing that the reason for the reduced labeling was not a reduction in cell viability.
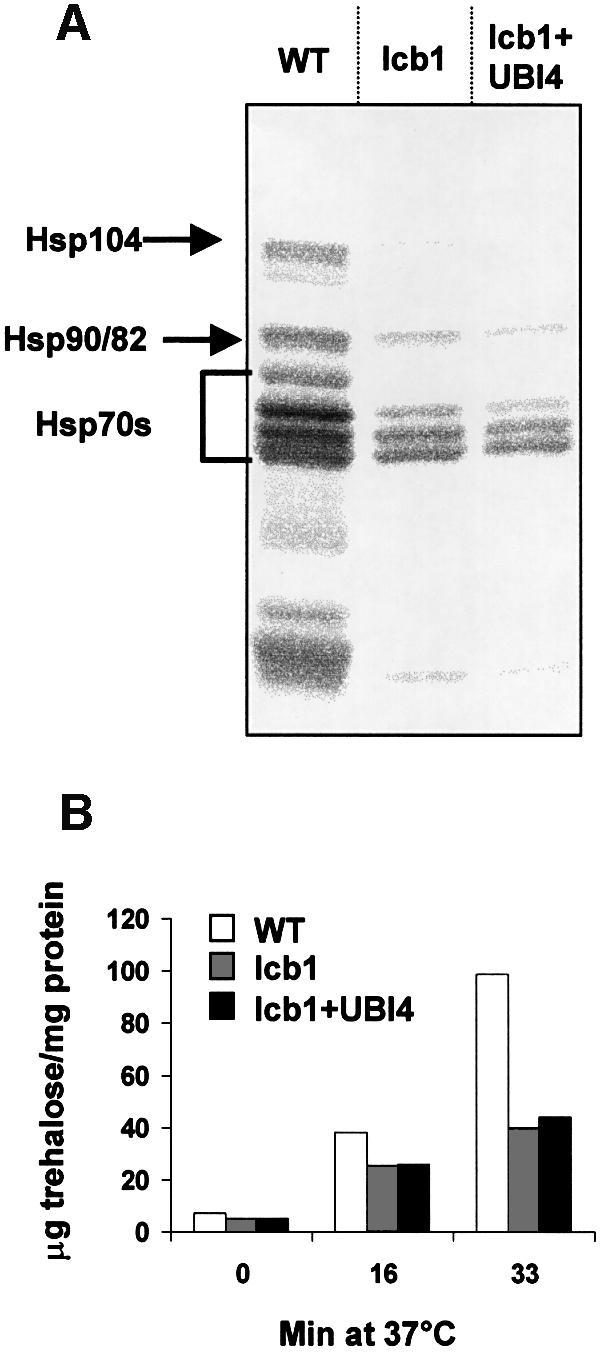
Fig. 4. Heat shock protein synthesis and sustained trehalose accumulation are defective in the lcb1-100 cells. (A) Cells actively dividing at 24°C were transferred to 44°C. The production of Hsps was assessed after [35S]Met/Cys labeling, followed by extraction, separation by SDS–PAGE and PhosphorImager analysis of labeled proteins. Hsp bands are indicated. (B) WT and lcb1-100 (lcb1) cells bearing a plasmid without insert or lcb1-100 cells overexpressing UBI4 (lcb1+UBI4) were shifted from 24 to 37°C, aliquots of cells were collected at the indicated times, cell extracts were prepared and trehalose contents were determined. Similar results were obtained in two independent experiments.
Heat shock causes the accumulation of another thermoprotectant in yeast, the non-reducing disaccharide trehalose (Attfield, 1987). Heat stress survival of the lcb1-100 mutant by UBI4 overexpression could be due to increased trehalose synthesis. Trehalose accumulation upon heat shock was determined in WT cells and in lcb1-100 mutant with or without a UBI4 plasmid (Figure 4B). Upon incubation for 16 min at 37°C, the level of trehalose increased markedly and to a similar extent in all cells, whereas after this, only WT cells continued to accumulate trehalose significantly. The initial induction of trehalose upon temperature shift has been shown to be independent of new protein synthesis (Neves and François, 1992). This could explain why we find a similar induction of trehalose after a short incubation at 37°C. Upon longer incubations, the continued increase in trehalose would require induction of enzymes involved in trehalose production. Our results show that the lcb1-100 cells are defective for sustained trehalose accumulation and for induction of Hsps and that these defects are not restored upon UBI4 overexpression, even though these cells are resistant to heat stress. Therefore, the ability to synthesize trehalose or Hsps does not correlate with the ability of UBI4 to suppress the lcb1-100 mutation. Therefore, we predicted that the capacity of a cell to degrade misfolded and/or aggegated proteins using the proteasome–ubiquitin pathway must be the most important factor allowing resistance to heat stress in the absence of Hsps and sustained trehalose accumulation.
Heat shock-induced protein degradation and ubiquitination is restored in lcb1-100 mutant overexpressing UBI4
To test whether UBI4 overexpression affects the rate of protein turnover in the lcb1-100 mutant cells, protein degradation after heat shock at 37°C was determined in these cells and compared with WT cells. Cells grown at 24°C were shifted to 37°C, pulse-labeled with a mix of [35S]methionine and [35S]cysteine, then chased in presence of cycloheximide to prevent re-incorporation of radioactive amino acids released from proteins. At the indicated time, aliquots of cells were taken and protein degradation was determined as the percentage of incorporated radioactivity converted into TCA-soluble fragments (Figure 5A). At 37°C, protein degradation in WT cells exceeded that in the lcb1-100 mutant by ∼2- to 3-fold. Overexpression of UBI4 in the lcb1-100 cells restored protein degradation upon heat treatment to the wild-type level. These results suggest that UBI4 overexpression may allow the lcb1-100 mutant cells to survive a heat stress by increasing the degradation of misfolded proteins via the ubiquitin–proteasome pathway.
