PANTHER version 6: protein sequence and function evolution data with expanded representation of biological pathways (original) (raw)
Abstract
PANTHER is a freely available, comprehensive software system for relating protein sequence evolution to the evolution of specific protein functions and biological roles. Since 2005, there have been three main improvements to PANTHER. First, the sequences used to create evolutionary trees are carefully selected to provide coverage of phylogenetic as well as functional information. Second, PANTHER is now a member of the InterPro Consortium, and the PANTHER hidden markov Models (HMMs) are distributed as part of InterProScan. Third, we have dramatically expanded the number of pathways associated with subfamilies in PANTHER. Pathways provide a detailed, structured representation of protein function in the context of biological reaction networks. PANTHER pathways were generated using the emerging Systems Biology Markup Language (SBML) standard using pathway network editing software called CellDesigner. The pathway collection currently contains ∼1500 reactions in 130 pathways, curated by expert biologists with authorship attribution. The curation environment is designed to be easy to use, and the number of pathways is growing steadily. Because the reaction participants are linked to subfamilies and corresponding HMMs, reactions can be inferred across numerous different organisms. The HMMs can be downloaded by FTP, and tools for analyzing data in the context of pathways and function ontologies are available at http://www.pantherdb.org.
INTRODUCTION
PANTHER is a publicly available database that relates protein sequence evolution to evolution of protein functions and biological roles (1,2). The data have been generated by computational algorithms and, crucially, expert biologist curation, using an extensive software system for associating ontology terms with phylogenetically-defined subfamilies of proteins. Protein family trees are constructed computationally from sequence data. Nodes in the tree, corresponding to common ancestors of extant family members, are annotated with their inferred functions and roles in biological processes and pathways, based on experiments performed on extant proteins. These inferences are made by expert biologists. These annotated nodes define protein subfamilies, each of which is represented by a hidden Markov model (HMM) (3,4) to allow classification of newly discovered protein sequences.
One of the major recent updates is the improvement of the PANTHER pathway curation software module, resulting in a steady increase in the number of pathways available. This has provided PANTHER with a much richer description of protein function for an increasing number of proteins. There are various related efforts in curating biological pathways, such as the Signal Transduction Knowledge Environment (STKE) (5), Kyoto Encyclopedia of Genes and Genomics (KEGG) (6), MetaCyc (7), Functional Relationship Explorer (FREX) (8) and Reactome (9). KEGG and MetaCyc have collected mostly metabolic pathways, while STKE and Reactome contain well curated, publicly available data on signaling pathways. STKE provides probably the most comprehensive description of signaling pathways, but these pathways are not connected to computationally-accessible data of the participating molecules, such as sequence accession numbers.
There are several common goals of these pathway efforts. The first goal is to make pathways accessible to computation. In the current post-genome-sequencing era, with the large amount of genomic information, data analysis largely relies on computation. It is crucial to make pathways that were traditionally represented in the review literature as simple diagrams, readable by computers. The second goal is to use a standard format to store the pathway information so that the data can be transferred among different databases or shared widely across different software packages. The third goal is to make these pathways accessible to biologists. The PANTHER pathway curation software module has been used to date by 20 expert biologists to represent 130 pathways. All PANTHER pathways are associated to protein sequences in the PANTHER library of protein families and subfamilies. In this way, the pathways are linked directly to molecular evolutionary data, including protein phylogenies, statistical models for protein functional conservation and divergence, and comparative genomics data (1,2). Because PANTHER family and subfamily models have been used to classify all known and predicted protein coding genes in the human, mouse, rat and Drosophila genomes (and code is available for classification of genes from any other organism), users can make use of the PANTHER web services (10) to analyze genomic data in the context of pathways.
IMPROVEMENTS TO PANTHER FAMILIES AND SUBFAMILIES
The process for defining protein families and subfamilies has been described previously (1,2). For PANTHER version 6, we followed the same process, with two main improvements. The first improvement is in the source of protein sequences used to build the protein family trees. The second improvement is the use of a computer-assisted manual curation step to better define the protein family clusters.