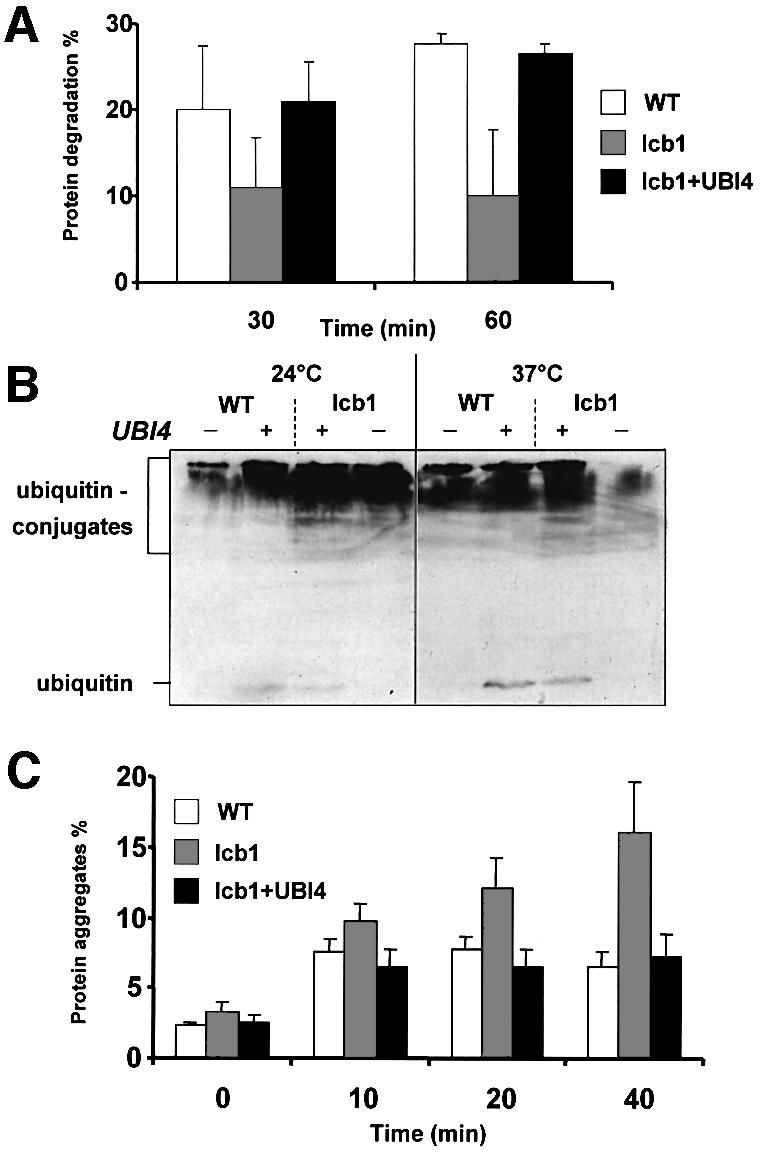
Fig. 5. Increased protein degradation and removal of protein aggregates in the lcb1-100 strain overexpressing UBI4. (A) WT, lcb1-100 (lcb1) and lcb1-100 cells overexpressing UBI4 (lcb1+UBI4) were pulse- labeled at 37°C with [35S]Met/Cys and the rate of protein degradation was measured during the chase period. The data shown are mean values and standard errors obtained from four independent experiments are shown. (B) WT, lcb1-100 (lcb1) cells bearing or not the UBI4 plasmid (UBI4) were grown at 24°C, then either kept at 24°C or shifted at 37°C for 1 h and cell extracts were prepared. Equal amount of proteins were applied to a 15% SDS–PAGE gel and probed with an anti-ubiquitin antibody. Free ubiquitin and high-molecular-weight ubiquitin–protein conjugates are indicated. (C) The determination of the percentage of aggregated [35S]Met/Cys labeled proteins was assessed in WT, lcb1-100 (lcb1) and lcb1-100 cells bearing the UBI4 plasmid (lcb1+UBI4) after heat shock at 37°C for the indicated times. Protein aggregates were identified by their sedimentation at 15 000 g for 15 min in glycerol and non-ionic detergent at physiological salt concentrations. Total and aggregated labeled proteins were quantified by liquid scintillation counting.
The conjugation of polyubiquitin chains to short-lived or damaged proteins marks them for subsequent degradation by the proteasome. To test if this response is defective in the lcb1-100 mutant, we compared the changes in the levels of ubiquitylated proteins after a shift to 37°C in WT and lcb1-100 cells bearing a UBI4 plasmid or not. Cells grown at 24°C were shifted for 1 h at 37°C to induce heat stress or kept at 24°C and the total level of ubiquitylated proteins was determined by western blotting with anti-ubiquitin antibody (Figure 5B). The lcb1-100 mutant displayed an increase in protein ubiquitylation at 24°C compared with WT cells, showing that these mutant cells already accumulate ubiquitin conjugates at 24°C without heat stress, which is consistent with our results showing greater expression of the UBI4-lacZ construct in lcb1-100 cells than in WT cells at 24°C (Figure 3B). This result suggests that even without heat shock, the lcb1-100 mutant cells may accumulate more misfolded and/or aggregated proteins that would need to be degraded via the ubquitin–proteasome pathway. After heat shock at 37°C, WT cells and the lcb1-100 mutant overexpressing UBI4 displayed an increase in ubiquitylated proteins, but in the lcb1-100 mutant cells where protein degradation was low (Figure 5A), the content of ubiquitylated proteins decreased (Figure 5B). Free ubiquitin was difficult to detect under conditions where UBI4 was not overexpressed. Therefore, we cannot rule out that a lack of free ubiquitin is a possible cause of cell death in the lcb1-100 mutant cells at 37°C. However, our results show that lcb1-100 cells are able to induce the UBI4 gene at 37°C (Figure 3B) and that protein ubiquitylation is not defective in these mutants cells. These results suggest that there probably is some free ubiquitin in the lcb1-100 mutant cells at 37°C but that the ubiquitin level is not sufficient to respond to the quantity of accumulated, misfolded and/or aggregated proteins that need to be degraded via the ubiquitin–proteasome pathway. It was previously shown that the essential function of UBI4 is to provide ubiquitin under conditions of stress (Finley et al., 1987). Therefore, overexpression of UBI4 in the lcb1-100 mutant cells allows a higher synthesis of free ubiquitin upon heat stress and permits the cells to survive at 37°C via degradation of abnormal proteins presumably by the ubiquitin–proteasome pathway.
Accumulation of protein aggregates in the lcb1-100 mutant is reduced by UBI4 overexpression
To determine whether the lcb1-100 mutant cells accumulate protein aggregates due to the lack of induction of Hsps, the percentage of aggregated proteins upon heat shock was analyzed in the lcb1-100 mutant and compared with WT cells. Cells were pulse-labeled with a mix of [35S]methionine and [35S]cysteine at 24°C and then heat shocked at 37°C for the indicated time. Whole-cell extracts, containing glycerol and non-ionic detergent, of WT and lcb1-100 cells were subjected to centrifugation to separate protein aggregates, which sedimented at 15 000 g and the percentage of aggregated proteins was determined (Figure 5C). The amount of aggregated proteins increased in WT cells following heat stress and then remained stable at 7%, whereas in the lcb1-100 strain the rate of protein aggregates constantly increased after the shift to 37°C and reached 16% after 40 min incubation at 37°C. This result indicates that the lcb1-100 mutant cells accumulate aggregated proteins upon heat shock and these aggregated proteins could be responsible for the heat sensitivity displayed by the lcb1-100 cells. To test if UBI4 overexpression, which restores heat shock resistance of the lcb1-100 mutant, abrogated the accumulation of protein aggregates upon heat shock, the same experiment was performed in the lcb1-100 cells transformed by the UBI4 plasmid (Figure 5C). The lcb1-100 cells overexpressing UBI4 showed only a small accumulation of aggregated proteins similar to WT cells upon heat shock. This result shows that UBI4 overexpression could function through the removal of denatured proteins before or after they have aggregated. If this is true, then degradation of the denatured/aggregated proteins should be required for the suppression.
The proteasome is required for UBI4-dependent suppression of lcb1-100
The proteasome is an important cellular protein degradation system that recognizes ubiquitylated proteins and functions in cellular quality control by degrading misfolded, unassembled or damaged proteins that could otherwise form potentially toxic aggregates (Ciechanover et al., 2000). The proteasome is a multi-enzyme complex consisting of a number of different protease subunits. In yeast cells, PRE1 and PRE2 genes encode two well-characterized subunits of the proteasome. The pre1-1 mutant strain is severely deficient in cytoplasmic proteolysis, accumulates ubiquityated proteins and shows reduced growth at 37°C (Figure 6A) (Heinemeyer et al., 1991). To determine if the proteasome is required for UBI4 suppression of lcb1-100, we constructed a double mutant strain lcb1-100 pre1-1; this strain is viable at 24°C, but does not grow at 37°C. We then analyzed survival at 37°C of the lcb1-100 pre1-1 strain with or without UBI4 overexpression. UBI4 overexpression was not able to suppress the temperature-sensitive growth defect of the lcb1-100 pre1-1 double mutant strain in contrast to the single lcb1-100 mutant cells (Figure 6A). This result suggests that the heat shock resistance and survival of the lcb1-100 cells due to UBI4 overexpression depends on the correct function of the cytoplasmic proteasome.