Selection of sequences to build protein family trees
One of the changes in version 6 of the PANTHER library is the use of UniProt (11) sequences as training sequences. The change helped us map better to the InterPro models (12,13), and facilitated the integration process of PANTHER to InterPro. For the current version 6.1 of the PANTHER library, the input was the set of all UniProt sequences from 53 different species. These organisms were chosen to ensure that (i) human genes would be covered to the greatest extent possible (nine mammalian species), (ii) all model organism databases with literature-curated protein function information were included (mouse, rat, zebrafish, Caenorhabditis elegans, fruit fly, cellular slime mold, budding yeast, Arabidopsis, Escherichia coli and fission yeast), and (iii) a phylogenetically diverse set of other vertebrates (chicken, frog and puffer fish), invertebrates (mosquito and Entamoeba), plants (Chlamydomonas, kidney bean, maize, potato and rice), fungi (Neurospora and Pneumocystis), protists (Leishmania, Plasmodium and Tetrahymena) and prokaryotes (20 different species).
Curator-defined protein families
Selected sequences were clustered into single-linkage clusters, and a dendrogram was built for each cluster using the UPGMA algorithm as described previously (1). Once we have the dendrograms, we need to divide the dendrogram into protein family clusters. Most clustering methods use a single similarity threshold across the dendrogram. However, pairwise BLAST score thresholds do not necessarily correlate well with the number of positions in a multiple alignment that are reliably aligned (which is the critical parameter for correctly inferring the family tree structure). Therefore, we introduce additional information at this stage, and suggest cuts of the tree based on ‘labels’ given to individual sequences. This algorithm has been published (14). The basicidea is to choose cuts of the tree that correlate best with the labels on the sequences: each subtree should contain as many sequences as possible having the same label, while minimizing the number of sequences with other labels. We can obtain these labels from many sources, but for PANTHER version 6.1 we obtained these labels from families in the previous release (version 5.1) of PANTHER. The computationally defined clusters were manually reviewed and edited if necessary by curators. If a cluster contained sequences of diverse functions or domain architectures, it would be divided into smaller clusters based on the following criteria:
- InterPro classification (12,13);
- previous PANTHER classification;
- protein definition and Gene Ontology (GO) classification;
- curator judgement.
The final clusters after the curation were regarded as family clusters.
IMPROVED AVAILABILITY OF PANTHER DATA
Data available by FTP
An FTP site (ftp://ftp.pantherdb.org/) was created to support the download of PANTHER data. The following data are available for download:
- The entire PANTHER library, including all family and subfamily HMMs in HMMER format.
- A PANTHER library annotation file containing the subfamily name, molecular fuction, biological process and pathway for every subfamily.
- PANTHER HMM scoring tool, which can be used to classify any protein sequence against PANTHER.
- A file containing the mapping of PANTHER families and subfamilies to InterPro models.
- The Systems Biology Markup Language (SBML) files for PANTHER pathways, and a file showing the association of sequences to the pathways.
- The pre-calculated HMM scoring results for the complete proteomes derived from the human, mouse, rat and fruit fly genomes, as well as all of UniProt.
PANTHER integration into InterPro
PANTHER is now an InterPro member database (13). PANTHER subfamily HMMs provide a level of specificity that was previously missing from most of InterPro. To date, 1135 PANTHER models have been integrated by InterPro curators, and integration is now accelerating. The InterProScan software (15,16), freely available at http://www.ebi.ac.uk/InterProScan/, scores query protein sequences against the entire PANTHER HMM library, not just the integrated models.
PANTHER PATHWAY CURATION SOFTWARE MODULE
PANTHER also contains a module for curation of biochemical pathways. This module is similar in many respects to the curation infrastructure and data model developed by both the Reactome and EcoCyc databases. It differs mainly in three ways from these other databases. First, the pathways are used to generate an ontology structure, in which ‘biological macromolecules’ (proteins, genes and RNAs) are treated as ontology terms, or classes, rather than specific instances. This means that more than one protein—from the same organism or a different organism—can potentially play the same given role in a pathway. In contrast, EcoCyc instantiates separate reactions for each protein, and can only represent catalysis reactions, not more generalized reactions such as binding events. Reactome also instantiates separate reactions, but only in different organisms, using computational predictions of orthology. The second main difference is the use of the CellDesigner software (17) for all of the pathway diagrams. This means that users can view a pathway diagram that has an exact, one-to-one correspondence with the computational, ‘back-end’ representation. CellDesigner has also proved to be an unsurpassed curation tool: biologist curators simply draw the pathway using a palette of symbols, and CellDesigner automatically creates a computational representation that is compatible with the SBML standard (18). The third major difference is that the curation software is designed to be simple enough to be used directly by bench biologists after a brief training course. All other pathway databases we are aware of employ highly trained curators, who of course cannot be experts in all areas of biology. The current set of PANTHER pathways have been curated by 20 different external experts from the scientific community; they must only have demonstrated their expertise with publications in the relevant field.
PANTHER pathway ontology
The PANTHER pathway ontology uses controlled vocabulary to describe pathways and their components. Below is a list of four key classes included in the ontology. The relationships among the four classes are illustrated in Figure 1.
Figure 1.
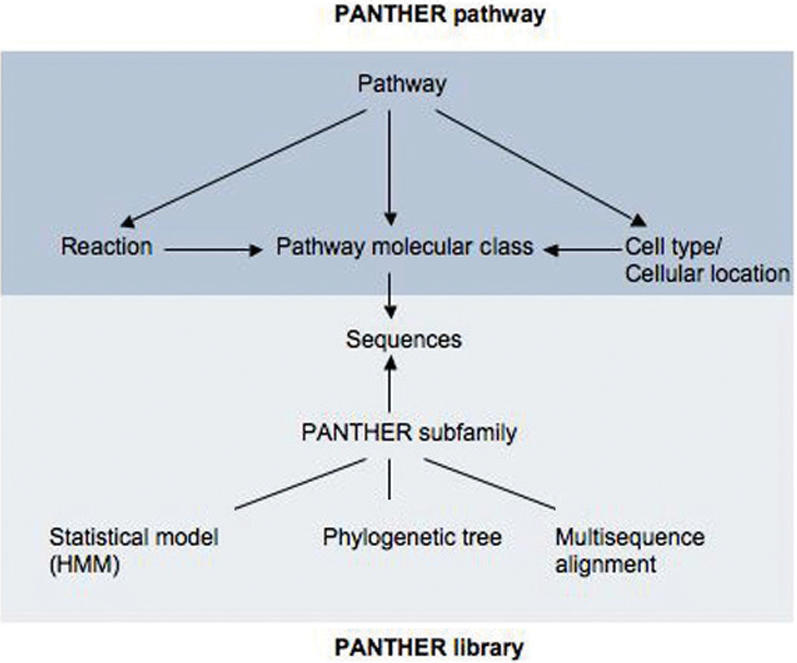
PANTHER pathway ontology and its relationship with PANTHER family and subfamily library. Each arrow indicates a has_part relationship. In the diagram, pathway has_part molecule classes, reactions and compartments, reaction contains molecule classes, and compartment has_part molecule classes. Since reaction and pathway can be across different compartment, their relationship to it is through molecule classes. The pathway is linked to PANTHER family/subfamily library through the association of molecule classes with sequences in the library. Since these sequences belong to the subfamilies, which are represented by statistical models (HMM), phylogenetic trees, and multisequence alignment (MSA), pathway is indirectly linked to the information.
Pathway class
A pathway is a collection of biological molecules, and the reactions in which they participate. Each PANTHER pathway includes only well-characterized and documented reactions and relationships. The scope of the pathways is similar to those documented in textbooks or review articles. The pathways are as representative and inclusive as possible, especially for regulatory pathways. Unless a pathway is sufficiently well-established to appear in a textbook, we require at least 3 references to support the overall structure and boundaries of the pathway.
Molecule class
Each molecule class represents a specific class of molecules that play the same mechanistic role within a pathway. The molecule subclasses are those supported by CellDesigner: proteins, genes/DNA, RNA, small organic or inorganic molecules and ions. If a molecule is a protein, gene, or transcribed RNA, it is associated with protein sequences in the PANTHER protein family trees by manual curation (see below). In these cases, a molecule is typically a group of homologous proteins across various organisms that participate in the same specific biochemical reactions within the pathway, e.g. the molecule ‘JAK’ in the pathway ‘JAK–STAT pathway’ includes JAK1, JAK2, JAK3 and TYK2 in vertebrates (Figure 2).
Figure 2.