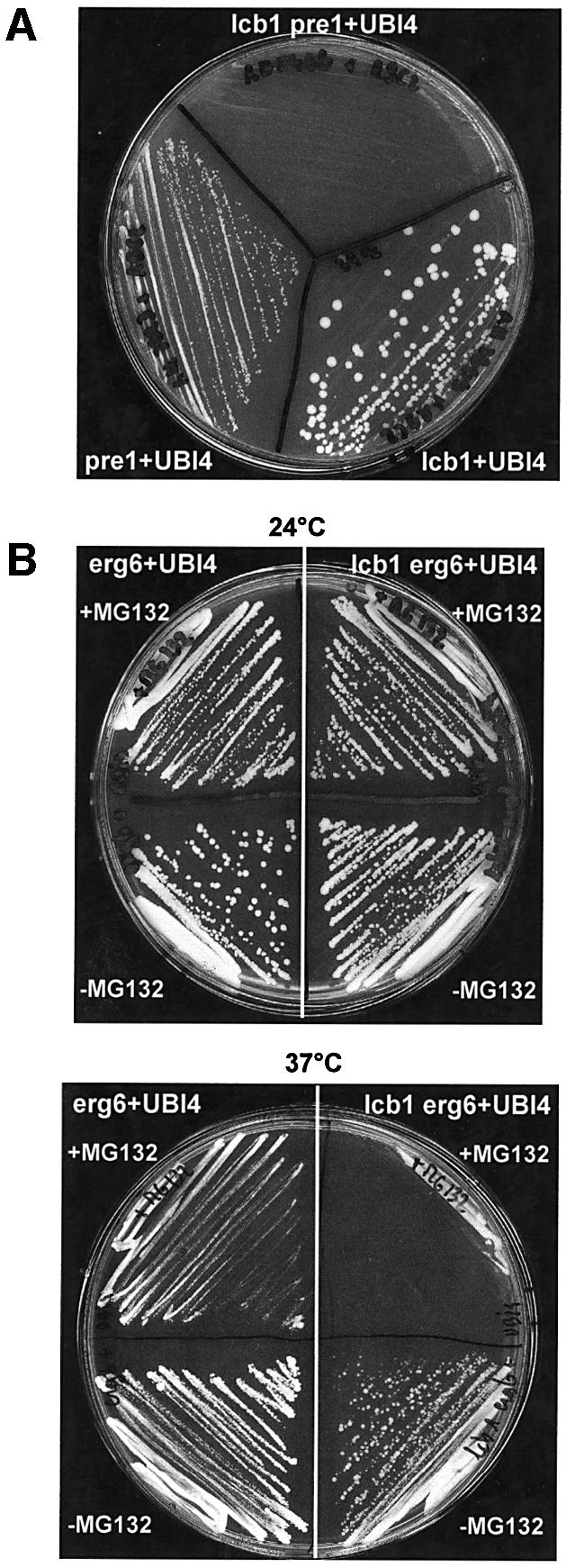
Fig. 6. The proteasome is required for UBI4 suppression of lcb1-100. (A) The pre1-1 strain is mutated in one subunit of the proteasome complex. Single mutant strains lcb1-100 and pre1-1 and the double mutant strain lcb1-100 pre1-1 were transformed with a high copy number UBI4 plasmid and after growth on SD selective medium at 24°C were tested for growth on YPUAD at 37°C. (B) erg6 and lcb1-100 erg6 mutant cells bearing the UBI4 overexpression plasmid were plated onto YPUAD medium (–MG132, lower part) and on YPUAD containing the proteasome inhibitor MG132 (+MG132, upper part) and tested for growth at 24°C and 37°C.
The recent identification of selective proteasome inhibitors such as the peptide aldehyde MG132 allowed us to further analyze the role of the ubiquitin–proteasome pathway in the UBI4 suppression of the lcb1-100 heat stress defect. MG132 cannot enter wild-type yeast cells, so it is essential to use yeast strains with increased membrane permeability such as the erg6 (ise1) mutant (Emter et al., 2002). In this mutant, MG132 blocks the rapid breakdown of proteins by the ubiquitin–proteasome pathway (Lee and Goldberg, 1998). Heat stress survival of the erg6 and lcb1-100 erg6 mutant strains transformed or not by the UBI4 plasmid were analyzed by plating the different strains at 37°C on plates containing 50 µM of MG132. The erg6 mutant strain like WT strains was able to grow at both 24 and 37°C. The double mutant strain lcb1-100 erg6 showed a clear defect in survival at 37°C, but was resistant to this temperature when overexpressing UBI4, meaning that it has the same phenotype as the lcb1-100 mutant strain and could be used to test the effect of the MG132 inhibitor (Figure 6B). The erg6 mutant strain grew at 37°C in presence of 50 µM MG132, whereas the double mutant strain lcb1-100 erg6 was defective for growth even with UBI4 overexpression (Figure 6C). This result confirms that the UBI4 suppression of the lcb1-100 heat stress defect requires the ubiquitin–proteasome degradation pathway.
In summary, we show that Hsp induction upon heat shock is defective in cells lacking serine palmitoyl transferase activity. Both the Hsf1p- and Msn2/4p-dependent stress pathways are dependent upon serine palmitoyl transferase activity. However, the expression of UBI4 is not completely abolished in cells deficient for these two stress pathways (Simon et al., 1999) or in lcb1-100 mutant cells. These results suggest that UBI4 expression is controlled by additional factor(s) whose activation could be independent of sphingoid base synthesis.
We have shown that the lack of Hsp induction, which presumably is the cause of a hypersensitivity to heat shock, can be overcome by increased expression of ubiquitin. The function of ubiquitin in this process requires the proteasome, because proteasome mutants and inhibitors abrogate the ability of ubiquitin to restore heat shock resistance. This suggests that the major essential function of Hsp induction at high temperature is to help refold denatured and/or aggregated proteins. Removal of these misfolded or aggregated proteins by ubiquitin-dependent proteasomal degradation is also sufficient to render cells resistant to heat shock. This shows that it is the removal of the aberrant proteins and not their refolding that is essential to recover from heat shock. This is consistent with the recent finding that aggregates formed from two non-disease-related proteins are substantially cytotoxic (Bucciantini et al., 2002). Our results also show that yeast cells can survive with substantially reduced levels of sphingolipid biosynthesis provided that they overexpress ubiquitin. This suggests that one of the major essential functions of the ceramide synthesis pathway is to control the expression of proteins involved in removal or refolding of denatured or aggregated cytoplasmic proteins.
Materials and methods
Plasmids and yeast strains
Previously described plasmids used in this study were pSH24 (PKC1), YEp195-PKH1, YEp195-PKH2 (Friant et al., 2000, 2001), pKN32 (gift from S.K.Lemmon), YEp352-UBI4 (gift from M.Ellison) and YEplac181-UBI4, bearing the UBI4 gene on high-copy number plasmids, and plasmid YEp112-CUP1-Ub containing a synthetic yeast ubiquitin gene under the control of the CUP1 promoter (gift from M.Hochstrasser) (Hochstrasser et al., 1991; Nelson and Lemmon, 1993; Prendergast et al., 1995), the YEplac195 plasmid containing TPS1, TPS2, TPS3 and TSL1 genes (kindly provided by J.M.Thevelein; Bell et al., 1998), pKAT6 (YEp24-HSC82) and pYSGal104 (pRS316-pGAL1-HSP104) (kind gifts from S.Lindquist; Nathan and Lindquist, 1995; Lindquist and Kim, 1996), YEp434-A4 (SSA4), YEp351-SSA2, YCp50-GAL1-SSA1, pZJHSE2-137 containing an HSE, HSE2 from SSA1 promoter fused to LacZ (kind gifts from E.A.Craig (Slater and Craig, 1987), The UBI4-lacZ and the HSP26-LacZ plasmids (kindly provided by T.Schmelzle), the pUKC414 vector containing the HSP26 promoter fused to LacZ (S.Christodoulou, P.Bossier, C.Stokes and M.F.Tuite, unpublished) and the UBI4-lacZ plasmid (Tanaka et al., 1988).
The yeast strains used in this study were RH448 (WT), RH3802 and RH3809 (lcb1-100) (Friant et al., 2000), RH3804 (Mat_α lcb1-100 trp1 leu2 ura3 lys2 bar1), RH3323 (Mata pre1-1 his3 his4 lys2 ura3 leu2 bar1), RH5404 (Mat_α pre1-1 lcb1-100 lys2 ura3 leu2), RH4237 (Mat_α erg6::LEU2 his4 lys2 ura3 leu2 bar1), RH4727 (Mat_α erg6::LEU2 lcb1-100 his4 ura3 leu2 bar1), and W303 derivatives (from F.Estruch) W303–1A (Mata ade2-1 can1-100, his3-11,15, leu2-3 112, ura3-1, trp1-1), ΔCTD (W303–1A Δ_CTD::URA3), msn2 msn4 (W303–1A msn2_-Δ_3::HIS3 msn4_Δ::URA3), msn2 msn4 ΔCTD (W303–1A msn2_-Δ_3::HIS3 msn4_Δ::TRP1 HSF(1–583)::URA3_), Δhsf (W303–1A tetO::HSF1:: KanMX4), Δhsf msn2/4 (W303–1A msn2_-Δ_3::HIS3 msn4_Δ::URA3_ tetO::HSF1::KanMX4) (Amoros and Estruch, 2001).
UBI4 suppression of the lcb1-100 mutation
The RH3809 (lcb1-100) strain carrying a temperature-sensitive allele of the LCB1 gene was transformed with pKN32, a YEp24-based plasmid bearing UBI4, the polyubiquitin gene. This transformant could grow at 37°C showing that UBI4 is a high copy suppressor of the lcb1-100 mutation. The lcb1-100 mutant strains RH3802, RH3804 and RH3809 were also transformed by other plasmids bearing the UBI4 gene (YEp352-UBI4 and YEplac181-UBI4) and tested for growth at 37°C, to ensure that the suppressor effect observed was due to overexpression of UBI4 gene. UBI4 rescues growth at 37°C only in high copy number and is not able to suppress the viability defect associated with a lcb1::URA3 strain. RH3804 strain was also transformed by YEp112-CUP1-Ub plasmid containing a synthetic yeast ubiquitin gene under the control of the CUP1 promoter and this transformant was able to grow on YPUAD plates containing CuSO4 (0.1 mM final concentration) at 37°C.
Viability assay
Mid log-phase cultures of WT (RH448) and lcb1-100 (RH3809) cells overexpressing UBI4 or not were grown in YPUAD at 24°C and an aliquot was shifted to 44°C. Samples were taken at the times indicated in duplicate, and diluted onto ice-cold YPUAD, and immediately plated onto YPUAD agar to assess cell viability (Martinez-Pastor et al., 1996). Viability was expressed as a percentage of viable cells relative to the initial colony-forming units, measured at 24°C before the heat shock. The viability experiments were repeated twice, yielding similar results.
Sphingolipid analysis
[3H]_myo_inositol and [3H]DHS labeling of yeast cells was performed for 30 min at 24°C or 37°C after a 15 min preincubation at the corresponding temperature. The lipids were extracted, treated with base to identify sphingolipids, and analyzed by TLC and PhosphorImaging as described (Zanolari et al., 2000).
Liquid β-galactosidase assay
A liquid β-galactosidase assay was performed as described previously with slight modifications (Miller, 1972; Guarente, 1983). For each sample, time, OD420 and protein concentration, using a Bradford assay kit (Bio-Rad), was determined. The values reported are the average of three independent measurements for WT and lcb1-100 experiments and of two independent measurements for experiments with UBI4 overexpression respectively.
Heat shock protein labeling
Strains were grown exponentially in SD medium at 24°C, shifted for 15 min at 44°C to induce heat shock protein synthesis and pulse-labeled with [35S]methionine/[35S]cysteine mix (EasyTag EXPRESS[35S] mix from NEN) for 10 min, followed by 1 min chase, prior to protein extraction, proteins were resolved by SDS–PAGE analysis essentially as described previously (Miller et al., 1979).