Reactions involving the Janus kinase (JAK, in yellow) in the JAK–STAT signaling pathway (accession no. P00038), and the phylogenetic tree for the JAK2 subfamily (accession no. PTHR23256:SF46,SF49,SF282,SF283)—JAK kinases comprise a monophyletic tree of four subfamilies: JAK1, JAK2, JAK3 and TYK2.
Typically, we capture the following information for each protein, gene/DNA or transcribed RNA molecule class:
- Name—the name that appears on the pathway diagram. It is usually an acronym or a short version of the full name, e.g. MAPK.
- Full name—the complete, more descriptive version of the name, e.g. Mitogen-activated protein kinase.
- Synonyms—all other names used to describe the molecule class, e.g. MAP kinase.
- Definition—a short description of the molecule class.
- Reference—literature references, usually OMIM entries or review articles, are captured at this level to support the involvement of the molecule class in the pathway. However, it is not a requirement. More references are captured when sequences are associated to the molecule class.
Reaction class and relationships
The pathway ontology captures reactions among various molecule classes, again using the set of molecular state transition classes from CellDesigner. Typical examples include transition, transport, complex formation/dissociation, catalysis, transcriptional activation/inhibition, etc.
Based on the reactions, we derive relationships among various molecule classes. For example, if a kinase catalyzes a transition of a protein from a non-phosphorylated state to a phosphorylated state, the kinase is upstream_of, and phosphorylates the protein. Typical relationships include: upstream, downstream, phosphorylates, dephosphorylates, acetylates, ubiquitinates and methylates.
Cell type or subcellular component
Each biochemical reaction is generally associated with a particular cell type or subcellular component where the reaction takes place. Currently, the cell type or component is free text entered by the curator, but we are in the process of enforcing the use of cellular component ontology terms from the GO.
Pathway diagrams
Each PANTHER pathway has a corresponding pathway diagram created by the curator using CellDesigner. These are illustrations that:
- graphically represent pathways while capturing structured data;
- use controlled graphic notation to illustrate molecules, and reactions in pathways;
- capture comprehensive molecular events of the pathways;
- are stored in a standard format so that the data can be easily parsed, or shared by different software packages.
Curators of PANTHER pathways have used a special pathway editing tool, CellDesigner (17,19). The software specializes in capturing molecular level events for pathways, and storing the data in SBML format (18).
Protein sequence association
If a pathway molecule is a protein, gene or transcript, it is associated with protein sequences in the PANTHER database that are used to build the PANTHER protein family trees, and the family and subfamily HMM models (2). This process is manual, and is very similar to the process of GO annotation performed by a number of GO Consortium member databases (20). For each annotation, a confidence code must be selected from the list defined by the GO Consortium (20) and a PubMed identifier of the source of the literature references is provided as evidence. Curators are allowed to associate orthologous or even paralogous sequences to the molecule class without experimental evidence, using the inferred by sequence similarity (ISS) evidence code.
Literature references
References are captured at three levels. First, each pathway as a whole requires a reference. For signaling pathways, at least three references, usually review papers, are required in order to provide a more objective view of the scope of the pathway. For metabolic pathways, a textbook reference is usually sufficient. Second, references are often associated to each molecule class in the pathway. Most of these references are OMIM records (21) or review papers. Third, references are provided to support association of specific protein sequences with a particular molecule class, e.g. the SWISS-PROT sequence P53_HUMAN is annotated as an instance of the molecule class ‘P53’ appearing in the pathway class ‘P53 pathway’. These are usually research papers that report the experimental evidence that a particular protein or gene participates in the reactions represented in the pathway diagram.
PANTHER PATHWAY CURATION PROCESS
The PANTHER pathway curation process was carefully designed to capture molecular events and biochemical reactions of the pathways (Figure 3). Curators were recruited from various universities across the nation with significant expertise and experience in the field of the pathways they were to curate. The candidates had to meet at least one of the following three criteria in order to curate a signaling pathway: (i) their current research project involves the pathway, (ii) they are an author of a review paper about the pathway, (iii) their PhD thesis was focused on the pathway.
Figure 3.

Schematic illustration of PANTHER pathway building process.