Extraction and assay of trehalose
Measurement of trehalose was performed on early log phase cells (0.4–0.5 OD600 units/ml) grown on YPUAD at 24°C and transferred to a prewarmed large flask at 37°C for the indicated times, by a protocol similar to that described previously (Lee and Goldberg, 1998) except that trehalose extraction was for 10 min at 95°C, enzymatic trehalose digestion for 3–4 h and that total proteins were extracted using the NaOH/2-mercaptoethanol and TCA procedure (Horvath and Riezman, 1994), dissolved in 0.1 N NaOH/1% SDS, and protein was determined using the detergent compatible procedure (Pierce). Trehalose induction experiments were repeated twice with essentially identical results.
Total protein degradation
Measurement of total protein degradation was performed as described previously with the following slight changes (Lee et al., 1996). Cells were grown to optical density at 600 nm 0.4 to 0.6 at 24°C in SD media and 2.5 OD600 units of cells were pulsed with 0.2 mCi [35S]methionine/[35S]cysteine mix (EasyTag EXPRESS[35S] mix from NEN) for 3 min as described. Chase was initiated by adding a 1/100 volume of a mixture of 0.3% methionine/cysteine in 0.3 M (NH4)2SO4 and cycloheximide (0.5 mg/ml). At indicated time intervals after shifting the cells to 37°C, aliquots were removed and analyzed as described before (Lee et al., 1996). The rate of protein degradation is expressed as the percentage of incorporated radioactivity converted into acid-soluble fragments from the cells during the chase period normalized to the total amount of cells in each sample.
Ubiquitin–protein conjugates determination
Analysis of ubiquitin–protein conjugates was performed as described previously with the following slight changes (Lee et al., 1996). Cells were grown to 0.6–0.8 OD600 units/ml at 24°C in YPUAD media, 1 OD600 was shifted to 37°C for 1 h or kept at 24°C and cells were harvested and lysed by vortexing with glass beads in 10 mM Tris–HCl pH 7.4, 1 mM EDTA–2% SDS buffer for 3 min at 4°C. Extracts were analyzed by SDS–PAGE and western blot analysis with anti-ubiquitin antibody (Zymed Laboratories Inc.) using ECL protocols (Amersham Biosciences).
Protein aggregate analysis
Cells were grown to a density of 0.5 × 106 cells/ml in synthetic SD media containing 0.5% yeast extract and 40 mg/l of the appropriate amino acids. 1.2 × 108 cells were harvested and washed in 10 ml SD without yeast extract. 3 × 107 cells per time point in a total volume of 0.5 ml SD were labeled with 0.2 mCi [35S]methionine/[35S]cysteine mix (EasyTag EXPRESS[35S] mix from NEN) for 10 min at 24°C. 5 µl 100× chase mix (0.3% methionine and cysteine, 0.3 M (NH4)2SO4) were added and cells were heat shocked for the indicated time points at 37°C. Heat shock was terminated by adding NaF and NaN3 to a final concentration of 8 mM and cooling on ice. Cells were washed with ice-cold glycerol buffer (1 mM EDTA, 150 mM KCl, 1 mM EGTA, 50 mM HEPES, 20% glycerol, 0.5% Triton X-100 pH 7.4) and resuspended in 0.2 ml ice-cold glycerol buffer containing 1 mM PMSF. Glass beads were added and lysis was performed as described (Miller et al., 1979). Cell debris was spun down at 3000 g at 4°C. 1/20 of total lysate was removed and 19/20 were centrifuged at 15 000 g for 15 min at 4°C. The supernatant was removed and the total or pellet fractions were analyzed by either 7.5% SDS–PAGE or liquid scintillation counting using a Packard Scintillation Counter (Packard Instrument Company, USA). The data shown represent the average of five individual experiments.
Acknowledgments
Acknowledgements
This work was supported by grants from the Swiss National Science Foundation and the Human Frontier Science Program Organization (to H.R.), from the Association pour la Recherche sur le Cancer (to S.F.) and a long-term fellowship from Human Frontier Science Program Organization (to S.F.). We thank E.A.Craig, S.Lindquist, J.M.Thevelein, M.Ellison, S.K.Lemmon, M.Hochstrasser and T.Schmelzle for plasmids, F.Estruch for strains, B.Vallée for technical help and advice, Claudio De Virgilio for advice, E.I.Pécheur and members of the Riezman laboratory for helpful comments on the manuscript.