The curation process is divided into two phases, which is schematically illustrated in Figure 3. Before the curation process begins, the biologist is given a detailed manual describing the process, and a real-time, interactive training presentation remotely via WebEx and telephone. In the pathway diagram and ontology generation step, a curator draws a pathway using CellDesigner. The pathway is reviewed by the Curation Coordinator to ensure that the proper symbols have been used for each biological molecule (macromolecule or small molecule), and for the transitions between molecular states (e.g. from a two free monomers to a bound heterodimer, or from extracellular to intracellular). Literature references must be provided for the pathway. The CellDesigner file that is created during this step adheres to SBML format, so all reactions can be read by a standard SBML parser. We have developed a parser that reads the SBML and uses the information to create a pathway ontology, which is then stored in the PANTHER Pathway curation database, implemented in Oracle. In the sequence annotation step, the curator works with a direct web interface to the curation database. The interface displays each of the ontology classes (terms) that correspond to a protein, mRNA or gene, e.g. P53 (protein) or JAK (gene). At this point, these are simply the terms the curator had used to name the classes in the CellDesigner pathway diagram. During the sequence annotation stage, the curator associates each term with individual protein sequences that are instances of the class as described above.
STATISTICS FOR THE CURRENT VERSION OF PANTHER
The PANTHER library of protein families and subfamilies (version 6.1) contains 221 609 UniProt sequences from 53 organisms, grouped into 5546 families. These families are divided further into 24 561 subfamilies.
The current PANTHER Pathway (version 1.3) contains 130 pathways, 2089 different protein, gene/DNA and RNA molecule classes. Most of the pathways are signaling pathways. There are 19 132 UniProt training sequences directly associated with the pathway components, using 3831 references.
Acknowledgments
We would like to thank Dr Hiroaki Kitano at the Systems Biology Institute in Japan for providing us with the CellDesigner source code for our development of the CellDesigner Lite applet for viewing pathways over the web, Akira Funahashi and Yukiko Mitsuoka at Systems Biology Institute in Japan for all the technical support they provide on CellDesigner software. We thank Orna Avsian-Kretchmer, Anita Bandrowski, Ami Bhatt, Kevin Corbit, Colin Davidson, Robert Del Vecchio, Adam Douglass, Caroline Heckman, Karen Ho, Janet Iwasa, Erica Jackson, Richard Ledwidge, Arnold Levine, Mark McCormick, Larisa Nonn, Rachel Ozer, Anand Sethuraman, Kevin Slep, Leon Su, Christina Wilson and Jianbo Yue for their expert curation of pathways and library. Funding to pay the Open Access publication charges for this article was provided by SRI International.
Conflict of interest statement. None declared.
REFERENCES
- 1.Mi H., Lazareva-Ulitsky B., Loo R., Kejariwal A., Vandergriff J., Rabkin S., Guo N., Muruganujan A., Doremieux O., Campbell M.J., et al. The PANTHER database of protein families, subfamilies, functions and pathways. Nucleic Acids Res. 2005;33:D284–D288. doi: 10.1093/nar/gki078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thomas P.D., Campbell M.J., Kejariwal A., Mi H., Karlak B., Daverman R., Diemer K., Muruganujan A., Narechania A. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res. 2003;13:2129–2141. doi: 10.1101/gr.772403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Krogh A., Brown M., Mian I.S., Sjolander K., Haussler D. Hidden Markov models in computational biology. Applications to protein modeling. J. Mol. Biol. 1994;235:1501–1531. doi: 10.1006/jmbi.1994.1104. [DOI] [PubMed] [Google Scholar]
- 4.Eddy S.R. Hidden Markov models. Curr. Opin. Struct. Biol. 1996;6:361–365. doi: 10.1016/s0959-440x(96)80056-x. [DOI] [PubMed] [Google Scholar]