References
- Amoros M. and Estruch,F. (2001) Hsf1p and Msn2/4p cooperate in the expression of Saccharomyces cerevisiae genes HSP26 and HSP104 in a gene- and stress type-dependent manner. Mol. Microbiol., 39, 1523–1532. [DOI] [PubMed] [Google Scholar]
- Attfield P.V. (1987) Trehalose accumulates in Saccharomyces cerevisiae during exposure to agents that induce heat shock response. FEBS Lett., 225, 259–263. [DOI] [PubMed] [Google Scholar]
- Bell W., Sun,W., Hohmann,S., Wera,S., Reinders,A., De Virgilio,C., Wiemken,A. and Thevelein,J.M. (1998) Composition and functional analysis of the Saccharomyces cerevisiae trehalose synthase complex. J. Biol. Chem., 273, 33311–33319. [DOI] [PubMed] [Google Scholar]
- Boy-Marcotte E., Lagniel,G., Perrot,M., Bussereau,F., Boudsocq,A., Jacquet,M. and Labarre,J. (1999) The heat shock response in yeast: differential regulations and contributions of the Msn2p/Msn4p and Hsf1p regulons. Mol. Microbiol., 33, 274–283. [DOI] [PubMed] [Google Scholar]
- Bucciantini M. et al. (2002) Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature, 416, 507–511. [DOI] [PubMed] [Google Scholar]
- Buede R., Rinker-Schaffer,C., Pinto,W.J., Lester,R.L. and Dickson,R.C. (1991) Cloning and characterization of LCB1, a Saccharomyces gene required for biosynthesis of the long-chain base component of sphingolipids. J. Bacteriol., 173, 4325–4332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L., Watt,R. and Piper,P.W. (1994) Polyubiquitin gene expression contributes to oxidative stress resistance in respiratory yeast (Saccharomyces cerevisiae). Mol. Gen. Genet., 243, 358–362. [DOI] [PubMed] [Google Scholar]
- Chung N., Jenkins,G., Hannun,Y.A., Heitman,J. and Obeid,L.M. (2000) Sphingolipids signal heat-stress-induced ubiquitin-dependent proteolysis. J. Biol. Chem., 275, 17229–17232. [DOI] [PubMed] [Google Scholar]
- Ciechanover A. (1994) The ubiquitin–proteasome proteolytic pathway. Cell, 79, 13–21. [DOI] [PubMed] [Google Scholar]
- Ciechanover A., Orian,A. and Schwartz,A.L. (2000) Ubiquitin-mediated proteolysis: biological regulation via destruction. BioEssays, 22, 442–451. [DOI] [PubMed] [Google Scholar]
- Dickson R.C., Nagiec,E.E., Skrzypek,M., Tillman,P., Wells,G.B. and Lester,R.L. (1997) Sphingolipids are potential heat stress signals in Saccharomyces. J. Biol. Chem., 272, 30196–30200. [DOI] [PubMed] [Google Scholar]
- Emter R., Heese-Peck,A. and Kralli,A. (2002) ERG6 and PDR5 regulate small lipophilic drug accumulation in yeast cells via distinct mechanisms. FEBS Lett., 521, 57–61. [DOI] [PubMed] [Google Scholar]
- Finley D., Ozkaynak,E. and Varshavsky,A. (1987) The yeast polyubiquitin gene is essential for resistance to high temperatures, starvation and other stresses. Cell, 48, 1035–1046. [DOI] [PubMed] [Google Scholar]
- Fraser J., Luu,H.A., Neculcea,J., Thomas,D.Y. and Storms,R.K. (1991) Ubiquitin gene expression: response to environmental changes. Curr. Genet., 20, 17–23. [DOI] [PubMed] [Google Scholar]
- Friant S., Zanolari,B. and Riezman,H. (2000) Increased protein kinase or decreased PP2A activity bypasses sphingoid base requirement in endocytosis. EMBO J., 19, 2834–2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friant S., Lombardi,R., Schmelzle,T., Hall,M.N. and Riezman,H. (2001) Sphingoid base signaling via Pkh kinases is required for endocytosis in yeast. EMBO J., 20, 6783–6792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover J.R. and Lindquist,S. (1998) Hsp104, Hsp70 and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell, 94, 73–82. [DOI] [PubMed] [Google Scholar]
- Guarente L. (1983) Yeast promoters and lacZ fusions designed to study expression of cloned genes in yeast. Methods Enzymol., 101, 181–191. [DOI] [PubMed] [Google Scholar]
- Heinemeyer W., Kleinschmidt,J.A., Saidowsky,J., Escher,C. and Wolf,D.H. (1991) Proteinase yscE, the yeast proteasome/multicatalytic–multifunctional proteinase: mutants unravel its function in stress induced proteolysis and uncover its necessity for cell survival. EMBO J., 10, 555–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrick J.P. and Hartl,F.U. (1993) Molecular chaperone functions of heat-shock proteins. Annu. Rev. Biochem., 62, 349–384. [DOI] [PubMed] [Google Scholar]
- Hochstrasser M., Ellison,M.J., Chau,V. and Varshavsky,A. (1991) The short-lived MAT alpha 2 transcriptional regulator is ubiquitinated in vivo. Proc. Natl Acad. Sci. USA, 88, 4606–4610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath A. and Riezman,H. (1994) Rapid protein extraction from yeast. Yeast, 10, 1305–1310. [DOI] [PubMed] [Google Scholar]
- Irie K., Takase,M., Lee,K.S., Levin,D.E., Araki,H., Matsumoto,K. and Oshima,Y. (1993) MKK1 and MKK2, which encode Saccharomyces cerevisiae mitogen-activated protein kinase-kinase homologs, function in the pathway mediated by protein kinase C. Mol. Cell. Biol., 13, 3076–3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins G.M. and Hannun,Y.A. (2001) Role for de novo sphingoid base biosynthesis in the heat-induced transient cell cycle arrest of Saccharomyces cerevisiae. J. Biol. Chem., 276, 8574–8581. [DOI] [PubMed] [Google Scholar]
- Jenkins G.M., Richards,A., Wahl,T., Mao,C., Obeid,L. and Hannun,Y. (1997) Involvement of yeast sphingolipids in the heat stress response of Saccharomyces cerevisiae. J. Biol. Chem., 272, 32566–32572. [DOI] [PubMed] [Google Scholar]
- Jungmann J., Reins,H.A., Schobert,C. and Jentsch,S. (1993) Resistance to cadmium mediated by ubiquitin-dependent proteolysis. Nature, 361, 369–371. [DOI] [PubMed] [Google Scholar]
- Kamada Y., Jung,U.S., Piotrowski,J. and Levin,D.E. (1995) The protein kinase C-activated MAP kinase pathway of Saccharomyces cerevisiae mediates a novel aspect of the heat shock response. Genes Dev., 9, 1559–1571. [DOI] [PubMed] [Google Scholar]
- Lee D.H. and Goldberg,A.L. (1998) Proteasome inhibitors cause induction of heat shock proteins and trehalose, which together confer thermotolerance in Saccharomyces cerevisiae. Mol. Cell. Biol., 18, 30–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D.H., Sherman,M.Y. and Goldberg,A.L. (1996) Involvement of the molecular chaperone Ydj1 in the ubiquitin-dependent degradation of short-lived and abnormal proteins in Saccharomyces cerevisiae. Mol. Cell. Biol., 16, 4773–4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K.S. and Levin,D.E. (1992) Dominant mutations in a gene encoding a putative protein kinase (BCK1) bypass the requirement for a Saccharomyces cerevisiae protein kinase C homolog. Mol. Cell. Biol., 12, 172–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K.S., Irie,K., Gotoh,Y., Watanabe,Y., Araki,H., Nishida,E., Matsumoto,K. and Levin,D.E. (1993) A yeast mitogen-activated protein kinase homolog (Mpk1p) mediates signalling by protein kinase C. Mol. Cell. Biol., 13, 3067–3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindquist S. (1981) Regulation of protein synthesis during heat shock. Nature, 293, 311–314. [DOI] [PubMed] [Google Scholar]
- Lindquist S. and Kim,G. (1996) Heat-shock protein 104 expression is sufficient for thermotolerance in yeast. Proc. Natl Acad. Sci. USA, 93, 5301–5306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Pastor M.T., Marchler,G., Schuller,C., Marchler-Bauer,A., Ruis,H. and Estruch,F. (1996) The Saccharomyces cerevisiae zinc finger proteins Msn2p and Msn4p are required for transcriptional induction through the stress response element (STRE). EMBO J., 15, 2227–2235. [PMC free article] [PubMed] [Google Scholar]
- Miller J.H. (1972) Experiments in Molecular Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbour, NY. [Google Scholar]
- Miller M.J., Xuong,N.H. and Geiduschek,E.P. (1979) A response of protein synthesis to temperature shift in the yeast Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA, 76, 5222–5225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan D.F. and Lindquist,S. (1995) Mutational analysis of Hsp90 function: interactions with a steroid receptor and a protein kinase. Mol. Cell. Biol., 15, 3917–3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson K.K. and Lemmon,S.K. (1993) Suppressors of clathrin deficiency: overexpression of ubiquitin rescues lethal strains of clathrin-deficient Saccharomyces cerevisiae. Mol. Cell. Biol., 13, 521–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neves M.J. and François,J. (1992) On the mechanism by which a heat shock induces trehalose accumulation in Saccharomyces cerevisiae. Biochem. J., 288, 859–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka H., Tanaka,K., Hirano,H., Fujiwara,T., Kohno,H., Umikawa,M., Mino,A. and Takai,Y. (1995) A downstream target of RHO1 small GTP-binding protein is PKC1, a homolog of protein kinase C, which leads to activation of the MAP kinase cascade in Saccharomyces cerevisiae. EMBO J., 14, 5931–5938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsell D.A. and Lindquist,S. (1993) The function of heat-shock proteins in stress tolerance: degradation and reactivation of damaged proteins. Annu. Rev. Genet., 27, 437–496. [DOI] [PubMed] [Google Scholar]
- Patton J.L., Srinivasan,B., Dickson,R.C. and Lester,R.L. (1992) Phenotypes of sphingolipid-dependent strains of Saccharomyces cerevisiae. J. Bacteriol., 174, 7180–7184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prendergast J.A., Ptak,C., Arnason,T.G. and Ellison,M.J. (1995) Increased ubiquitin expression suppresses the cell cycle defect associated with the yeast ubiquitin conjugating enzyme, CDC34 (UBC3). Evidence for a noncovalent interaction between CDC34 and ubiquitin. J. Biol. Chem., 270, 9347–9352. [DOI] [PubMed] [Google Scholar]
- Ruis H. and Schuller,C. (1995) Stress signaling in yeast. BioEssays, 17, 959–965. [DOI] [PubMed] [Google Scholar]
- Simon J.R., Treger,J.M. and McEntee,K. (1999) Multiple independent regulatory pathways control UBI4 expression after heat shock in Saccharomyces cerevisiae. Mol. Microbiol., 31, 823–832. [DOI] [PubMed] [Google Scholar]
- Slater M.R. and Craig,E.A. (1987) Transcriptional regulation of an hsp70 heat shock gene in the yeast Saccharomyces cerevisiae. Mol. Cell. Biol., 7, 1906–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith B.J. and Yaffe,M.P. (1991) Uncoupling thermotolerance from the induction of heat shock proteins. Proc. Natl Acad. Sci. USA, 88, 11091–11094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K., Matsumoto,K. and Toh-e,A. (1988) Dual regulation of the expression of the polyubiquitin gene by cyclic AMP and heat shock in yeast. EMBO J., 7, 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanolari B., Friant,S., Funato,K., Sutterlin,C., Stevenson,B.J. and Riezman,H. (2000) Sphingoid base synthesis requirement for endocytosis in Saccharomyces cerevisiae. EMBO J., 19, 2824–2833. [DOI] [PMC free article] [PubMed] [Google Scholar]