- 5.Gough N.R. Science's signal transduction knowledge environment: the connections maps database. Ann. N. Y. Acad. Sci. 2002;971:585–587. doi: 10.1111/j.1749-6632.2002.tb04532.x. [DOI] [PubMed] [Google Scholar]
- 6.Kanehisa M., Goto S., Hattori M., Aoki-Kinoshita K.F., Itoh M., Kawashima S., Katayama T., Araki M., Hirakawa M. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 2006;34:D354–D357. doi: 10.1093/nar/gkj102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caspi R., Foerster H., Fulcher C.A., Hopkinson R., Ingraham J., Kaipa P., Krummenacker M., Paley S., Pick J., Rhee S.Y., et al. MetaCyc: a multiorganism database of metabolic pathways and enzymes. Nucleic Acids Res. 2006;34:D511–D516. doi: 10.1093/nar/gkj128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fukuda K.I., Yamagata Y., Takagi T. FREX: a query interface for biological processes with hierarchical and recursive structures. In Silico Biol. 2004;4:63–79. [PubMed] [Google Scholar]
- 9.Joshi-Tope G., Gillespie M., Vastrik I., D'Eustachio P., Schmidt E., de Bono B., Jassal B., Gopinath G.R., Wu G.R., Matthews L., et al. Reactome: a knowledgebase of biological pathways. Nucleic Acids Res. 2005;33:D428–D432. doi: 10.1093/nar/gki072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thomas P.D., Kejariwal A., Guo N., Mi H., Campbell M.J., Muruganujan A., Lazareva-Ulitsky B. Applications for protein sequence-function evolution data: mRNA/protein expression analysis and coding SNP scoring tools. Nucleic Acids Res. 2006;34:W645–W650. doi: 10.1093/nar/gkl229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu C.H., Apweiler R., Bairoch A., Natale D.A., Barker W.C., Boeckmann B., Ferro S., Gasteiger E., Huang H., Lopez R., et al. The Universal Protein Resource (UniProt): an expanding universe of protein information. Nucleic Acids Res. 2006;34:D187–D191. doi: 10.1093/nar/gkj161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mulder N.J., Apweiler R., Attwood T.K., Bairoch A., Bateman A., Binns D., Biswas M., Bradley P., Bork P., Bucher P., et al. InterPro: an integrated documentation resource for protein families, domains and functional sites. Brief Bioinform. 2002;3:225–235. doi: 10.1093/bib/3.3.225. [DOI] [PubMed] [Google Scholar]
- 13.Mulder N.J., Apweiler R., Attwood T.K., Bairoch A., Bateman A., Binns D., Bradley P., Bork P., Bucher P., Cerutti L., et al. InterPro, progress and status in 2005. Nucleic Acids Res. 2005;33:D201–D205. doi: 10.1093/nar/gki106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lazareva-Ulitsky B., Diemer K., Thomas P.D. On the quality of tree-based protein classification. Bioinformatics. 2005;21:1876–1890. doi: 10.1093/bioinformatics/bti244. [DOI] [PubMed] [Google Scholar]
- 15.Zdobnov E.M., Apweiler R. InterProScan—an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001;17:847–848. doi: 10.1093/bioinformatics/17.9.847. [DOI] [PubMed] [Google Scholar]
- 16.Quevillon E., Silventoinen V., Pillai S., Harte N., Mulder N., Apweiler R., Lopez R. InterProScan: protein domains identifier. Nucleic Acids Res. 2005;33:W116–W120. doi: 10.1093/nar/gki442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kitano H. A graphical notation for biochemical networks. Biosilico. 2003;1:169–176. [Google Scholar]
- 18.Hucka M., Finney A., Sauro H.M., Bolouri H., Doyle J.C., Kitano H., Arkin A.P., Bornstein B.J., Bray D., Cornish-Bowden A., et al. The systems biology markup language (SBML): a medium for representation and exchange of biochemical network models. Bioinformatics. 2003;19:524–531. doi: 10.1093/bioinformatics/btg015. [DOI] [PubMed] [Google Scholar]
- 19.Kitano H., Funahashi A., Matsuoka Y., Oda K. Using process diagrams for the graphical representation of biological networks. Nat. Biotechnol. 2005;23:961–966. doi: 10.1038/nbt1111. [DOI] [PubMed] [Google Scholar]
- 20.Gene Ontology Consortium. The Gene Ontology (GO) project in 2006. Nucleic Acids Res. 2006;34:D322–D326. doi: 10.1093/nar/gkj021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hamosh A., Scott A.F., Amberger J.S., Bocchini C.A., McKusick V.A. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005;33:D514–D517. doi: 10.1093/nar/gki033. [DOI] [PMC free article] [PubMed] [Google Scholar]